
Metal-free ionic liquid-derived electrocatalyst for high-performance oxygen reduction in acidic and alkaline electrolytes†
K.
Elumeeva
*,
N.
Fechler
,
T. P.
Fellinger
and
M.
Antonietti
Colloid Chemistry Department, Max-Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: karina.elumeeva@mpikg.mpg.de
First published on 16th July 2014
Abstract
We report herein a new simplified approach to metal-free N-doped carbon aerogels made from ionic liquids by a bottom-up “salt templating” strategy, which are known to be promising electrocatalytic materials. An optimized pore transport system as well as a high catalytically active surface allowed the ionic liquid-derived carbons to possess favourably high electrocatalytic performance in oxygen reduction under alkaline and, not found for similar materials made by other pathways, under technically more relevant acidic conditions. Even in acid, the performance favourably compares with a commercial Pt-based catalyst.
The development of low-cost, durable and high-performance alternatives to the current precious platinum-based electrocatalysts can accelerate the successful commercialization of fuel cells (FCs) as an alternative power source. The hindered oxygen reduction reaction (ORR) on the cathode is the major bottleneck of FCs and requires around ten times more Pt than the hydrogen oxidation on the anode.1–3 Despite numerous efforts, creation of cheap and viable catalyst is still highly desirable. Various types of non-precious ORR electrocatalysts have been proposed in the recent years, and currently nitrogen-doped carbon-based materials have the best chances to replace precious-metal containing analogues.4–7 On the one hand this is due to their high electrical conductivity, their tolerance to harsh conditions, as well as their inertness against catalyst poisoning. On the other hand, sufficiently high concentrations of active sites can be generated by chemical means while the pore transport characteristics can be optimized through variation of the precursors as well as synthesis parameters.1,3,8 In order to approach the activity level of a Pt-based catalyst, N-doped carbons (NCs) can be additionally modified by introduction of different non-precious metals, such as Fe, Co, Mn,9–11 as well as by incorporation of non-metal heteroatoms, i.e. S, P, B, F.12–16 It is supposed that incorporation of non-metal heteroatoms into the graphene-like structure can cause drastic changes in charge localization and, thus the appearance of positively charged species participating in molecular oxygen chemisorption and reduction.4,6,17,18 However, the nature of active sites and factors influencing the ORR activity of metal-free electrocatalysts still remain to be clarified and are widely discussed in the literature. Furthermore, the efficient synthesis of these materials remains challenging.
Today, extensive work focusses on metal-free electrocatalysts based on carbon blacks,15 graphene,17,19,20 carbon nanotubes4,7,21 and other carbon modifications.22–24 For an excellent review on the subject, we want to point at ref. 25.
Nevertheless, most of these metal-free catalysts are able to reduce oxygen only under alkaline conditions. Performance of such materials was found to be very low under the practically more relevant acidic conditions,13,14 which is problematic for the development of metal-free FCs based on classical proton-exchange membranes.2,26
Our previous findings showed that nonvolatile ionic liquids (ILs) can be successfully utilized as NCs precursor.27,28 Depending on the synthesis approach, it is possible to obtain the ionic liquid-derived NCs with micro-, meso- and macroporous structure.24,29,30 A number of metal-free catalysts recently reported and with promising activities miss a clear diffusion-limited current regime i.e. show kinetic limitation over the whole potential range.4,17,31,32 Such behaviour is often seen for graphene based catalysts and likely to result from mass transport issues. This correlation between the morphology of self-supporting NC-catalysts and their ORR performance suggests that the optimization of transport pore system along with increase of the catalytically active surface area can provide optimized catalytic activity.
One of the recent achievements that allowed us to simplify the synthesis and at the same time tune the pore structure of nanocarbons is the “salt templating” method.29,33 It was observed that use of non-carbonizable salt melts as a solvent and simultaneously as a template provides NCs with different morphology ranging from microporous to aerogel-like materials. Herein, we therefore employed the bottom-up technique of salt templating to obtain metal-free NC electrocatalysts from ionic liquids. It will be shown that outstanding ORR performance with a well-defined diffusion limited region is found in both acidic and alkaline media, on a competitive level with Pt-based commercial catalysts. These NCs possess an aerogel-like structure with surface areas up to 1800 m2 g−1, and are capable to catalyze a four-electron oxygen reduction to water in acid and base. The new metal-free IL-derived ORR electrocatalyst obtained in this work possess high potential to be applied in fuel cells.
The simple one-pot synthesis involved the carbonization of ionic liquid 1-ethyl-3-methylimidazolium dicyanamide (Emim-dca, Fig. S1†) mixed with the salt melt (NaCl/ZnCl2, denoted as NZ) at 1000 °C in nitrogen atmosphere. Then, highly porous carbon products were washed with water and dried in vacuum. Using this synthesis approach it is possible to achieve comparably high carbonization yields of up to 45 wt%. It is worth noting that the salts could be easily captured and recovered, if needed. The carbon samples under investigation are named as NC-NZ-X, where X is the mass ratio of salt melt (NZ) to IL. General characteristics of NCs can be found in Table 1.
| Sample | N content [wt%] | C/N ratio | S BET [m2 g−1] | Pore volume [cm3 g−1] |
|---|---|---|---|---|
| a ΔE1/2 is the negative shift of half-wave potential in comparison with commercial 20% Pt/C catalyst. | ||||
| NC-NZ-13 | 3.8 | 20.4 | 1550 | 2.1 |
| NC-NZ-10 | 3.7 | 18.6 | 1770 | 2.9 |
| NC-NZ-6 | 4.5 | 16.3 | 1410 | 1.2 |
As depicted in the scanning electron microscopy (SEM) images (Fig. 1a–c), a striking correlation between the morphology of the final NCs and the initial amount of salt melt used for their production is observed. In the case of NC-NZ-6, a more compact structure with small pores is formed (Fig. 1a). Further increase of salt melt amount results in formation of aerogel-like NCs which are constituted of connected small primary nanoparticles (with less than 20 nm diameter) creating an extended pore transport system. These observations are in accordance with the nitrogen sorption data shown in Fig. 1d.
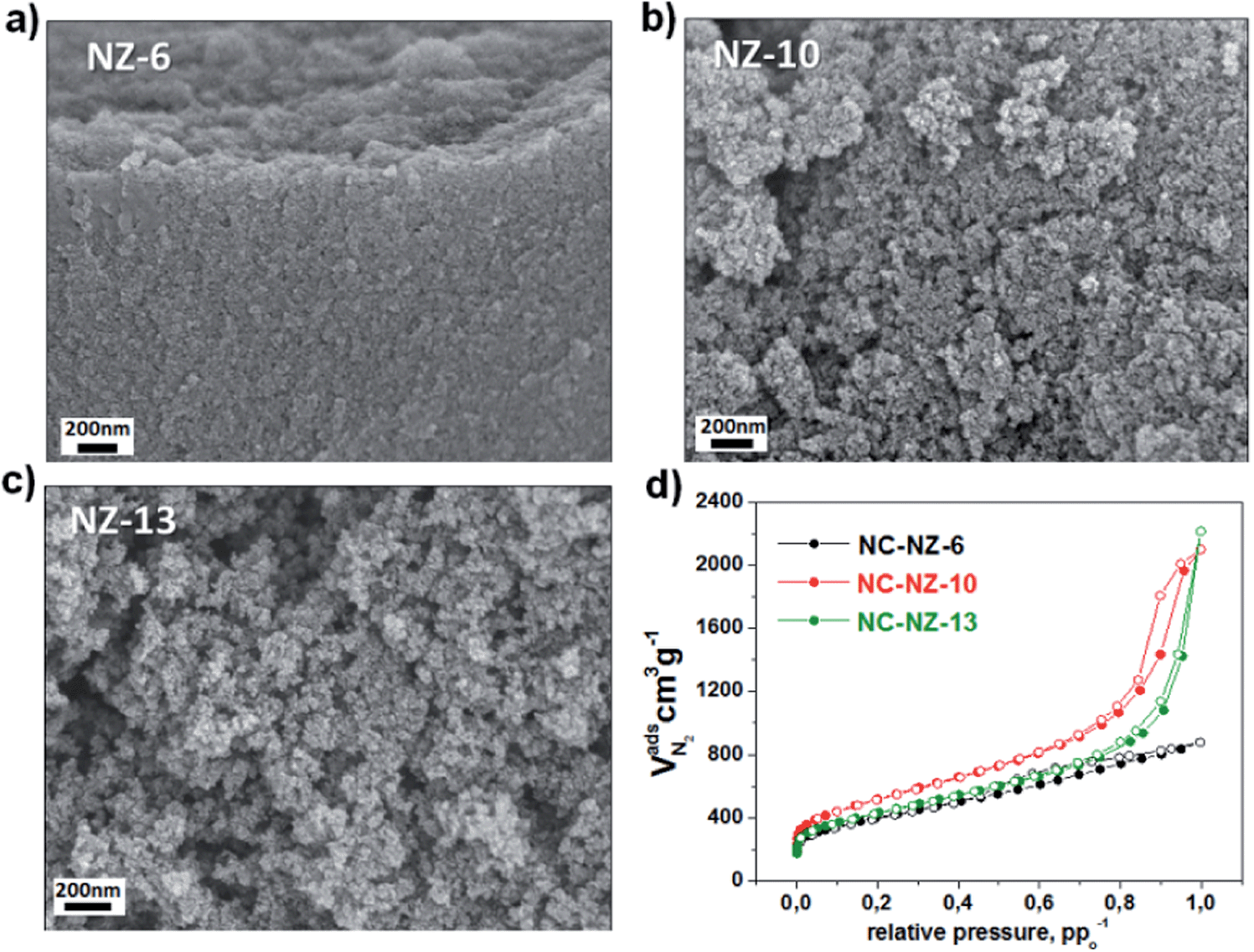 | ||
Fig. 1 SEM images (a–c) and nitrogen sorption isotherms (d) of NCs obtained using different mass ratios of ionic liquid and NZ salt melt: (a) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6; (b) 1:10; (c) 1:13. :6; (b) 1:10; (c) 1:13. | ||
In general, for all samples a strong nitrogen uptake at low relative pressure due to a micropore contribution is observed. This means that the primary nanoparticles are microporous as such. Furthermore, in each case the particles possess a comparable micropore size (Fig. S2†). The more pronounced gas uptake at high relative pressures for the NC-NZ-10 and NC-NZ-13 points to the larger interstitial room between the particles, as compared to NC-NZ-6. In other words: in NC-NZ-10 the larger transport pore system is in the upper mesopore region, while NC-NZ-13 is rather macroporous. Essentially, with increasing salt amount the connectivity of the particles (inter-particle distance) increases while the inherent porosity of the particles itself (intra-particular) is not affected. This is important for a following explanation of the high activity of NC-NZ-13. The surface areas of all NCs, calculated according to the Brunauer–Emmett–Teller (BET) model, are found to be in the range of 1410–1770 m2 g−1 (Table 1), thus “salt templating” allows the direct synthesis of high surface area carbons. The morphology change with increasing NZ/IL ratio can be explained by the course of phase separation during the IL carbonization in the presence of excess solvent – the salt melt. When the concentration of carbon nuclei is high, i.e. at lower amounts of salt, macroscopic phase separation cannot take place, and their fast agglomeration leads to the formation of a continuous porous solid phase with only little transport porosity. Lower carbon concentrations lead to partial demixing from a pure salt melt phase, which constitutes later the visible transport pore system.
The bulk elemental analysis showed overall nitrogen content of 3.8 wt% (Table 1). Furthermore, the surface species of the NCs were studied using X-ray photoelectron spectroscopy analysis (XPS) which shows the additional presence of oxygen (6.9 at%) and zinc (2.0 at%) (Fig. 2a). Comparable sample series in our lab with even higher Zn-contents show in all cases no specific influence of Zn2+/ZnO on the electrochemical performance. Compared to former reports the influence of Zn on the carbon nanostructure and type of nitrogen-sites is obvious, however a direct chemical contribution of Zn-species to the ORR performance can to our opinion be neglected.34
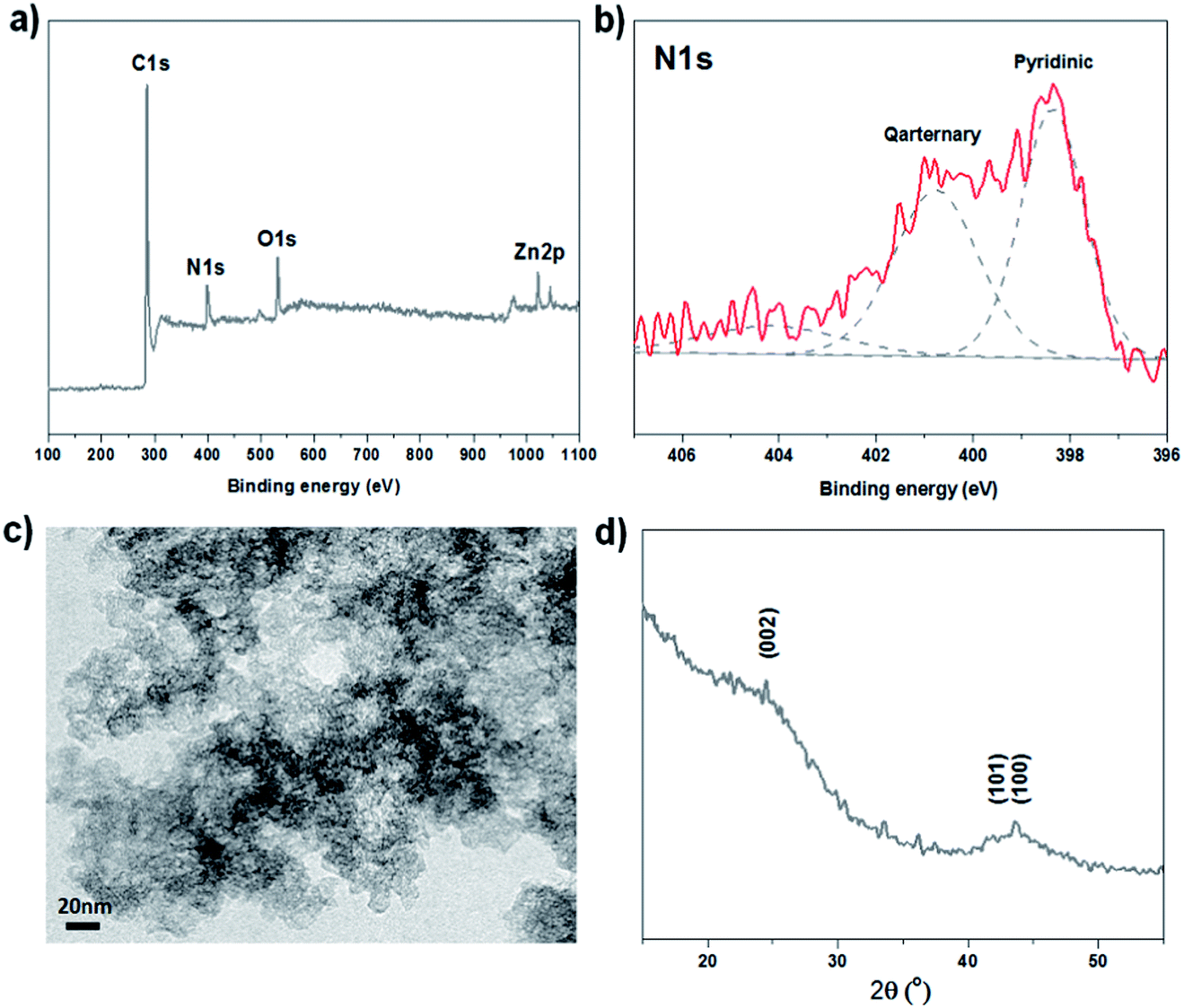 | ||
| Fig. 2 Main structural characterization of NC-NZ-13: (a) XPS survey spectrum; (b) high resolution XPS spectrum of N 1s region; (c) high resolution TEM image showing aerogel-like structure in the nanometer range; (d) XRD pattern. | ||
According to high resolution N 1s spectra, NC-NZ-13 contains pyridinic (at 398.5 eV), quaternary (400.9 eV) and oxidized (402–405 eV) nitrogen species.5,27 The additional presence of pyrrolic nitrogen (at 400.5 eV) cannot be excluded since the signal is very close to the quaternary species. Interestingly, the relative amount of pyridinic and graphitic nitrogen is opposite to the bulk pyrolysis product of the ionic liquid.27 Coordination of zinc throughout the synthesis may stabilize nitrogen edge-sites and therefore also vary the catalytic activity. The nominal surface N concentration was found to be 3.9 at% (Fig. 2b). With regard to the catalyst application, it is to emphasize that no further transition metals like Fe were detected, which is also in accordance with the data obtained by inductively coupled plasma optical emission spectrometry (ICP-OES) and energy dispersive X-ray spectroscopy (EDX, Fig. S3†).
The high resolution transmission electron microscopy (HR TEM) images of NC-NZ-13 (Fig. 2c) reveal the aerogel-like structure formed by agglomeration of small carbon particles. This is consistent with wide angle X-ray diffraction (XRD) data (Fig. 2d) with two broad peaks of low intensity which can be ascribed to the (002) and (100) planes of graphitic carbon. Such high surface area carbons have only very small stacking heights, thus explaining the width of their peaks due to the nanostructure.
The ORR activity of NC-NZ samples was investigated using rotating disk/ring-disk electrode (RDE/RRDE) measurements in a three-electrode electrochemical cell. A Pt wire was used as counter electrode and Ag/AgCl (sat. KCl) electrode as a reference. The cyclic voltammograms (CVs) as well as linear sweep voltammetry (LSV) curves recorded in nitrogen and oxygen saturated 0.05 M H2SO4 and 0.1 M KOH solutions are presented in Fig. 3. For comparison, the commercial 20% Pt/C catalyst with the same load was additionally tested. While the CVs recorded in N2 saturated acidic and alkaline solution (Fig. S4†) reveal the absence of any redox processes, the steep slopes indicates the high electrical conductivity typical for ionic liquid-derived N-doped carbons.24,30 However, when NC-NZ catalysts were tested in O2 saturated H2SO4 and KOH, pronounced cathodic ORR peaks are observed with maxima at around 0.3 V and −0.2 V (vs. Ag/AgCl), respectively. To further investigate the ORR kinetics, RDE/RRDE measurements were also performed. The comparative LSV polarization curves of NCs obtained at 1600 r.p.m. (Fig. 3a and b) show the progressive decrease of kinetic limitation. While NC-NZ-6 shows no diffusion limited current, especially in acidic electrolyte, the catalysts synthesized with higher salt amounts reach diffusion limited currents. This is often not achieved by carbon catalysts. Those trends are observed in both alkaline and acidic electrolytes, and we assign this to successively improved accessibility of the active sites, which are in addition less dependent on pH as other n-doped carbons. In alkaline electrolyte NC-NZ-10 and NC-NZ-13 show outstanding activity with a half-wave potential equal to the 20 wt% platinum catalyst at the same loading. In acidic medium, due to the salt melt synthesis, we have an improved activity as compared to silica-templated material24 and generally high performance for a non-metal catalyst.13,14 RDE measurements of NC-NZ-13 catalytic activity at different rotation speeds revealed an electron transfer number of ∼4 calculated from the slopes of Koutecky–Levich (K–L) plots in the potential range of 0.1 V to 0.25 V (H2SO4) and −0.175 V to −0.25 V (KOH).35,36 The KL plots are shown in the ESI (Fig. S5 and S6†). This indicates that the ORR on NC-NZ-13 in both solutions proceeds via the favourable direct four-electron (4e) process.37 The 4e reduction of oxygen to water is also obtained for NC-NZ-10 in KOH. However, in acid conditions the electron transfer number for this sample was calculated to be ∼3.7. Because of the strong dependence of the diffusion limited current on the porosity rather than being related to surface area and surface chemistry of the catalysts alone, we questioned the adequateness of Koutecky–Levich (K–L) analysis for the NZ-samples and performed RRDE measurements to learn more about the mechanism.
 | ||
| Fig. 3 Electrocatalytic activity of NC-NZ catalysts: (a) LSV polarization curves in O2 saturated 0.05 M H2SO4; (b) LSV polarization curves in O2 saturated 0.1 M KOH. LSV curves were recorded at scan rate 10 mV s−1 and at rotation speed 1600 r.p.m.; (c) and (d) hydrogen peroxide yield plots obtained in O2 saturated 0.05 M H2SO4 and 0.1 M KOH, respectively. Inserts – electron transferred number of NC-NZ-13 at different potentials determined from the corresponding RRDE data. For all the RRDE measurements, the catalyst loading is around 0.21 mg cm−2 (including Pt/C catalyst). | ||
In fact, the ring currents mimic the trend in Levich currents and K–L analysis, which indicates the influence of the pore system on the electron transfer number. We would like to highlight that NC-NZ-13 in alkaline medium over the whole potential range produces less than 2.5% H2O2, which is especially at low overpotentials even less than the commercial platinum catalyst (Fig. 3c and d). Moreover, according to RRDE data the transferred electron number was found to be close to 4 at different potentials in both acid and base solutions.
This exceptional electrocatalytic activity of NC-NZ-13 in both electrolytes is attributed to the combination of the aerogel-like structure with an improved pore transport system together with high catalytically active surface. Increased concentration of accessible active sites results from the small size of primary particles along with the relatively little stacking of the graphitic carbon constituting them (the specific surface area can be recalculated into an apparent average stacking height of on average only two graphene layers). Furthermore, the aerogel-like morphology provides a wide interstitial pore system which decreases the diffusion limitations while the particle interconnection is still sufficient to provide high electrical conductivity at the same time. This obviously leads to exceeding of the theoretically possible value of limiting current at a fixed rotation speed, as is clearly observed for NC-NZ-13 in KOH (Fig. 3b). Speculatively, we can attribute that to non-diffusive and more convective mass transport, a suggestion also supported by sheer observation of such high performing electrodes by the naked eye.
Comparing the 3 carbons of our series, NZ-6 to NZ-13, with each other, we see that the activity is not strongly related to the specific surface area, but more directly to the absolute amount of salt throughout the synthesis, i.e. it follows the natural series NZ-6–>NZ-13. This speaks rather directly for the beneficial exposure of the appropriate nitrogen-sites with increasing salt concentration, that are the pyridinic or neighbouring multi-pyridinic sites driving the ORR activity, both in alkaline and acidic conditions.
The ORR active sites of metal-free NCs are classically proposed to appear due to the charge redistribution in carbon atoms induced by the nitrogen doping.4,17,18 The type of nitrogen species incorporated into graphitic carbon network can strongly affect the catalytic performance.17,38 In a very recent paper,39 it was shown by a combination of careful electrochemical measurement and density functional theory that a good descriptor for ORR activity is the adsorption free energy of intermediates, resulting for a variety of species in a “volcano plot”. This also means that, as in metals, both the absolute HOMO energy as well as specific chemical binding motifs have influence on the electrochemical activity. Here, we have to assume that the high concentration of pyridinic N species in the presented NC-NZ-systems is the reason of their high catalytic activity and the ability to drive ORR through 4e mechanism:5,17 this is the identified major chemical difference between salt-melt synthesized carbons and corresponding hard template versions. All based on the same IL-precursors. This would result in the exciting consequence that especially under acidic conditions it is a protonated/proton-sponge version of the pyridinic sites driving higher adsorption free energy of the (usually oppositely charged) oxygenated intermediates. However, the action of N-doping type is still a controversial subject and remains under discussion.6
Besides the high electrocatalytic activity in both media, chronoamperometry further reveals also a comparably high stability, here representatively shown for the best material NC-NZ-13 (Fig. 4). In both H2SO4 and KOH, the carbon-sample showed superior stability compared to the commercial Pt–C standard. Essentially, this further points to the perspective that the herein presented metal-free IL-derived carbons prepared by “salt templating” are highly attractive alternatives to current (precious metal) catalysts in both alkaline and -even more important- acidic electrolytes.
 | ||
| Fig. 4 Chronoamperometric response for ORR on NC-NZ-13 and Pt/C electrodes: (a) in O2-saturated 0.05 M H2SO4 at 0.4 V; (b) in O2-saturated 0.1 M KOH at −0.2 V. Rotation speed, 1600 r.p.m. | ||
Conclusions
In summary, a metal-free electrocatalyst based on ionic-liquid derived N-doped carbons was synthesized which showed in comparison to commercial Pt/C catalyst an exceptional ORR activity and high durability, in the present case in both acid and base electrolytes. Taking advantage of ionic liquid as convenient N-doped carbon precursors, we obtained N-doped carbon aerogels by applying the recently developed “salt template” technique. The described significant improvement of electrocatalytic performance is to our opinion a result of an increased relative amount of open edge nitrogen sites, e.g. pyridinic and multi-pyridinic entities, optimized pore transport system as well as high catalytically active surface area due to the small size of primary particles constituting the aerogel. Specifically, this enabled the generation of well-defined limited currents indicative of low kinetic hindrances. Moreover, the metal-free electrocatalysts developed in this work are capable to catalyze the direct 4e oxygen reduction to water in acidic and alkaline electrolytes in a very clean way. Essentially, this make the IL-derived N-doped carbon aerogels prepared with the salt templating approach highly attractive candidates for future fuel cell catalysts and related energy material applications.Methods
Catalyst preparation and characterization
Emim-dca was purchased from IoLiTec with a purity of >98%. Sodium chloride and zinc chloride (both 99%) were acquired from Sigma Aldrich. All chemicals were used without further purification.The salt mixtures were freshly prepared prior to the synthesis by grinding sodium chloride and zinc chloride (42 mol%). In a typical synthesis Emim-dca (1 g) was thoroughly mixed with the salt mixture (6 g, 10 g, 13 g) prior to the calcination process. The resulting mixtures were placed in ceramic crucibles and heated to 1000 °C with a heating rate of 2.5 K min−1 in a Nabertherm N7/H Chamber Oven. After holding this temperature for 1 h, the samples were allowed to cool to room temperature. All steps were carried out under a constant flow of nitrogen. In order to remove the residual salt porogen, the materials were washed in water for several hours and finally filtrated and dried in vacuum.
TEM images and EDX spectra were obtained using TECNAI G220 S-TWIN transmission electron microscope with LaB6-cathode. Elemental analysis for nitrogen and carbon was accomplished as combustion analysis using a Vario Micro device and for transition metals using Vista-MPX Simultaneous ICP-OES. SEM images were obtained on a JSM7500F (Jeol) instrument. WAXS-patterns were measured on a Bruker D8 Advance instrument using Cu-Kα -radiation. Nitrogen sorption measurements were accomplished with N2 at 77 K after degassing the samples at 150 °C under vacuum for 20 hours using a Quantachrome Quadrasorb SI porosimeter. The apparent surface area was calculated by applying the Brunauer–Emmett–Teller (BET) model to the isotherm data points of the adsorption branch in the relative pressure range p/p0 < 0.3. The pore size distribution was calculated from N2 sorption data using the nonlocal density functional theory (NLDFT) equilibrium model method for slit pores provided by Quantachrome data reduction software QuadraWin Version 5.05. XPS-measurements were made with Thermo ESCALAB 250 spectrometer with a monochromatized Al-Kα X-ray source (200 W).
Electrochemical measurements
The electrocatalyst ink was prepared using 5 mg of catalyst powder, 95 μl of 5% Nafion® solution and 350 μl of pure ethanol. After sonication, 5 μl aliquot was deposited onto well-polished ring-disk electrode (diameter, 5.7 mm; Gamry instrument) and dried in air. The final catalyst loads were 0.21 mgcat cm−2. The same loading of commercial 20 wt% Pt on carbon black (Alfa Aesar) has been used as a reference.The RDE and RRDE measurements were conducted in nitrogen and oxygen saturated 0.1 KOH and 0.05 M H2SO4 solutions using the Ag/AgCl (vs. saturated KCl) electrode as the reference electrode and a platinum wire as the counter electrode. To minimize the influence of the capacitive current, the LSV polarization curves recorded in oxygen saturated solution have been normalized to the curves obtained after N2 purging. The CVs were recorded at scan rate of 100 mV s−1 and the LSV curves – at the scan rate of 10 mV s−1 and different rotation rates. In RRDE experiments, the ring potential was set to 1.3 V for acidic and 0.5 V for alkaline electrolyte. The 4e selectivity was evaluated from the RRDE data according to the equation:
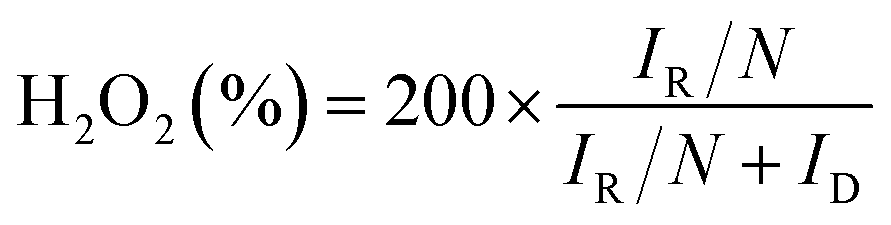

The chronoamperometric measurements were performed in oxygen saturated solutions at 1600 r.p.m. at 0.4 V in H2SO4 and −0.2 V in KOH.
References
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114 CAS.
- M. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- E. J. Biddinger, D. von Deak and U. S. Ozkan, Top. Catal., 2009, 52, 1566 CrossRef CAS.
- M. Zhang and L. Dai, Nano Energy, 2012, 1, 514 CrossRef CAS PubMed.
- A. Morozan, P. Jeqou, M. Pinault, S. Campidelli, B. Jousselme and S. Palacin, ChemSusChem, 2012, 5, 647 CrossRef CAS PubMed.
- C. Médard, M. Lefèvre, J. P. Dodelet, F. Jaouen and G. Lindbergh, Electrochim. Acta, 2006, 51, 3202 CrossRef PubMed.
- K. Parvez, S. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. Feng and K. Muellen, ACS Nano, 2012, 6, 9541 CrossRef CAS PubMed.
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770 CrossRef CAS PubMed.
- Y. M. Tan, C. F. Xu, G. X. Chen, X. L. Fang, N. F. Zheng and Q. J. Xie, Adv. Funct. Mater., 2012, 22, 4584 CrossRef CAS.
- Z. Liu, H. Nie, Z. Yang, J. Zhang, Z. Jin, L. Yang, X. Zhubing and S. Huang, The Royal Society of Chemistry, 2013, 5, 3283 CAS.
- C. H. Choi, S. H. Park and S. I. Woo, J. Mater. Chem., 2012, 22, 12107 RSC.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6(8), 7084 CrossRef CAS PubMed.
- X. Sun, P. Song, Y. Zhang, C. Liu and W. Xing, Sci. Rep., 2013, 3, 2505–2511 Search PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110–3116 CrossRef CAS PubMed.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5(7), 7936 CAS.
- X. Fan, W. T. Zheng and J. L. Kuo, The Royal Society of Chemistry, 2013, 3, 5498 CAS.
- S. Wang, L. Z. Xia, A. Roy, A. D. W. Chang, J. B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209 CrossRef CAS PubMed.
- I. Y. Jeon, H. J. Choi, M. Choi, J. M. Seo, S. M. Jung and M. J. Kim, Sci. Rep., 2013, 3, 1 Search PubMed.
- S. Wang, E. Iyyamperumal, A. Roy, Y. Xue, D. Yu and L. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756 CrossRef CAS PubMed.
- S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593 CrossRef CAS PubMed.
- S. A. Wohlgemuth, T. P. Fellinger, P. Jaker and M. Antonietti, J. Mater. Chem. A, 2013, 1, 4002 CAS.
- W. Yang, T. P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec, J. G. Ji and S. Z. Qiao, Small, 2012, 23, 3550–3566 CrossRef PubMed.
- M. Dresselhaus, G. Crabtree and M. Buchanan, Basic Research Needs for the Hydrogen Economy, U.S. Department of Energy, Washington, DC, 2003 Search PubMed.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87 CrossRef CAS PubMed.
- J. Y. Yuan, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5003 CrossRef CAS.
- N. Fechler, T. P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75 CrossRef CAS PubMed.
- T. P. Fellinger, A. Thomas, J. Yuan and M. Antonietti, Adv. Mater., 2013, 25, 5838 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS PubMed.
- Y. Zhang, K. Fugane, T. Mori, L. Niu and J. Ye, J. Mater. Chem. A, 2012, 22, 6575 RSC.
- N. Fechler, S. A. Wohlgemuth, T. P. Fellinger, P. Jaker and M. Antonietti, J. Mater. Chem. A, 2013, 1, 9418 CAS.
- E. Proetti, F. Jaouen, M. Lefevre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1995, 99, 3411 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: Fundamentals and applications, Wiley, 2011 Search PubMed.
- C. Song and J. Zhang, PEM Fuel Cell Electrocatalysts and Catalyst Layers, Springer, 2008 Search PubMed.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170 CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mh00123k |
| This journal is © The Royal Society of Chemistry 2014 |