
Self-assembly of size-tunable supramolecular nanoparticle clusters in a microfluidic channel†
Carmen
Stoffelen
,
Rajesh
Munirathinam
,
Willem
Verboom
and
Jurriaan
Huskens
*
Molecular Nanofabrication group, MESA+ Institute for Nanotechnology, University of Twente, P. O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: j.huskens@utwente.nl; Fax: +31-534894645
First published on 21st August 2014
Abstract
Supramolecular nanoparticle clusters (SNPCs) have been formed in a microfluidic device by controlling the diffusive mixing of the constituting supramolecular building blocks. Cluster formation between ligand-functionalized silica nanoparticles, dendrimers, and poly(ethylene glycol) (PEG) stopper molecules is induced by the ternary charge-transfer complex formation between cucurbit[8]uril, methyl viologen and naphthol. The resulting SNPC size depends strongly on the stoichiometry of the host and guest binding partners, the competition between multivalent and monovalent naphthol entities, and the microfluidic flow conditions. Variation of the PEG length leads to modulation of its diffusion rate and thus to an additional kinetic control parameter of the SNPC formation process.
Conceptual insightsClusters of nanoparticles, also called network aggregates, form an important class of materials, as they promise the development of multifunctional materials by a toolbox approach. This work integrates a microfluidic assembly strategy for such materials with host–guest recognition between the building blocks to elucidate and deconvolute effects of thermodynamic driving force vs. kinetic control parameters and of diffusion of the components on the formation and stabilization of the core and shell of these materials. The main lessons learned here are: (i) the implementation of a rather slow molecular recognition motif onto multivalent and nanoparticle-based building blocks leads to kinetically controlled cluster self-assembly; (ii) the concept of size control by building block stoichiometry, as is common for soft nanoparticles made under thermodynamical control, is upheld under kinetic control, (iii) microfluidic assembly allows careful control over numbers and local concentrations of interacting building blocks and thus leads to stable and size-controlled clusters, (iv) the fraction of nanoparticles incorporated in clusters is determined by the in-diffusion time of molecular components into the nanoparticle stream, (v) if the diffusion of cluster-terminating stopper molecules is slower than that of the multivalent crosslinking component, leading to an effective temporal decoupling of cluster growth and termination, the cluster size is increased. These design and assembly rules will promote the further development of non-covalent synthesis of well-defined, complex materials. |
Self-assembly is the process by which individual components organize into ordered structures spontaneously, and it is a central theme of nanoscience and nanotechnology.1,2 Self-assembly enables the formation of functional materials in nano- or micrometer dimensions, exclusively based on non-covalent forces such as van der Waals interactions, hydrogen bonding, electrostatic interactions, magnetic interactions and coordination chemistry.3,4 Supramolecular host–guest chemistry offers the ability to engineer desired structures based on a blend of different non-covalent interactions, which thus enables the formation of stable, yet reversible constructs. Inclusion complexes formed by the interaction of cucurbit[8]uril or cucurbit[10]uril with appropriately sized guest molecules are known to lead to high binding affinities, and their size allows the inclusion of two guests leading to ternary complexes.5 Besides supramolecular binding motifs, polymers such as poly(ethylene glycol) (PEG) can be used as structure-directing agents that support the formation of self-assembled functional materials.6 Using heteroternary host–guest complexes and guest-modified PEG, we recently reported the formation of size-tuneable7 and dual responsive8 supramolecular nanoparticles (SNPs) mediated by the interaction of CB[8], methyl viologen (MV), and naphthol (Np), or of CB[8], MV, and azobenzene, respectively.
The modification of inorganic nanoparticles with host and guest moieties has been an established strategy to fabricate nanoparticle networks in aqueous solution.9–11 For example, the clustering of β-cyclodextrin-functionalized silver nanoparticles with aromatic guest molecules provides a sensitive detection method of different isomers by eye.12 The optical, magnetic and biological properties of clustered materials have been shown to depend strongly on their size and conformation.13
The ultimate goal of chemists is to control the properties of a structure prior to synthesis. This requires superior control over the fabrication of the resulting structures, especially in self-assembly processes, because the formation of supramolecular host–guest structures is strongly dependent on thermodynamic and kinetic parameters. We have recently shown that the assembly kinetics of adamantane/cyclodextrin nanoparticle network aggregates can be varied by the flow conditions in a multi-inlet vortex mixer.14 In contrast to the turbulent flow conditions in such a mixer, uniform and well-defined laminar flow is observed within microfluidic reactors.
Microfluidics deals with the manipulation of small volumes of liquids in a microscale channel, and its large applicability and versatility have allowed it to grow into a powerful interdisciplinary technology.15 The micrometer device dimensions provide a large surface-to-volume ratio, rapid heat transfer, low Reynolds numbers and unique mass transport properties which are beneficial in applications such as chemical synthesis,16 medical diagnostics,17 high throughput biological assays,18 controlled synthesis of nano-materials19 and the controlled self-assembly of various nanostructures.20–26 In microfluidic devices, liquid streams from different inlets flow in parallel, and the mixing of the interacting components in those streams is driven by lateral molecular diffusion. This diffusive mixing is strongly dependent on the diffusion properties of the building blocks and the residence time in the microfluidic device, and can therefore be directly regulated by the flow rates of the ingoing fluid streams. For electrostatic interactions, the formation of soft NPs was controlled using hydrodynamic flow focussing, by tuning the residence time, temperature and pressure.27,28 The assembly of gold and iron oxide nanoparticles onto silica nanoparticles has been controlled by varying the flow rates within a multistep microfluidic device.29 The self-assembly of porphyrin architectures has been shown recently to be regulated by the flow field in a micro-flow environment.30
Only few studies have been described in literature in which microfluidic technology is merged with the field of supramolecular host–guest chemistry for the fabrication of large self-assembled structures. Tseng and co-workers have used a digital microreactor to prepare an assortment of cell-targeting and DNA encapsulated SNPs based on the interaction of β-cyclodextrin and adamantane, providing a better dispersity and higher reproducibility than in batch processes.31,32 Using the same host–guest chemistry, Thompson and co-workers have described the variation of polymeric supramolecular nanoparticle properties (diameter, polydispersity and zeta-potential) by controlling the velocity of the two mixing inlet streams within a microfluidic reactor.33 The groups of Scherman and Abell have shown the formation of monodisperse supramolecular microcapsules self-assembled within microfluidic droplets using the ternary interaction between CB[8], and MV and Np building blocks.34 To the best of our knowledge, the diffusive mixing omnipresent in microfluidics has not been used so far to control the formation of host–guest nanoparticle network aggregates.
Here, we present the size-controlled self-assembly of aggregates of nanoparticles induced by host–guest interactions, so-called supramolecular nanoparticle clusters (SNPCs), mediated by multiple heteroternary host–guest interactions using CB[8] as a host within a microfluidic device (Fig. 1a). As interacting guest-modified building blocks, we use silica nanoparticles functionalized with MV (SiO2-MV), Np-terminated poly(amido amine) (Np8-PAMAM) dendrimer, and Np-terminated poly(ethylene glycol) (Np-PEG) (Fig. 1b). In the presence of CB[8], cluster formation is induced by multivalent interactions forming between the SiO2-MV and the multivalent dendrimer and CB[8], whereas Np-PEG terminates the aggregation by monovalent interaction at the surrounding cluster shell (Fig. 1a). Aggregate formation was carried out comparing mixing in a batch reactor and diffusive mixing within a microfluidic device (Fig. 1a). By placing the silica nanoparticles in one stream and the, non-interacting, molecular components in the other, assembly formation is controlled by the diffusion of the molecular components into the nanoparticle stream, as exemplified by the diffusion profiles assumed in the absence of complexation (Fig. 1d). The size tunability of the supramolecular clusters was evaluated by modulation of the competition between the mono- and multivalent Np-bearing components, and by changing the mixing time of the interacting host and guest binding partners in the microreactor.
 | ||
| Fig. 1 (a) Schematic illustration of microfluidic assisted supramolecular network aggregation of silica nanoparticles, dendrimers and PEG stopper molecules, mediated by the ternary complex formation between MV, Np, and CB[8]. (b) Supramolecular building blocks involved in SNPC formation: CB[8], Np-PEG (Mw 1000 or 5000), Np8-PAMAM, and SiO2-MV. (c) Ternary complex formation by inclusion of MV in CB[8], followed by inclusion of Np. (d) Schematic illustration of the particle (SiO2-MV, red) and molecular (here shown for CB[8] in green, similar for Np-PEG and Np8-PAMAM) diffusion profiles in time over the width of the microfluidic reactor (top view). | ||
To this end, MV-functionalized silica nanoparticles (SiO2-MV) were synthesized according to an adapted, previously reported procedure for the functionalization of transferrin-decorated NPs (Fig. S1, ESI†).35 Bare silica NPs were aminated using 3-(aminopropyl)-triethoxysilane (APTES), followed by reaction with an excess of bifunctional succinimide-maleimide hexa(ethylene glycol) linker. The terminal maleimide group was used to functionalize the particles with thiol-modified MV, followed by backfilling the unreacted maleimide groups with mercaptoethanol. Dynamic light scattering (DLS) and scanning electron microscopy (SEM) showed an average NP size of 61 nm and 52 nm, respectively (Fig. S2a and b, ESI†). The density of guest moieties attached onto the particle surface, important for stoichiometry control during supramolecular assembly, was determined by UV/Vis-spectroscopy to be 5.3 μmol MV per g of NPs (∼900 MV/NP, approx. 1 MV per 12 nm2) (Fig. S2c, ESI†). This coverage is 3.6 times higher than reported for the transferrin-functionalized silica nanoparticles, which might be attributed to the bulkier transferrin protein compared to MV. This density ensures that the guest moieties have sufficient space for supramolecular host–guest complexation with CB[8] and Np.
The formation of SNPCs in solution was studied using a 2 μM concentration of CB[8], MV and Np, while keeping the molecular recognition moieties in an equimolar 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio. The relative concentrations of the monovalent Np-PEG and the multivalent Np8-PAMAM were varied during self-assembly, while keeping the overall concentration of Np moieties constant. SNPC formation was first tested in bulk, by vigorous shaking of the SiO2-MV dispersion with a pre-mixed aqueous solution of CB[8], Np-PEG (Mw = 5000 g mol−1) and Np8-PAMAM, using either 25% or 50% Np from Np8-PAMAM. Instantaneous SNPC formation was observed 10 min after mixing as witnessed by a drastic increase of the average hydrodynamic cluster size by DLS (Fig. S3, ESI†). At the same time, the size distributions of the SNPCs were very broad (ranging from 143 nm to 441 nm and from 776 nm to 1813 nm for the 25% and 50% Np8-PAMAM samples, respectively). Our earlier work7 on SNPs with the same heteroternary motif had shown these to be dynamic but slowly equilibrating over the course of days. In contrast, the current batch-formed SNPCs showed complete cluster sedimentation after 14 h. Apparently, the turbulence and fluid motion from stirring have led to uncontrolled and irreproducible NP aggregation.36 We assume that, in contrast to the fully molecular SNPs, the SNPCs targeted here cannot equilibrate because of the higher valency and the slower diffusion of the SiO2-MV NPs compared to the MV-polymer used in the soft SNPs. Therefore, kinetic control is deemed mandatory to achieve stable SNPCs.
:1:1 ratio. The relative concentrations of the monovalent Np-PEG and the multivalent Np8-PAMAM were varied during self-assembly, while keeping the overall concentration of Np moieties constant. SNPC formation was first tested in bulk, by vigorous shaking of the SiO2-MV dispersion with a pre-mixed aqueous solution of CB[8], Np-PEG (Mw = 5000 g mol−1) and Np8-PAMAM, using either 25% or 50% Np from Np8-PAMAM. Instantaneous SNPC formation was observed 10 min after mixing as witnessed by a drastic increase of the average hydrodynamic cluster size by DLS (Fig. S3, ESI†). At the same time, the size distributions of the SNPCs were very broad (ranging from 143 nm to 441 nm and from 776 nm to 1813 nm for the 25% and 50% Np8-PAMAM samples, respectively). Our earlier work7 on SNPs with the same heteroternary motif had shown these to be dynamic but slowly equilibrating over the course of days. In contrast, the current batch-formed SNPCs showed complete cluster sedimentation after 14 h. Apparently, the turbulence and fluid motion from stirring have led to uncontrolled and irreproducible NP aggregation.36 We assume that, in contrast to the fully molecular SNPs, the SNPCs targeted here cannot equilibrate because of the higher valency and the slower diffusion of the SiO2-MV NPs compared to the MV-polymer used in the soft SNPs. Therefore, kinetic control is deemed mandatory to achieve stable SNPCs.
Subsequently, network aggregation of SiO2-MV was evaluated in the confined environment of a microreactor. A flow of the SiO2-MV dispersion was put in laminar contact with a flow of a premixed solution of CB[8], Np-PEG and Np8-PAMAM within a 65 cm long two-inlet microreactor (Fig. 1a). The diffusion coefficients and the time required for the different SNPC components to diffuse over the distance of the microreactor were evaluated by the Stokes–Einstein and the Einstein–Smoluchowski-equations, respectively (Table S1, ESI†), assuming absence of complexation. Given the dimensions of the device and the flow rates used here, the diffusion rate of SiO2-MV is too small to expect appreciable penetration of the particles into the other stream. In contrast, the diffusion of Np8-PAMAM, Np-PEG (Mw 1000), and CB[8] are comparable, allowing these molecules to diffuse into the complete NP flow within approx. 35 s (Fig. 1d). The diffusion rate of Np-PEG (Mw 5000) is calculated to be somewhat lower. Based on these results, we decided to start clustering experiments with a residence time of 60 s.
In contrast to the experiments performed in bulk, distinct SNPCs were observed at a residence time of 60 s. DLS and SEM show clearly that the resulting cluster size can be tuned over a large dynamic range by varying the ratio of the two Np components while keeping the overall stoichiometry of MV/Np/CB[8] constant (Fig. 2). By increasing the amount of Np from Np8-PAMAM from 25% to 62.5%, while decreasing the amount of Np from Np-PEG (Mw 5000) from 75% to 37.5%, an increase in SNPC size from 125 ± 10 nm to 370 ± 100 nm is observed by DLS, while SEM indicates a similar tuning range from 65 ± 20 nm to 256 ± 146 nm. Furthermore the ratio of the monovalent and multivalent binding partners does not significantly influence the uniformity of the formed aggregates: DLS shows polydispersity values of the observed SNPCs between 0.3 and 0.4 irrespective of the sample (Table S2, ESI†). In contrast, individual unaggregated particles were observed by carrying out SNPC formation in the absence of Np8-PAMAM (Fig. S4, ESI†), whereas huge, uncontrolled aggregates were observed by SEM (Fig. S5, ESI†) upon mixing SiO2-MV, CB[8] and Np8-PAMAM in the absence of Np-PEG. Generally for DLS analysis, the clustering solution was collected from the outlet of the microreactor for 40 min to obtain sufficient sample. In contrast, all SEM samples were prepared by collecting the as-assembled SNPC solution directly from the microreactor onto the substrate. To assess the validity of the DLS results, SEM control experiments were carried out on samples collected for 40 min. These showed that the observed cluster sizes do not vary between the instantaneous measurements and the measurements prepared from collecting over 40 min (Fig. S6, ESI†) and up to 3 h. The good correlation between the DLS and SEM data, as well as this control, confirm therefore the strong kinetic trapping already mentioned above. In contrast, all SNPCs showed sedimentation overnight from the suspension. This is not unexpected as even the initial, non-aggregated SiO2-MV NPs start to sediment within a few hours. In another control experiment, SiO2-MV and both Np-bearing components (62.5% Np from Np8-PAMAM) were used, but now with CB[7] instead of CB[8]. The smaller CB[7] is too small to include both the MV and Np guest moieties in its cavity, and consequently the hetero-ternary complex cannot be formed. Indeed, no SNPC formation was observed by DLS or SEM (Fig. S7, ESI†) in this case. A similar control was performed using bare silica nanoparticles instead of the MV-functionalized ones. Also in this case, only unaggregated NPs were observed (Fig. S8, ESI†). Summarizing, SNPCs can be formed within a microfluidic reactor in which diffusive mixing leads to controllable aggregation, their formation is based on specific ternary complex formation, and the size of the resulting clusters is strongly dependent on the ratio between the mono- and multivalent Np guest species. The dependence of the resulting SNPC size on the fraction of Np8-PAMAM in the Np-bearing building blocks can be ascribed to the competition between monovalent and multivalent host–guest interactions in the presence of CB[8] and SiO2-MV. The amount of inter- and intramolecular binding events increases with the increased content of multivalent dendrimers, which leads to larger SNPC structures. The monovalent Np-PEG is essential to terminate the network formation and the accompanying uncontrolled aggregation as it blocks interaction sites and shields the crosslinked cores by the sterics of the PEG chains.
 | ||
| Fig. 2 Size determination of SNPCs induced by ternary host–guest interactions (2 μM of interacting moieties, 60 s residence time). (a) Average NP cluster diameter by DLS (light grey bars) and SEM (dark grey bars). SEM images (b)–(e) of the resulting SNPCs, as a function of the Np content derived from Np8-PAMAM ((b) 25%, (c) 37.5%, (d) 50%, (e) 62.5%) used during microfluidic assisted NP network aggregation. | ||
To investigate the effect of residence time and thus the diffusion time of the components on the size and kinetics of the assembly formation, clustering experiments were carried out with SiO2-MV, CB[8], Np8-PAMAM, and Np-PEG (5000 g mol−1) at a constant 62.5% Np derived from Np8-PAMAM. As seen in Fig. 3a, the cluster sizes observed by DLS varied strongly and in a non-trivial manner: a maximum in cluster size was observed at a 30 s residence time. The non-linear trend is tentatively explained by the calculated diffusion times of the different molecular building blocks.
 | ||
| Fig. 3 SNPC formation as a function of assembly conditions: (a) SNPC size by assembly of SiO2-MV, CB[8], Np8-PAMAM, and Np-PEG (Mw 5000) at 2 μM 1:1:1 CB[8]/MV/Np (62.5% Np from Np8-PAMAM) as a function of residence time by DLS direct after SNCP assembly (light grey) and DLS 60 min after starting SNPC formation (gray). (b) SNPC size and (c) fraction of unbound SiO2-MV NPs, by assembly of SiO2-MV, CB[8], Np8-PAMAM, and Np-PEG (Mw 1000) at 2 μM 1:1:1 CB[8]/MV/Np (50% Np from Np8-PAMAM) as a function of residence time, as measured by DLS (light grey) and SEM (grey bars including all observed SiO2-MV NPs and dark grey bars excluding unbound SiO2-MV NPs). | ||
The diffusion time of Np-PEG (5000 g mol−1) is about 2.5 times slower than the diffusion time of Np8-PAMAM and CB[8] (Table S1, ESI†), which may result in less controlled cluster formation at the shorter residence times due to faster diffusion of the crosslinking Np8-PAMAM compared to the cluster-stabilizing Np-PEG. For example, by using a residence time of 30 s, CB[8] and the dendrimer can diffuse over almost the complete width of the microreactor, whereas the monovalent Np-PEG (5000 g mol−1) can only penetrate for approx. 25% into the other fluid stream. In contrast, at 60 s residence time, all components are able to penetrate the full NP stream explaining the observed size control at this residence time.
In order to eliminate this diffusion effect, we used Np-PEG with a Mw of 1000 instead of the earlier used 5000. The shorter Np-PEG polymer has a diffusion time that is very similar to Np8-PAMAM and CB[8] (Table S1, ESI†). Upon cluster formation using Np-PEG (1000 g mol−1), Np8-PAMAM, CB[8] and SiO2-MV, a monotonous dependence of SNPC size with residence time is now observed (Fig. 3b and S9, ESI†), both with DLS and SEM. This confirms that the equal diffusion times of the molecular components govern the simultaneous assembly and termination of the clusters. Furthermore, DLS shows that the PDI strongly depends on the mixing time and decreases upon increase of the overall residence time of the components within the microreactor (Table S3, ESI†). Nevertheless, the DLS size measurements only provide averages of cluster sizes, whereas the SEM images provide more detail on the size distribution. Many images show the presence of unaggregated particles in the SNPC samples, which prompted us to evaluate their fraction as a function of residence time (Fig. 3c). At short residence times, the diffusion of the molecular components is insufficient to pass the whole width of the microfluidic channel. Consequently, not all SiO2-MV NPs are integrated in SNPCs. This is confirmed by the results shown in Fig. 3c: the fraction of unaggregated NPs decreases strongly, from 30 to 5%, upon increasing the residence time from 15 to 60 s. By excluding the non-assembled NPs from the formed SNPCs, correction of the size determinations by SEM is possible. As is visible in Fig. 3b, a clear SNPC size increase and a decrease in the SNPC size distribution is observed by excluding the unbound SiO2-MV. These effects are more pronounced for the samples prepared with lower residence times, since the fractions of unbound NPs are higher in this case.
Because SNPC formation is dependent on the diffusion of the molecular components into the NP stream, we decided to investigate the effect of the concentration of these components on the assembly process. Therefore, the stoichiometry of CB[8] and Np with respect to MV was varied under constant flow conditions (residence time 30 s), while keeping the content of Np from Np8-PAMAM constant at 50%. By increasing the CB[8]/MV/Np ratio from 1:1:1 to 3:1:3, the overall observed size of the SNPCs increased from 180 ± 46 nm to 354 ± 60 nm and from 123 ± 58 nm to 205 ± 70 nm by DLS and SEM, respectively (Fig. 4 and S10, ESI†). Further increase of the CB[8]/MV/Np ratio to 5:1:5 led to large SNPCs and visible precipitation (Fig. S10d, ESI†). The PDI of the samples increased upon increase of the ratio of CB[8] and Np with respect to MV (Table S4, ESI†). Irrespective of the composition though, the samples showed the presence of 10% of unbound SiO2-MV NPs. The latter result confirms that the extent of unaggregated NPs is solely dependent on the diffusion rates of the molecular components and thus on the penetration depth of the molecular components into the NP stream (Fig. 5a). On the other hand, this result and the observed increase of the SNPC size by increase of the concentrations of the molecular components (Fig. 5b) indicate that the molecular fraction of multivalent Np8-PAMAM, and the concentrations of the components are not fully incorporated into the SNPCs, since complete use at the 1:1:1 stoichiometry should have been seen in an independence of aggregate size with further increase of the concentration and a depletion of the molecular components and thus a lower penetration depth (and higher fraction of unbound SiO2-MV NPs) at lower concentrations.
 | ||
| Fig. 4 SNPC size by assembly of SiO2-MV, CB[8], Np8-PAMAM, and Np-PEG (Mw 1000) at different CB[8]/MV/Np ratios (50% Np from Np8-PAMAM) as measured by DLS (light grey bars) and SEM (grey bars including all observed SiO2-MV NPs and dark grey bars excluding unbound SiO2-MV NPs). | ||
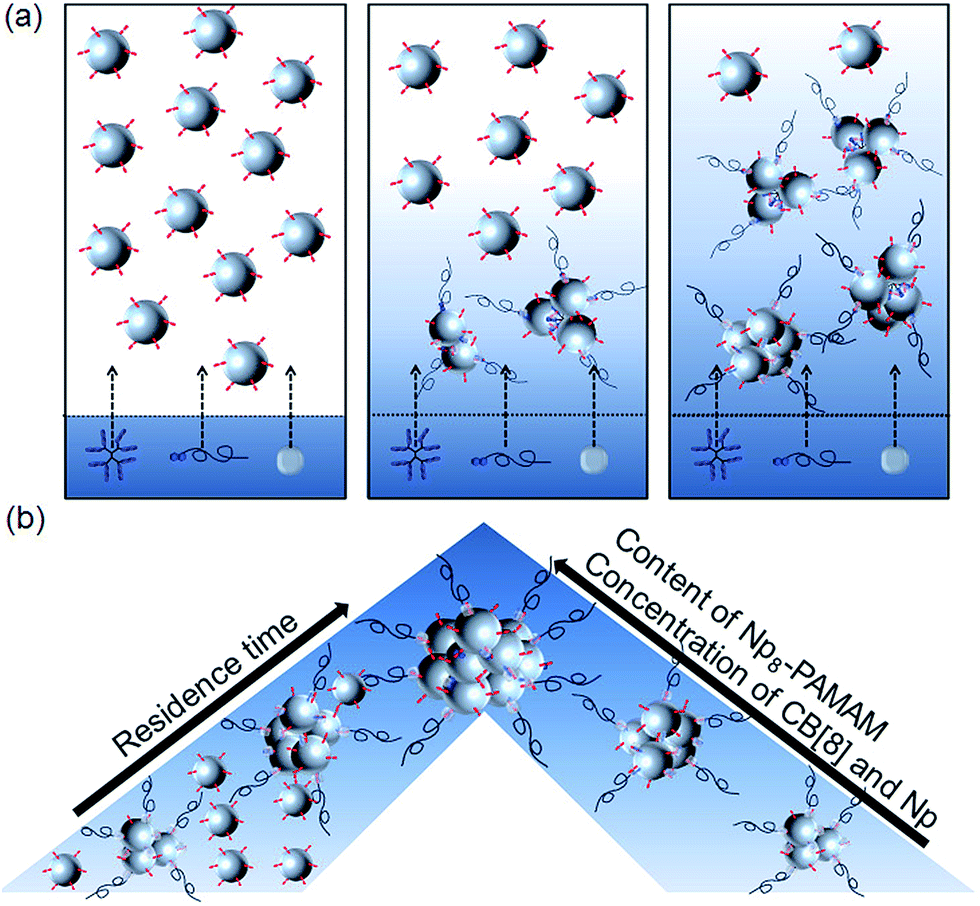 | ||
| Fig. 5 (a) Schematic illustration of SNPC formation inside the microfluidic reactor depending on the diffusion of Np8-PAMAM, Np-PEG and CB[8] within the SiO2-MV stream. (b) Dependence of SNPC size and of the fraction of unbound SiO2-MV on the residence time, the relative fraction of multivalent Np8-PAMAM, and the concentrations of the molecular components. | ||
Conclusions
In conclusion, we have shown that the microfluidic assisted formation of SNPCs is driven by the ternary host–guest interaction of CB[8], MV and Np within a microfluidic reactor. As schematically shown in Fig. 5a, the diffusive mixing is strongly determined by the diffusion profile of the molecular components CB[8], Np8-PAMAM, and Np-PEG into the SiO2-MV NP stream. The size of the resulting SNPCs is strongly depending on the content of multivalent interacting Np dendrimers, the residence time of the interacting building blocks within the microchannel, and the stoichiometry of the ternary host–guest binding partners (Fig. 5b). The residence time variation as well as the discrepancy between clustering experiments carried out in bulk and in the microfluidic reactor show that the supramolecular host–guest assembly is to a large extent kinetically controlled. Interestingly, the molecular weight of the PEG stopper provides an additional kinetic control parameter to the assembly process because a longer length leads to slower diffusion into the NP stream and a concomitantly later termination of the clusters. Overall, the combination of microfluidic technology with the design and control parameters provided by supramolecular chemistry provides an advanced platform for the tunable assembly of NP network aggregates, which may prove beneficial for the preparation of functional nanostructures for materials engineering.Acknowledgements
This work was supported by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO-CW; Vici grant 700.58.443 to JH) and NanoNext NL (RM). We gratefully acknowledge Jan Eijkel for fruitful discussions.Notes and references
- B. A. Grzybowski, C. E. Wilmer, J. Kim, K. P. Browne and K. J. M. Bishop, Soft Matter, 2009, 5, 1110 RSC.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591 CrossRef CAS PubMed.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600 CrossRef CAS PubMed.
- M. D. Yilmaz and J. Huskens, Soft Matter, 2012, 8, 11768 RSC.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213 RSC.
- F. Cheng, Z. Tao, J. Liang and J. Chen, Chem. Mater., 2007, 20, 667 CrossRef.
- C. Stoffelen and J. Huskens, Chem. Commun., 2013, 49, 6740 RSC.
- C. Stoffelen, J. Voskuhl, P. Jonkheijm and J. Huskens, Angew. Chem., Int. Ed., 2014, 53, 3400 CrossRef CAS PubMed.
- D. Patra, F. Ozdemir, O. R. Miranda, B. Samanta, A. Sanyal and V. M. Rotello, Langmuir, 2009, 25, 13852 CrossRef CAS PubMed.
- A. B. Descalzo, R. Martínez-Máñez, F. Sancenón, K. Hoffmann and K. Rurack, Angew. Chem., Int. Ed., 2006, 45, 5924 CrossRef CAS PubMed.
- H. Li, D.-X. Chen, Y.-L. Sun, Y. B. Zheng, L.-L. Tan, P. S. Weiss and Y.-W. Yang, J. Am. Chem. Soc., 2012, 135, 1570 CrossRef PubMed.
- X. Chen, S. G. Parker, G. Zou, W. Su and Q. Zhang, ACS Nano, 2010, 4, 6387 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293 CrossRef PubMed.
- R. Mejia-Ariza and J. Huskens, J. Mater. Chem. B, 2014, 2, 210 RSC.
- G. M. Whitesides, Nature, 2006, 442, 368 CrossRef CAS PubMed.
- K. S. Elvira, X. C. i Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905 CrossRef CAS PubMed.
- P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tam and B. H. Weigl, Nature, 2006, 442, 412 CrossRef CAS PubMed.
- B. Zheng, J. D. Tice and R. F. Ismagilov, Anal. Chem., 2004, 76, 4977 CrossRef CAS PubMed.
- Y. Song, J. Hormes and C. S. S. R. Kumar, Small, 2008, 4, 698 CrossRef CAS PubMed.
- M. Numata and T. Kozawa, Chem.–Eur. J., 2013, 19, 12629 CrossRef CAS PubMed.
- F. S. Majedi, M. M. Hasani-Sadrabadi, S. Hojjati Emami, M. A. Shokrgozar, J. J. VanDersarl, E. Dashtimoghadam, A. Bertsch and P. Renaud, Lab Chip, 2013, 13, 204 RSC.
- M. Lu, Y.-P. Ho, C. L. Grigsby, A. A. Nawaz, K. W. Leong and T. J. Huang, ACS Nano, 2013, 8, 332 CrossRef PubMed.
- J. He, L. Wang, Z. Wei, Y. Yang, C. Wang, X. Han and Z. Nie, ACS Appl. Mater. Interfaces, 2013, 5, 9746 CAS.
- Z. Yu, C.-F. Wang, L. Ling, L. Chen and S. Chen, Angew. Chem., Int. Ed., 2012, 51, 2375 CrossRef CAS PubMed.
- C.-W. Wang, D. Sinton and M. G. Moffitt, J. Am. Chem. Soc., 2011, 133, 18853 CrossRef CAS PubMed.
- G. Schabas, H. Yusuf, M. G. Moffitt and D. Sinton, Langmuir, 2008, 24, 637 CrossRef CAS PubMed.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906 CrossRef CAS PubMed.
- G. Tresset, C. Marculescu, A. Salonen, M. Ni and C. Iliescu, Anal. Chem., 2013, 85, 5850 CrossRef CAS PubMed.
- N. Hassan, V. Cabuil and A. Abou-Hassan, Angew. Chem., Int. Ed., 2013, 52, 1994 CrossRef CAS PubMed.
- M. Numata and T. Kozawa, Chem.–Eur. J., 2014, 20, 6234 CrossRef CAS PubMed.
- K. Liu, H. Wang, K.-J. Chen, F. Guo, W.-Y. Lin, Y.-C. Chen, D. L. Phung, H.-R. Tseng and C. K.-F. Shen, Nanotechnology, 2010, 21, 445603 CrossRef PubMed.
- H. Wang, K. Liu, K.-J. Chen, Y. Lu, S. Wang, W.-Y. Lin, F. Guo, K.-i. Kamei, Y.-C. Chen, M. Ohashi, M. Wang, M. A. Garcia, X.-Z. Zhao, C. K. F. Shen and H.-R. Tseng, ACS Nano, 2010, 4, 6235 CrossRef CAS PubMed.
- A. Kulkarni, R. VerHeul, K. DeFrees, C. J. Collins, R. A. Schuldt, A. Vlahu and D. H. Thompson, Biomater. Sci., 2013, 1, 1029 RSC.
- J. Zhang, R. J. Coulston, S. T. Jones, J. Geng, O. A. Scherman and C. Abell, Science, 2012, 335, 690 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137 CrossRef CAS PubMed.
- M. Elimelech, J. Gregory, X. Jia, R. A. Williams, J. Gregory, X. Jia and R. A. Williams, in Particle Deposition & Aggregation, ed. M. Elimelech, J. Gregory, X. Jia, R. A. Williams, J. Gregory, X. Jia and R. A. Williams, Butterworth-Heinemann, Woburn, 1995, pp. 157 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mh00103f |
| This journal is © The Royal Society of Chemistry 2014 |