
Flexibility versus rigidity: what determines the stability of zeolite frameworks? A case study†
E.
Verheyen
a,
L.
Joos
b,
C.
Martineau
c,
C. J.
Dawson
d,
C.
Weidenthaler
e,
W.
Schmidt
e,
R.
Yuan
a,
E.
Breynaert
a,
V.
Van Speybroeck
b,
M.
Waroquier
b,
F.
Taulelle
ac,
M. M. J.
Treacy
d,
J. A.
Martens
a and
C. E. A.
Kirschhock
*a
aCentre for Surface Chemistry and Catalysis, KU Leuven, Kasteelpark Arenberg 23–box 2461, B-3001 Leuven, Belgium. E-mail: Christine.Kirschhock@biw.kuleuven.be
bCenter for Molecular Modeling, Ghent University, B-9052 Zwijnaarde, Belgium
cTectospin, Institut Lavoisier de Versailles, UMR CNRS 8180 Université de Versailles St Quentin en Yvelines, 45 Avenue des Etats Unis, Versailles Cedex, France
dDepartment of Physics, Arizona State University, P.O. Box 871504, Tempe, USA
eMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhem-Platz 1, Mülheim an der Ruhr, Germany
First published on 19th August 2014
Abstract
All silica COK-14/-COK-14 with OKO topology is the first case of a zeolite which reversibly transforms from a systematically interrupted to a fully connected state and back. Analysis of the opening/closing behavior allowed the study of entropy and framework flexibility as determinants for the stability of zeolite topologies, which, until now, has been experimentally inaccessible. Interconversion of the all-silica COK-14 zeolite with fully connected OKO topology and its -COK-14 variant with systematic framework interruption was investigated using high-temperature XRD, thermogravimetric analysis, 29Si MAS NMR, nitrogen adsorption and a range of modelling techniques. Specific framework bonds in the OKO framework can be reversibly hydrolyzed and condensed. Structural silanols of the parent -COK-14, prepared by degermanation of the IM-12 zeolite, were condensed by heating at 923 K, and hydrolyzed again to the initial state by contacting the zeolite with warm water. Molecular modelling revealed an inversion of the relative stabilities for both variants depending on temperature and hydration. Condensation of the structural silanols in -COK-14 to COK-14 is entropy driven, mainly resulting from the release of water molecules. Framework reopening in the presence of water is spontaneous due to the high rigidity of the fully connected OKO framework. Isomorphous substitution was demonstrated as a viable option for stabilization of the fully connected OKO framework as this renders the closed framework flexible.
Conceptual insightsThe outstanding stability of zeolites is one of the reasons for their success as porous materials in industry. However, the assessment whether or not a hypothetical zeolite topology is stable and accessible to synthesis has until now largely neglected the impact of entropy and framework flexibility. Now, the newly discovered zeolites, -COK-14 and COK-14 with OKO topology, allow a detailed study of entropic and flexibility effects on the framework stability. COK-14 is the first zeolite which exists in its fully connected as well as in its systematically interrupted form and can be reversibly transformed between these states. This not only allows rational tuning of the porosity and hydrophilicity of this unique material but clearly highlights the importance of the framework flexibility. Fully connected siliceous COK-14 is demonstrated as a very rigid structure in its siliceous form. This renders it unstable at room temperature in the presence of humidity. An opening of Si–O–Si bonds occurs spontaneously, resulting in systematically arranged silanol groups and framework flexibility. Only at high temperatures the entropic disadvantage of the rigid, closed structure can be overcome through the release of water molecules by re-condensation. The hypothesis that entropic effects are essential to determine the feasibility of a zeolite framework is further highlighted by the observation that the presence of heteroatoms, like Al, with larger bond distances to oxygen compared to silicon, results in perfectly stable, fully closed COK-14 zeolites. It is not surprising that these heteroatoms lend flexibility to the structure as has been determined by theoretical analysis which explains the observation of the heteroatom containing OKO frameworks in the fully closed state. |
Introduction
Idealized zeolite frameworks exclusively contain fourfold connected T-atoms (T = Si, Al, P, etc.). In a fully connected zeolite framework all T-atoms share bridging oxygen atoms with neighboring T-sites. Real zeolites, however, may contain defect sites, lowering the T-atom connectivity. While these broken links are often statistically distributed, an interruption in four-connected frameworks can also occur periodically. Framework type codes assigned to such periodically interrupted frameworks are indicated by a leading dash, and are encountered in both naturally occurring zeolites (-CHI,1-LIT,2-PAR,3-RON,4-WEN5 and synthetic materials (-CLO,6,7-ITV,8-IRY,9-SRV10). The -COK-14 zeolite, having a systematically interrupted OKO framework, is a recent addition to the interrupted framework zeolite family (Fig. 1, top).11 | ||
| Fig. 1 Top: framework of -COK-14 and bottom: COK-14 (OKO framework type code), viewed along the 12-MR pores. Structural silanol groups in -COK-14, depicted with balls and sticks, protrude into the 12-MR channels (top). | ||
-COK-14 materials can be obtained by degermanation of the IM-12 zeolite11 (UTL topology12,13). The structure of germanosilicate IM-12 can be described as silicate layers connected by double four-ring (D4R) units. IM-12 with UTL topology has an intersecting two-dimensional channel system circumscribed by 12-membered rings (12-MRs) and 14-MRs. The D4R in IM-12 contains a germanate four-ring (Ge-4R) connected to a silicate four-ring (Si-4R).11 Acidic treatment converts the IM-12 zeolite to the -COK-14 material by selective removal of the Ge-4R, thereby creating a two-dimensional channel system with interconnecting 8-, 10- and 12-MRs (Fig. 1). While Ge removal leaves the silicate layers intact, the 4Rs connecting the layers split symmetrically into T-atom pairs carrying silanol groups, pointing to the 12-MRs of the -COK-14 structure (Fig. 1, top). These silanols can be reoriented and condensed resulting in COK-14 (Fig. 1, bottom). We investigated these framework annealing and disruption processes using X-ray diffraction, 29Si MAS NMR, thermal analysis and nitrogen adsorption porosimetry. Theoretical modelling assisted in rationalizing the observations based on the stability differences of the interrupted zeolite framework and its fully connected analogue.
Experimental section
The synthesis of -COK-14 was performed according to the original publication.11 In short, the parent -COK-14 sample, denoted as -COK-14(H1), was obtained by slurrying a freshly calcined IM-12 zeolite in concentrated hydrochloric acid (1 g IM-12 in 42.4 mL HCl, 12 M, from VWR) in a 60 mL polypropylene bottle for 2 days at 363 K. -COK-14(H1) was recovered by centrifugation and washed until the water reached pH 6, and dried overnight at 333 K.The first time condensed COK-14 sample, denoted as COK-14(C1), was obtained by heating -COK-14(H1) at 923 K under nitrogen flow for 6 h using a temperature ramp of 1 K min−1. To prevent back transformation to -COK-14, rehydration was carefully avoided during cooling, sample storage and manipulation.
COK-14(C1) was hydrolyzed to the interrupted -COK-14(H2) by suspending the powder in water at 333 K for 1 day.
COK-14(C2) was obtained by heating -COK-14(H2) again at 923 K, similar to COK-14(C1).
A detailed description of X-ray diffraction, 29Si MAS NMR, nitrogen adsorption, thermogravimetric analysis and computational approaches is available in the ESI.†
Results and discussion
Parent -COK-14, denoted as -COK-14(H1), and the fully connected COK-14 obtained by heating -COK-14(H1) at 923 K, denoted as COK-14(C1), have distinct XRD patterns (Fig. 2). Indexing of the powder XRD pattern of COK-14(C1) in space group C2/m resulted in a unit cell of a = 24.13 Å, b = 13.79 Å, and c = 12.30 Å and a monoclinic angle of 109.60° compared to a = 24.64 Å, b = 13.94 Å, c = 12.26 Å and β = 109.22° for -COK-14(H1). A listing of all h k l reflections, and corresponding 2 theta positions, d spacings and relative intensities, of -COK-14 and COK-14 in the 2–30° 2 theta range is provided in Table S1 in the ESI.†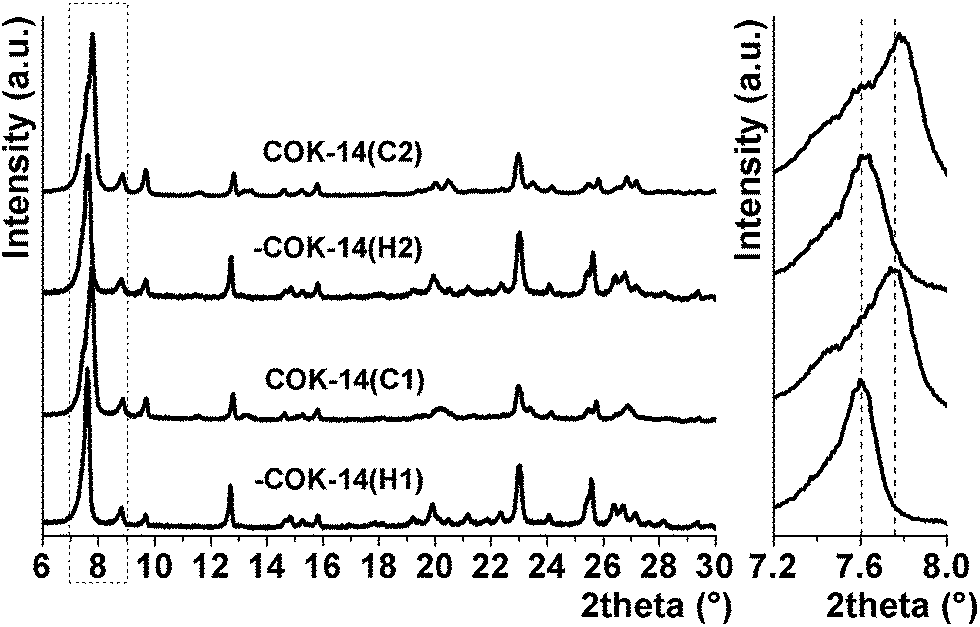 | ||
| Fig. 2 X-ray diffraction patterns of -COK-14(H1), COK-14(C1), -COK-14(H2) and COK-14(C2) zeolites. Magnification of the 7.2–8° 2 theta range highlights the shifting of the 2 0 0 reflection between 7.60 and 7.77° 2 theta (dashed lines) during transformation. | ||
The transformation of -COK-14 to COK-14 is revealed by the altered diffraction pattern, and is most clearly visible by the shift of the 2 0 0 reflection from 7.60 to 7.77° 2 theta (Fig. 2). Near to the 2 0 0 reflection in the 7.2 to 8° 2 theta range, the 0 0 1 and 1 1 0 reflections are present (Fig. 2). The shift of both reflections, resulting from the framework transformation, is documented in Fig. S1 and Table S1 in the ESI.†COK-14(C1) was slurried in warm water (333 K) to provoke transformation to -COK-14, denoted as -COK-14(H2), through hydrolysis of the layer-connecting 4Rs (Fig. 1). X-ray diffraction patterns of the original -COK-14(H1) sample and -COK-14(H2), obtained after condensation of -COK-14(H1) at 923 K and subsequent hydrolysis in water, were almost identical (Fig. 2), showing the reversibility of the transformation.
-COK-14(H2) was heated for the second time at 923 K. The XRD patterns of the resulting sample, denoted as COK-14(C2), and the first time condensed sample, COK-14(C1), strongly resembled each other (Fig. 2). The 2 0 0 reflection was fully shifted to 7.77° 2 theta. The remaining signal around 7.62° 2 theta resulted from the 0 0 1 reflection (Fig. S1 and Table S1†). It was observed that the diffraction pattern of COK-14(C2), obtained after condensation, hydrolysis and second condensation, improved in quality and resolution. This can be seen as indication that cycling between both states has a positive influence on the integrity of the overall structure.
Using a high temperature reaction chamber allowed to follow this framework transformation in situ and record XRD patterns at 50 K intervals while heating the sample from room temperature to 1073 K under a continuous flow of dry nitrogen. The structural transformation of -COK-14(H1) to COK-14(C1) was probed by monitoring the shift of the 2 0 0 reflection (Fig. 3). At temperatures up to 573 K, the 2 0 0 reflection was at 7.6° 2 theta, characteristic of the systematically interrupted framework. From 573 K onwards, the transformation gradually proceeded, as traced by the 2 0 0 reflection (Fig. 2). At 923 K, the fully connected framework was obtained and no changes were detected while further heating up to 1073 K. The position of the 2 0 0 reflection as a function of temperature for all recorded X-ray diffraction patterns is shown in Fig. S2 in the ESI.†
 | ||
| Fig. 3 X-ray diffraction patterns at the 2 0 0 reflection recorded at stepwise increased temperature, revealing transformation of -COK-14(H1) to COK-14(C1) in the temperature range 573–923 K. | ||
While XRD reflected the global state of the structure, 29Si MAS NMR allowed observation of the bonding state of the silicon atoms in the frameworks. The spectra of -COK-14(H1), COK-14(C1), and -COK-14(H2) are reported in Fig. 4. Chemical shifts and signal intensities of the quantitative 29Si MAS NMR spectra of -COK-14(H1) and COK-14(C1) are given in Table S2 in the ESI.† All spectra show signals in the regions of 4-fold and 3-fold connected silicon atoms (Q4 and Q3, respectively). In the broadened Q4 region of COK-14(C1) two maxima can be discerned, at δ = −109 and δ = −114 ppm, respectively. The interrupted frameworks -COK-14(H1) and -COK-14(H2) show a main signal centered at −111 ppm, which confirms the reversibility of the transformation.
 | ||
| Fig. 4 29Si MAS NMR spectra of -COK-14(H1), COK-14(C1) and -COK-14(H2). | ||
The Q3 regions allow quantitative determination of the content of silanol groups in the zeolites. The idealized unit cell composition of -COK-14 corresponds to Si68O132(OH)8,11 resulting in a concentration of 11.7% Q3 silicon atoms carrying an OH group (Q3 silicon atoms; δ ∼ −101 ppm). The experimental content of Q3 silicon atoms in -COK-14(H1), determined by 29Si MAS NMR, was 13%, slightly exceeding this theoretical value (Table S2†). Heating at 923 K, which according to XRD leads to the condensed COK-14(C1) with unit cell composition Si68O136, removed the majority of Q3 silicon atoms down to 4%. The residual Q3 silicon atoms could be due to defect sites already present in -COK-14(H1) before heating, or disruption of some bonds in the layers upon heating resulting in additional silanol groups, or due to incomplete condensation of the systematic silanol groups. Exposure to water at 333 K to form the interrupted -COK-14(H2) increases the Q3 silicon atom concentration at the expense of Q4, reaching a value of 16%, slightly exceeding the value of 13% for -COK-14(H1). The 29Si MAS NMR spectrum of -COK-14(H2) can be fitted as a linear combination of 77% -COK-14(H1), 21% COK-14(C1) and 2% Q3 silanols, as shown in Fig. S3 in the ESI.†-COK-14(H2) is therefore nearly fully reopened according to 29Si MAS NMR.
The structural changes associated with the reversible transformation of the -COK-14 to COK-14 zeolite are mirrored in the microporosity of the samples, as characterized by nitrogen adsorption (Fig. S4†). The specimens with structural hydroxyls, -COK-14(H1) and -COK-14(H2), have a similar micropore volume (0.146 and 0.145 mL g−1, respectively) and pore size (0.67 and 0.68 nm, respectively), substantially different from the fully connected sample COK-14(C1) having a pore volume of 0.160 mL g−1 and a pore size of 0.78 nm. The differences can be explained by silanol groups systematically pointing to the 12-MR channels of the interrupted framework (Fig. 1, top).
The physical and chemical water content of -COK-14 samples was quantified by thermogravimetric analysis (TGA) (Fig. 5). Upon heating -COK-14 samples three weight loss steps could be discerned. In the first step up to ca. 350 K, an arbitrary amount of physisorbed water molecules was removed. A subsequent weight loss in the temperature range 350–475 K on -COK-14(H1) and 350–600 K on -COK-14(H2) represented ca. 1.6 weight%, while the third, more gradual loss also represented a weight fraction of 1.6%. Transformation of the -COK-14(H1) and -COK-14(H2) samples to the condensed form of COK-14 after TGA was confirmed by XRD. The TGA pattern is consistent with the presence of 4 tightly bound water molecules per unit cell, bridging the structural silanol groups (Fig. 6). DFT calculations indicated the adsorption energy of such bridging water molecules to be as high as 152 kJ mol−1 for the first water molecule, hence explaining the high temperature necessary for desorption.11 The weight loss in the temperature range 350–475 K on -COK-14(H1) and 350–600 K on -COK-14(H2) of about 1.6 weight% corresponds to the desorption of these 4 tightly bound water molecules per unit cell. The additional further weight loss can be explained by the release of 4 water molecules per unit cell stemming from condensation of the structural silanols. When loosely held water is discarded, the idealized unit cell compositions of interrupted and fully condensed OKO zeolites correspond to T68O132(OH)8·4H2O, and T68O136, respectively, corresponding to a content of ca. 3.2 weight% of strongly physisorbed and chemical water, The TGA of -COK-14(H1) and -COK-14(H2) revealed such water contents.
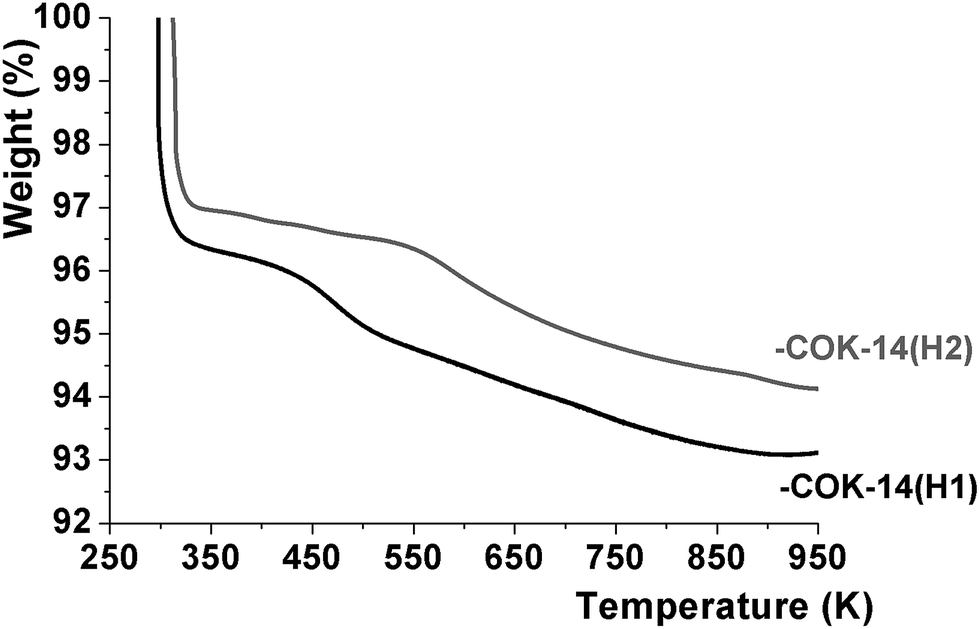 | ||
| Fig. 5 Thermogravimetric analysis of -COK-14(H1) and -COK-14(H2). | ||
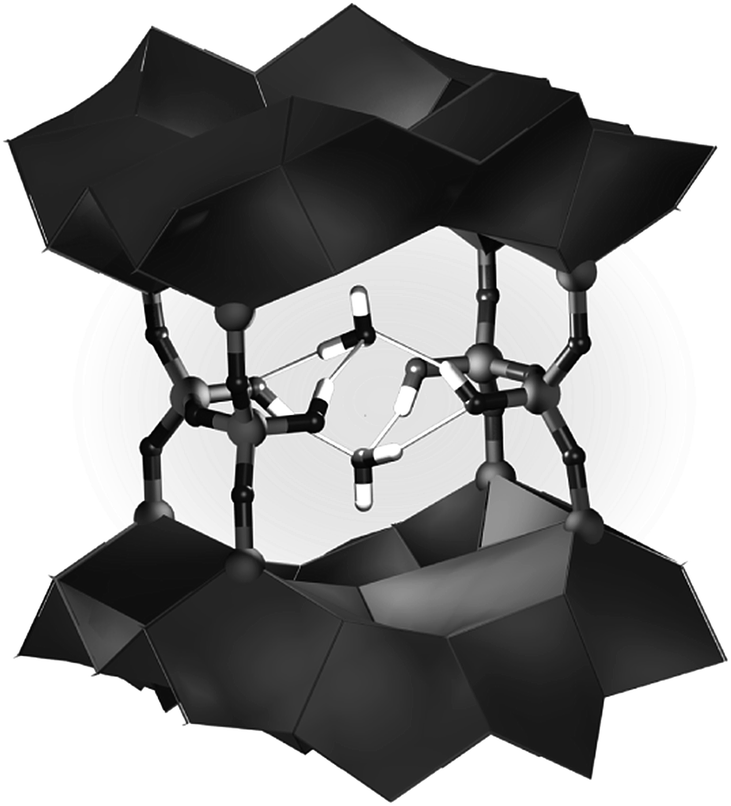 | ||
| Fig. 6 Framework fragment of -COK-14, illustrating the bridging of 4 silanol groups with 2 water molecules (DFT calculation at 0 K). | ||
Under ambient conditions, hydration of COK-14(C1) causes spontaneous interruption of the structure, but the process is slow. After 1 month, a partially reopened sample called -COK-14(H2-43%) was formed. The 29Si MAS NMR spectrum and XRD pattern are shown in Fig. S3 and S5 in the ESI.†-COK-14(H2-43%) was only partially converted as evidenced by the 2 0 0 reflection at 7.69° 2 theta. The 29Si MAS NMR spectrum could be fitted as a linear combination of 43% -COK-14(H1) and 57% COK-14(C1).
Until the discovery of -COK-14, the SSZ-74 zeolite (-SRV topology) was the only example of a purely siliceous framework with systematic framework interruption. While calcination of -SRV leads to irreversible loss of crystallinity,10 the interrupted framework of -COK-14 can thermally be transformed to the highly crystalline and fully connected COK-14 with OKO framework type code11 (Fig. 1, bottom). This contrasting behavior can be explained by the distribution of the silanol groups. In -COK-14, structural silanols are located pairwise, suitable for condensation reactions, while in SSZ-74 they are isolated. The large distance between the nearest silanol sites in the latter explains why this zeolite cannot be calcined beyond the stability temperature of its silanol groups. A range of modelling techniques was applied to gain more insight into this remarkable structural transformation.
A set of DFT calculations was performed to obtain quantitative insight into the various contributions of the free energy determining the framework transformation from -COK-14 to COK-14. The methodology is outlined in the computational section in the ESI.†Fig. 7 provides a summary of the energetic (ΔH) and entropic (TΔS) contributions to the free energy (ΔG). Since framework condensation involves the release of four water molecules per unit cell upon silanol condensation, energetic comparisons are made between the interrupted framework and the condensed state enclosing four water molecules. At 0 K (Fig. 7a), the electronic energy difference (ΔE0) including zero-point-energies reveals a 161 kJ mol−1 lower energy for the interrupted framework. Such an energy difference per unit cell is substantial and correlated with the high temperature required for framework condensation. Including thermal effects, however, is essential for the evaluation of the free energy profile during condensation. Since both enthalpy (Fig. 7, ΔH) and entropy (Fig. 7, TΔS) terms significantly contribute to the overall free energy (Fig. 7, ΔG) of the interrupted framework releasing water molecules upon condensation, full vibrational analysis was essential for calculating the various thermal contributions and entropy terms contributing to the free energy. At 300 K (Fig. 7b), the interrupted framework appears to be slightly less stable according to its free energy (4 kJ mol−1 higher energy compared to the condensed framework) including thermal corrections and the entropy term. The release of four water molecules upon framework condensation is entropically favored (TΔS < 0) and at 300 K, this contribution (TΔS = −148 kJ mol−1) is enough to achieve an overall more stable condensed state. Investigating the entropy terms of the framework itself revealed that the interrupted framework has a slightly higher entropy resulting from its higher degree of flexibility. The additional stabilization of the interrupted -COK-14 due to the four tightly bound water molecules per unit cell (Fig. 5 and 6) has not been taken into account in the DFT calculations. Including these bridging water molecules would probably demonstrate the thermodynamical preference of the periodically interrupted -COK-14 at 300 K, as experimentally observed. At 700 K (Fig. 7c), the entropy contribution (TΔS = −312 kJ mol−1) exceeds the enthalpic contribution (ΔH = 121 kJ mol−1) thus favoring framework condensation (ΔG = −191 kJ mol−1). The results indeed explain the observation that the framework can be annealed at elevated temperatures. Although this analysis contains some simplifying approximations, such as the harmonic oscillator approach and full recovery of all degrees of freedom, it allows assignment of the different factors determining the energetic evolution upon framework condensation. As shown in Fig. 7, the enthalpic term always favors the interrupted state, while the entropic contribution increases with temperature and favors the condensed framework. The framework transformation from the interrupted to the condensed framework is thereby entropy driven.
 | ||
| Fig. 7 Energy profiles of the condensed OKO framework enclosing four water molecules per unit cell (T68O136 + 4H2O) compared to the interrupted state of -COK-14 (T68O132(OH)8) at (a) 0 K, (b) 300 K and (c) 700 K. Energies are reported in kJ mol−1 per unit cell and with respect to the energy of the interrupted -COK-14 structure. | ||
In addition to the energetic approach discussed above, the potential of a zeolite for reversible framework condensation can be linked to the framework flexibility in combination with the systematic presence of vicinal silanol groups. The framework flexibility is essential to allow accommodation of small shifts in the T-atom positions resulting from the condensation of the silanol pairs.
Geometrically, the flexibility window of a zeolite can be assessed by representing the framework as a periodic assembly of rigid, regular SiO4 tetrahedra that have force-free ‘spherical’ joints at the tetrahedral corners where the oxygen atoms reside.14–16 Such idealized models allow the representation of all known zeolite frameworks within a limited density range, called the flexibility window, wherein strain is removed by rotation of the T–O–T linkages (T = Si, Ge, or Al). Although the Si–O bond length and the O–Si–O angle are strongly constrained in pure silicates, the variability of the Si–O–Si angles17 provides flexibility to 97% of the known frameworks. The remaining 3% can be obtained only when larger tetrahedra (such as AlO4 and GeO4) are incorporated at some sites. Compared to pure aluminosilicates, germanosilicates tend to be more flexible because germanates exhibit slightly ‘softer’, or less-rigid, tetrahedra, thereby allowing Ge–O–Ge angles in the range of 116–135°.
This geometric flexibility analysis reveals that the interrupted framework of -COK-14 is flexible when represented as an idealized pure silicate in its full symmetry, C2/m. In contrast, oxygen-neighbor overlap prevents flexibility in the fully 4-coordinated all-silica OKO framework of COK-14, even when its symmetry is reduced to P1. This result explains the observation that the siliceous fully condensed COK-14 is not stable and spontaneously transforms to -COK-14 upon contact with humidity. As an idealized, pure germanate however, COK-14 becomes flexible in C2 symmetry, although one of the Ge–O–Ge angles drops to 108.7°, which is small even for a germanate material. This observation provided an indication that flexibility can be restored to the silica-rich COK-14 framework by partial isomorphous substitution of Si by Ge. Indeed, the minimal requirement to restore flexibility in COK-14 is a reduction of the unit cell symmetry to P1 in combination with substitution of Si for Ge at either site 1 or site 9 to give a nominal unit cell composition of Si32Ge2O68 with a Si/Ge ratio of 16. The increased Ge–O distance relative to Si–O at these sites is sufficient to avert oxygen atom overlaps in the framework.
Framework energy calculations (GULP, SLC potential) not only support the conclusions of the geometric flexibility analysis, but also explain why the broken framework -COK-14 is the most stable geometry for this material in the siliceous form. The calculations indicate a lower energy (0.1435 eV/SiO2 relative to quartz) for the pure silicate form of COK-14 when optimized in the C2 space group symmetry, as compared to the C2/m symmetry (0.1518 eV/SiO2 relative to quartz). Even in the lower C2 symmetry, the structure can only be relaxed by substituting Si for Ge in more than 50 percent of the T6 sites (as labeled in the higher C2/m symmetry), which would indicate a maximum Si/Ge ratio of 16. The experimentally determined Si/Ge ratio of 110 for as made -COK-14 consequently is too high to render enough flexibility to the OKO framework.
This also implies that stabilization of COK-14 requires the introduction of heteroelements such as GeO4 or AlO4, thereby providing flexibility. Experimental evidence demonstrating this strategy for stabilization of COK-14 was recently reported.18 In contrast with the all-silica zeolite COK-14, the aluminosilicate version of this zeolite, obtained by framework condensation of Al-ALD aluminated -COK-14, did not return to its interrupted form upon hydration and remained stable under ambient conditions for at least 6 months.
Conclusions
COK-14/-COK-14 is the first reversible transformation encountered between a fully connected zeolite framework and an interrupted analogue, influencing hydrophilicity and porosity. Experimental evidence, in combination with two unrelated theoretical analysis methods evaluating respectively energetic and framework flexibility aspects, not only demonstrates and documents the mechanism of the framework condensation, but also explains the high temperature requirements for condensation of -COK-14 and the experimentally observed instability of all-silica, condensed framework zeolite COK-14. This information was exploited for formulating a strategy for stabilization of such frameworks by incorporation of suitable hetero-elements and allowed us to interpret and confirm recently published experimental results involving Al-ALD modified COK-14. These results show that introduction of trace amounts of germanium or aluminum provides stability to the condensed OKO framework by introduction of flexibility thereby broadening the potential for application of this recently discovered large pore zeolite in industrial applications. This work also demonstrates that the impact of entropy, including the framework flexibility, needs to be considered in assessment of framework feasibility and development of new zeolite syntheses.Acknowledgements
J.A.M. acknowledges the Flemish government for long-term structural funding (Methusalem). This work is part of a Belgian Interuniversity networking program IAP-PAI. E.B. and L.J. recognize FWO Vlaanderen for a postdoctoral fellowship and a mandate as aspirant, respectively. L.J. is a 2013–2014 Fulbright grantee and acknowledges the Vlaams Supercomputer Center for the computational resources. C.M. acknowledges financial support from the French ANR under contract ANR-12-JS08-008-01. V.V.S. and M.W. acknowledge funding from the Research Board of Ghent University (BOF) and from the European Research Council under the European Community's Seventh Framework Programme (FP7(2007–2013) ERC grant agreement number 240483). M.T. acknowledges support from the U.S. National Science Foundation, award #0835605.Notes and references
- V. Tazzoli, M. C. Domeneghetti, F. Mazzi and E. Cannillo, Eur. J. Mineral., 1995, 7, 1339 CrossRef CAS.
- Z. V. Pudovkina, L. P. Soloveva and Y. A. Pyatenko, Sov. Phys. Dokl., 1986, 31, 941 Search PubMed.
- N. Engel and K. Yvon, Z. Kristallogr., 1984, 169, 165 CrossRef CAS PubMed.
- G. Giuseppetti, F. Mazzi, C. Tadini and E. Galli, Neues Jahrb. Mineral., Monatsh., 1991, 7, 307 Search PubMed.
- H.-R. Wenk, Z. Kristallogr., 1973, 137, 113 CrossRef CAS PubMed.
- E. Estermann, L. B. McCusker, C. Baerlocher, A. Merrouche and H. Kessler, Nature, 1991, 352, 320 CrossRef.
- M. E. Davis, Nature, 1991, 352, 281 CrossRef.
- J. Sun, C. Bonneau, A. Cantín, A. Corma, M. J. Díaz-Cabañas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154 CrossRef CAS PubMed.
- A. Corma, M. J. Díaz-Cabañas, J. Jiang, M. Afeworki, D. L. Dorset, S. L. Soled and K. G. Strohmaier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13997 CrossRef CAS PubMed.
- C. Baerlocher, D. Xie, L. B. McCusker, S.-J. Hwang, I. Y. Chan, K. Ong, A. W. Burton and S. I. Zones, Nat. Mater., 2008, 7, 631 CrossRef CAS PubMed.
- E. Verheyen, L. Joos, K. Van Havenbergh, E. Breynaert, N. Kasian, E. Gobechiya, K. Houthoofd, C. Martineau, M. Hinterstein, F. Taulelle, V. Van Speybroeck, M. Waroquier, S. Bals, G. Van Tendeloo, C. E. A. Kirschhock and J. A. Martens, Nat. Mater., 2012, 11, 1059 CAS.
- J.-L. Paillaud, B. Harbuzaru, J. Patarin and N. Bats, Science, 2004, 304, 990 CrossRef CAS PubMed.
- A. Corma, M. J. Diaz-Cabanas, F. Rey, S. Nicolopoulus and K. Boulahya, Chem. Commun., 2004, 1356 RSC.
- A. Sartbaeva, S. A. Wells, M. M. J. Treacy and M. F. Thorpe, Nat. Mater., 2006, 5, 962 CrossRef CAS PubMed.
- V. Kapko, C. J. Dawson, M. M. J. Treacy and M. F. Thorpe, Phys. Chem. Chem. Phys., 2010, 12, 8531 RSC.
- M. M. Treacy, C. J. Dawson, V. Kapko and I. Rivin, Philos. Trans. R. Soc., A, 2013, 372, 20120036 CrossRef PubMed.
- C. J. Dawson, R. Sanchez-smith, P. Rez, M. O. Kee and M. M. J. Treacy, Chem. Mater., 2014, 26, 1523 CrossRef CAS.
- E. Verheyen, S. Pulinthanathu Sree, K. Thomas, J. Dendooven, M. De Prins, G. Vanbutsele, E. Breynaert, J.-P. Gilson, C. E. A. Kirschhock, C. Detavernier and J. A. Martens, Chem. Commun., 2014, 50, 4610 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: A detailed description of characterization techniques, X-ray diffraction, high temperature X-ray diffraction, 29Si MAS NMR, nitrogen adsorption and thermogravimetric analysis, and computational details. See DOI: 10.1039/c4mh00127c |
| This journal is © The Royal Society of Chemistry 2014 |