
A step forward in the sigma enigma: a role for chirality in the sigma1 receptor–ligand interaction?†
Daniela
Rossi‡
a,
Annamaria
Marra‡
a,
Marta
Rui
a,
Erik
Laurini
b,
Maurizio
Fermeglia
b,
Sabrina
Pricl
bc,
Dirk
Schepmann
d,
Bernhard
Wuensch
d,
Marco
Peviani
e,
Daniela
Curti
e and
Simona
Collina
*a
aDepartment of Drug Sciences, Medicinal Chemistry and Pharmaceutical Technology section, University of Pavia, Viale Taramelli 12, 27100 Pavia, Italy. E-mail: simona.collina@unipv.it; Fax: +39 0382-422975; Tel: +39 0382 987379
bMOSE – DEA-University of Trieste, Via Valerio 10, 34127 Trieste, Italy
cNational Interuniversity Consortium for Material Science and Technology (INSTM), Research Unit MOSE-DEA University of Trieste, Trieste, Italy
dInstitute of Pharmaceutical and Medicinal Chemistry, University of Muenster, Correnstrasse 48, 48149 Muenster, Germany
eDepartment of Biology and Biotechnology “L. Spallanzani”, Laboratory of Cellular and Molecular Neuropharmacology, University of Pavia, Via Ferrata 9, 27100 Pavia, Italy
First published on 18th September 2014
Abstract
In our recent research racemic RC-33 was identified as a potent and highly promising σ1 receptor agonist, showing excellent σ1 receptor affinity and promoting NGF-induced neurite outgrowth in PC12 cells at very low concentrations. Surprisingly, both its interaction with the biological target and its effect on neurite sprouting proved to be non-stereoselective. Starting from the observation that a hydrogen bond center in the scaffold of a σ1 ligand is an important pharmacophoric element for receptor/ligand interaction, we hypothesized that the absence of such pharmacophoric feature in the structure of RC-33 could be also responsible for the lack of enantioselectivity in its interaction with the target receptor. To verify our hypothesis, in this paper we evaluated – both in silico and in vitro – the ability of a series of enantiomeric arylalkylaminoalcohols and arylpyrrolidinols 1–5 to interact with the receptor. All these compounds are structurally related to RC-33 and are characterized by the presence of an –OH group as the additional pharmacophore feature. Interestingly, the results of our study show that the σ1 receptor exhibits enantiopreference toward compounds characterized by (S)-configuration at the stereogenic center bearing the aromatic moiety only when the alcoholic group is also present at that chiral center, thus supporting our original hypothesis.
Introduction
The sigma (σ) binding sites were originally defined and classified as opioid receptor subtypes.1 Later investigations demonstrated that σ receptors were distinct from opioid and phencyclidine analogues, and since then at least two distinct σ receptor subtypes, designated σ1 and σ2,2 have been pharmacologically characterized.3–5 In particular, the σ1 receptor subtype has been purified and cloned from several animal species and humans.6,7 σ1 receptors are ubiquitously expressed in mammalian tissues and highly distributed in the central nervous system (CNS),8–10 with the highest density found in the spinal cord, cerebellum, hippocampus, hypothalamus, midbrain, cerebral cortex, and pineal gland. Strong pharmacological evidence indicates that σ1 receptors are involved in the pathophysiology of all major CNS disorders,11 including mood disorders (anxiety12 and depression13), psychosis and schizophrenia,14 as well as drug addiction, pain,15 and neurodegenerative diseases such as Parkinson's, Alzheimer's, and amyotrophic lateral sclerosis.16 Moreover, from a biological perspective, the σ1 receptors reside in the endoplasmic reticulum (ER) at the ER–mitochondria interface,17 and they are unique ligand-regulated molecular chaperones18–20 that can translocate to the plasma membrane or to other subcellular compartments under stressful conditions and/or pharmacological manipulation.Ligands displaying preferential affinity for the σ1 receptor subtype are (+)-benzomorphans such as (+)-pentazocine and (+)-N-allylnormetazocine (NANM, SKF-10047), whereas haloperidol and 1,3-di-(2-tolyl)guanidine (DTG) exhibit high affinity for both receptor subtypes.21 Since (+)-pentazocine shows a very low affinity for the σ2 receptors, it represents the prototypical selective agonist used in its tritiated form to label σ1 receptors. Several compounds endowed with σ1 affinity and selectivity, characterized by different scaffolds, have been identified, e.g., arylalkylamines,22a–f benzooxazolones,23 and spirocyclic pyranopyrazoles,24 and different pharmacophore models for σ1 receptor ligands have been published. All these models share the common features of a basic amino group and at least two hydrophobic substituents at the basic nitrogenatom.25a–f However, the last-generation, three-dimensional (3D) pharmacophore models25c–f are characterized by an additional pharmacophore requirement: a heterogroup in the scaffold of the molecule that is able to form hydrogen bond interactions with the receptor counterpart. Actually, heteroatoms such as O or S are frequently present in very potent σ1 ligands, bridging the aromatic component and the classic alkyl or cycloalkyl intermediate spacer linked to the basic nitrogenatom.26,27
In this scenario, our group designed and synthesized a large number of very interesting σ1 receptor ligands.22a–c Among these, the most promising molecule is 1-[3-(1,1′-biphen)-4-yl]butylpiperidine (RC-33, Fig. 1), showing excellent σ1 receptor affinity and agonistic profile in its racemic form, as testified by its Kiσ1 value of 0.70 ± 0.3 nM and by the potentiation of the NGF-induced neurite outgrowth in the PC12 cell at very low concentrations.22c,d
 | ||
| Fig. 1 (R,S)-RC-33. | ||
Racemic resolution of RC-33 and isolation of the enantiomers revealed that (i) the interaction with the biological target is non-stereoselective (Kiσ1(S)-RC-33 = 1.9 ± 0.2 nM, Kiσ1(R)-RC-33 = 1.8 ± 0.1 nM) and (ii) the pharmacological activity is not dependent on the absolute configuration.22e,f The behavior of RC-33 is particularly surprising, since the enantioselectivity of the σ1 receptor is well documented.20 Starting from the observation that an important pharmacophoric element is missing in the RC-33 structure (i.e., a hydrogen bond donor or acceptor),25c–e we first hypothesized that the absence of such pharmacophoric feature could be responsible for the lack of enantioselectivity in the interaction with the biological target. Then, we verified our hypothesis by evaluating the ability of a series of enantiomeric arylalkylaminoalcohols and arylpyrrolidinols, structurally related to RC-33, to interact with σ1 receptors. Specifically, here we report and discuss in detail the results of the in silico study, synthesis, chiral resolution and biological evaluation of the arylalkylaminoalcohol 1 (Table 1), analogue of RC-33, complemented by the in silico and in vitro studies of other enantiomeric arylalkylaminoalcohol and arylpyrrolidinol derivatives (Table 1). These molecules were selected from a compound cohort previously prepared and characterized by us as analgesic agents with effects similar or higher than morphine but never evaluated as σ1 receptor ligands.28a–d
|
|
|||||
|---|---|---|---|---|---|
| Compound | Template | Ar | R1 | R2 | NR3R4 |
| (R,S)-1 | I | biphenyl-4yl | CH3 | H | N(CH2)5 |
| (R)-1 | |||||
| (S)-1 | |||||
| (R,S)-2 | I | naphth-2-yl | CH3 | H | N(CH3)2 |
| (R)-2 | |||||
| (S)-2 | |||||
| (R,S)-3 | I | 6-methoxy-naphth-2yl | CH3 | H | N(CH3)2 |
| (R)-3 | |||||
| (S)-3 | |||||
| (2R/S,3S/R)-4 | II | naphth-2yl | CH3 | CH3 | |
| (2R,3S)-4 | |||||
| (2S,3R)-4 | |||||
| (2R/S,3S/R)-5 | II | 6-methoxy-naphth-2yl | CH3 | CH3 | |
| (2R,3S)-5 | |||||
| (2S,3R)-5 | |||||

The final aim of our work is to understand how chirality may affect the σ1 receptor–ligand interaction and activity, thus contributing a step forward in unveiling the sigma-enigma. Indeed, even although our knowledge of the sigma receptors has evolved over the past 20 years, several aspects in the sigma field still remain rather obscure.
Results and discussion
Compound selection
With the aim of evaluating the role of a hydrogen bond center as an additional pharmacophore element in the stereoselective interaction with the σ1 receptor we designed compound 1, featuring an alcoholic function on the alkyl spacer bridging the aromatic ring to the basic nitrogenatom, as an analogue of the arylalkylamino derivative RC-33 (Table 1).For the purpose of comparison and discussion, we selected other structurally related alcoholic compounds from our library of chiral molecules synthesized over the years. Among these, we chose molecules 2 and 3 (template I, Table 1) as structurally related to 1, and the constrained arylpyrrolidinols 4 and 5 (template II, Table 1), being characterized by less conformational freedom.28a,c,d
Synthesis, chiral resolution and configurational assignment
For the synthesis of (R,S)-1 we planned to follow the methodology described in our previous work, with suitable modifications (Scheme 1).28a We started our synthetic approach with the synthesis of 4-piperidinyl butan-2-one (6), obtained via Michael addiction from a solution of piperidine and but-3-en-2-one in PEG 400 in good yield (62%). Concerning the synthesis of (R,S)-1, β-aminoketone 6 was added to the biphenyl anion, obtained by halogen–metal exchange between the aromatic substrate and tert-butyllitium (tert-BuLi) in anhydrous ethyl ether (Et2O) at −40 °C. After an acid–base work-up and purification by crystallization from methanol/water, (R,S)-1 was obtained as a white solid in good yield (68%). The final compound was characterized by 1H-NMR. | ||
| Scheme 1 Synthesis of (R,S)-1. Reagents and conditions: (a) t-BuLi, anhydrous Et2O, −40 °C to rt; (b) 4-piperidinyl butan-2-one (6), −78 °C to 0 °C; (c) H2O rt. | ||
In order to make (R,S)-1 suitable for biological assays, a small amount of this compound was obtained in its salt form as (R,S)-1·dl-tartrate.
Chiral resolution of (R,S)-1 was achieved using chiral high performance liquid chromatography (HPLC). To identify the best experimental condition for the subsequent scaling-up, a standard screening protocol for cellulose and amylose derived chiral stationary phases (CSPs) was applied to the Chiralcel OJ-H (4.6 mm diameter × 150 mm length, 5 μm), Chiralpak AS-H (4.6 mm diameter × 250 mm length, 5 μm) and Chiralpak IC (4.6 mm diameter × 250 mm length, 5 μm) columns, whose chiral selectors are cellulose tris-(4-methylbenzoate) (Chiralcel OJ-H) and amylosetris [(S)-α-methylbenzylcarbamate] (Chiralpak AS-H) coated on a silica gel substrate and cellulose tris (3,5-dichlorophenylcarbamate) immobilized on silica gel (Chiralpak IC). Elution conditions adopted included mixtures of n-heptane and polar modifiers (EtOH or 2-propanol), alcohols (MeOH, EtOH, and 2-propanol), and acetonitrile; in all cases 0.1% of diethylamine was added to the mobile phase; in the analysis with Chiralpak IC 0.3% of trifluoroacetic acid was also added. The best result, in terms of enantioselectivity (α) and resolution factor (RS), was obtained with Chiralcel OJ-H, eluting with MeOH/diethylamine (100/0.1, v/v), as clearly illustrated in Fig. 2 (tr1 = 6.99 min; tr2 = 9.59 min; α = 1.98; RS = 6.93).
 | ||
| Fig. 2 Analytical separation of (R,S)-1. Chromatographic conditions: Chiralcel OJ-H (4.6 mm × 150 mm, 5 μm), MeOH/diethylamine 100/0.1 (v/v), flow rate: 0.5 mL min−1, UV detector at 254 nm. | ||
These experimental conditions are characterized by the most important prerequisites for an economic and productive enantiomeric separation on a semi-preparative scale, such as high solubility of racemate and enantiomers in the eluent solvent, shortest retention times, and the use of a mobile phase consisting of a pure low-cost solvent, which ultimately facilitates workup and re-use of the mobile phase. Therefore, the analytical method was suitably transferred to the semi-preparative scale employing a Chiralcel OJ-H column (10 mm × 250 mm, 5 μm). In 17 cycles, 51 mg of (R,S)-1 were processed, yielding 22.1 mg of the first eluted enantiomer and 23.2 mg of the second eluted one, characterized by [α]20D values of +24.1 and −24.2, respectively (c: 0.5 in MeOH), along with 5.7 mg of an intermediate fraction as a mixture of the two enantiomers. Both enantiomers of 1 were obtained with a yield of about 87% and ee ≥ 99.9%, as evidenced by analytical control of the collected fractions.
The configuration assignment study of the resolved enantiomers of compound 1 was then performed comparing the electronic circular dichroism (ECD) curves of (+)-1 with that of (S)-(−)-2, whose absolute configuration was already assigned.28a The ECD spectra (reported in the ESI†) of both (+)-1 and (S)-(−)-2 evidenced a similar profile in the range of wavelength between 200 and 300 nm. In detail, both compounds show a negative Cotton Effect (CE) at about 210 nm [(+)-1: λmax 206.5 nm, Mol. CD −6.61; (S)-(−)-2: λmax 209.0 nm, Mol. CD −6.21] and a positive CE in the range 220–260 nm [(+)-1: λmax 253.5 nm, Mol. CD +2.60; (S)-(−)-2: λmax 224.0 nm, Mol. CD +16.80]. Based on these considerations, the absolute configuration (S) was assigned to (+)-1. Both enantiomers of 1 were finally converted into the corresponding tartrates [(R)-1·l-tartrate and (S)-1·d-tartrate, respectively], suitable for biological investigation.
Molecular modeling studies
Molecular Dynamics (MD) simulations were carried out to predict binding mode, affinity, and eventual stereoselective binding features of the selected compounds towards the σ1 receptors. To the purpose, both (R) and (S) enantiomers of compounds 1–5 were modeled and the relevant free energy of binding (ΔGbind) with the protein was estimated via MM/PBSA calculations29a,b using the optimized structure of the compounds in complex with our validated homology model of the σ1 receptor.30a,bTaking compounds (R)-1 and (S)-1 as a proof-of-concept, the analysis of the corresponding MD trajectories revealed that four major types of interactions are involved in the binding mode of both (R)-1 and (S)-1 to the σ1 receptor, as shown in Fig. 3A and B: (i) a permanent salt bridge is detected between the –NH+ moiety of the ligand piperidine ring and the COO− group of Asp126; (ii) the side chains of Arg119 and Trp121 are engaged in stabilizing π interactions with the biphenyl group of the ligands; (iii) several further hydrophobic interactions concur to stabilize compound/receptor binding, mainly via the side chains of the σ1 residues belonging to the hydrophobic pocket Ile128, Phe133, and Tyr173; and (iv) a hydrogen bond (HB) between the hydroxyl group of the compounds and the carboxylic chain of Glu172 conclusively anchors the ligand to the protein binding cavity.
 | ||
| Fig. 3 (A) Two dimensional schematic representation of postulated interactions between the σ1 receptor and 1, established by direct affinity measurements. The lines/arrows indicate proposed key interaction between the receptor and its ligand. (B) Modeled complex of the σ1 receptor with (S)-1 showing the key interactions proposed in the topographical interaction model depicted in part A. The main protein residues involved in these interactions are Arg119 (red), Trp121 (cyan), Asp126 (blue), Ile128 (forest green), Glu172 (yellow), and Tyr173 (magenta). The ligand is portrayed in sticks and balls and colored by element, while the protein residues mainly involved in the interaction with (S)-1 are highlighted as colored sticks and labeled. Salt bridge and H-bond interactions are shown as black lines. Water, ions, and counterions are not shown for clarity. | ||
The results of our modeling investigation predict that both enantiomers of 1 can be aptly accommodated within the σ1 binding site and establish similar networks of stabilizing interactions with the receptor.
To quantify the overall effect of these interactions, binding free energy calculations were applied and, according to our simulations, both molecules are endowed with similar affinities towards the biological target, with a slight preference of the receptor for the (S) enantiomer, as ΔGbind = −10.81 ± 0.22 kcal mol−1 for (R)-1 and ΔGbind = −11.09 ± 0.23 kcal mol−1 for (S)-1. The same trend was obtained for all other protein/ligand complexes considered, although compounds 2–5 showed lower affinities towards the σ1 receptor with respect to the biphenyl derivatives, as seen from the ΔGbind values listed in Table 2.
| Compounds | ΔHbind kcal mol−1 | −TΔS kcal mol−1 | ΔGbind kcal mol−1 | K iσ1(calcd)* |
|---|---|---|---|---|
| (R)-1 | −23.89 ± 0.09 | −13.08 ± 0.20 | −10.81 ± 0.22 | 12 nM |
| (S)-1 | −24.20 ± 0.08 | −13.11 ± 0.22 | −11.09 ± 0.23 | 7.5 nM |
| (R)-2 | −22.55 ± 0.11 | −12.83 ± 0.21 | −9.72 ± 0.24 | 76 nM |
| (S)-2 | −22.71 ± 0.12 | −12.79 ± 0.20 | −9.92 ± 0.23 | 54 nM |
| (R)-3 | −22.51 ± 0.13 | −12.81 ± 0.23 | −9.70 ± 0.26 | 78 nM |
| (S)-3 | −22.74 ± 0.10 | −12.86 ± 0.19 | −9.88 ± 0.21 | 57 nM |
| (2R,3S)-4 | −20.97 ± 0.08 | −11.88 ± 0.22 | −9.09 ± 0.23 | 219 nM |
| (2S,3R)-4 | −21.46 ± 0.09 | −11.93 ± 0.24 | −9.53 ± 0.26 | 104 nM |
| (2R,3S)-5 | −19.90 ± 0.10 | −11.79 ± 0.21 | −8.11 ± 0.23 | 1.1 μM |
| (2S,3R)-5 | −19.89 ± 0.12 | −11.70 ± 0.18 | −8.19 ± 0.22 | 998 nM |
To investigate in detail the reason for this behavior, deconvolution of the enthalpic component (ΔHbind) of the binding free energy into contributions from each protein residue was carried out. As shown in Fig. 4 for compounds (R)-1 and (S)-1, the stable salt bridge involving Asp126 is responsible for comparable stabilizing contribution of −2.15 kcal mol−1 and −2.19 kcal mol−1, respectively (average dynamic length (ADL) = 4.11 ± 0.05 Å for (R)-1 and ADL= 4.08 ± 0.06 Å for (S)-1). Furthermore, the substantial van der Waals and electrostatic interactions contributed, via the aforementioned π interaction, by Arg119 (−0.82 kcal mol−1 for (R)-1 and −0.88 kcal mol−1 for (S)-1) and Trp121 (−1.01 kcal mol−1 for (R)-1 and −0.98 kcal mol−1 for (S)-1), and by the residues belonging to the hydrophobic pocket Ile128, Phe133, and Tyr173 (with a clustered contribution of −3.08 kcal mol−1 for (R)-1 and −3.04 kcal mol−1 for (S)-1), also did not discriminate the affinity of the enantiomers for the receptor. In contrast, the stabilizing effects provided by the permanent hydrogen bond through interactions with Glu172 are dissimilar, as confirmed by the corresponding ADL (2.08 ± 0.06 Å for (R)-1 and 1.92 ± 0.04 Å for (S)-1, Fig. 4A) and, more importantly, the specific ΔHbind values (−1.01 kcal mol−1 for (R)-1 and −1.59 kcal mol−1 for (S)-1, Fig. 4B). This structural and energetical evidence explains the slightly higher affinity of the enantiomer with (S) configuration toward the σ1 receptor.
 | ||
| Fig. 4 (A) Comparison between the zoomed view of a MD representative snapshot of the hydrogen bond interaction between (R)-1 (purple) and (S)-1 (green) with the σ1 receptor residue Glu172. The compounds are portrayed as ball-and-stick, while the amino acid is depicted as stick and colored accordingly. (B) Per residue energy decomposition for the σ1 receptor in complex with (R)-1 (purple) and (S)-1 (green), showing those residues for which |ΔHbind| > 0.60 kcal mol−1. | ||
This per residue-based analysis allowed us to better understand and quantitatively explain the differences in affinity among all compounds of the series. As shown in Table 2, the derivatives 2 and 3 show a decrease in ΔGbind of about 1 kcal mol−1 compared to the best ligand 1. Based on the results of our computational approach both molecules are able to preserve all key interactions with the main σ1 residues involved in the binding site (Fig. S3A and B†). Nevertheless, taking the (S) enantiomers as reference for our considerations, the replacement of the piperidine portion of (S)-1 with a less bulky N,N-dimethyl group leads to a substantial reduction of the stabilizing contribution afforded by the σ1 amino acids Ile128, Phe133, and Tyr172 which constitute the typical hydrophobic pocket of the σ1 binding site (Fig. S4†). Actually, if the contribution of the other residues remained comparable to that of (S)-1, the clustered ΔHbind of these three residues (−1.36 kcal mol−1 for (S)-2 and −1.45 kcal mol−1 for (S)-3, respectively) would become significantly lower in comparison with the value of the piperidine derivative (−3.04 kcal mol−1).
Regarding the constrained derivatives (2S,3R)-4 and (2S,3R)-5, the increased structural rigidity leads to a different binding pose with respect to the compounds discussed above. As already shown in other studies on σ1 ligands,25f,30 to comply with the pharmacophoric requirements upon target binding these molecules must adopt a reverse orientation into the hydrophobic binding pocket (Fig. S3C and D†). Therefore, the arylpyrrolidinol derivative (2S,3R)-4 exhibits a moderate binding affinity (Kiσ1(calcd) = 104 nM, Table 2) since its naphthyl moiety can still be encased in the binding pocket by establishing favorable interactions with the involved σ1 residues. As a result of this binding pose, the interaction profile of Ile128, Phe133, and Tyr172 with (2S,3R)-4 is very similar to that of compound (S)-1 (Fig. S4†). However, the structure of this compound prevents it to establish other stabilizing interactions: indeed, we detected a drastic loss in the stabilization effect of the salt bridge with Asp126 and of the hydrogen bond with Glu172 (Fig. S4†). In addition, the contribution of the π interaction with Arg119 and Trp121 was completely abolished (Fig. S3†). Finally, the 6-OCH3 substituted compound (2S,3R)-5 ranks as the weakest σ1 binder of the series, with an estimated affinity in the μM range (Table 2). In fact, the steric hindrance of its methoxyl group prevents the molecule to penetrate deeply in the receptor binding pocket, (Fig. S3D†) with a consequent overall decrease of all binding stabilizing interactions (Fig. S4†).
Taken together, our in silico studies support our original hypothesis: actually, the presence of an extra pharmacophoric feature in compounds 1–5, missing in the original compound RC-33, leads to an additional interaction of the ligands with the σ1 receptor. Importantly, however, the presence of this feature does not afford a meaningful contribution in differentiating binding affinity of enantiomeric ligands. But, at the same time, it seems to be a potential key-requirement for the stereoselective compound interaction with the σ1 receptor. In fact, notwithstanding the interaction spectra of (R)- and (S)-1 reported in Fig. 4B differ, both qualitatively and quantitatively, from those obtained for (R)- and (S)-RC-33,22e the contribution afforded by each residue involved in ligand binding is somewhat lower in the case of 1 with respect to RC-33, ultimately resulting in an only slightly lower affinity of 1 for the receptor. Hence, the presence of the additional pharmacophore feature detected for the present series of compounds seems to play an orientational, rather than an energetic role, in the selectivity of σ1 for their (S) enantiomers.
Pharmacological evaluation
The affinities of (R,S)-1–3, (2R/S,3S/R)-4–5, (R)-1–3, (2R,3S)-4–5, (S)-1–3, and (2S,3R)-4–5 towards the σ1 and σ2 receptors were experimentally determined in radioligand receptor binding studies.In σ1receptor bindingassay the test compounds compete with a potent and selective radioligand (i.e. [3H]-(+)-pentazocine) for the respective binding site. Nonspecific binding was recorded in the presence of cold non-radiolabeled (+)-pentazocine in large excess. Membrane preparations from guinea pig cerebral cortex homogenates served as the receptor source. In the σ2 assay, membrane preparations of the rat liver served as the source for σ2 receptors. The nonselective radioligand [3H]DTG was employed in the σ2 assay because no σ2-selective radioligands are commercially available yet. To mask the σ1 receptors, an excess of non-tritiated (+)-pentazocine was added to the assay solution, while a high concentration of non-tritiated DTG was used to determine nonspecific binding. In Table 3 the σ1 and σ2 receptor affinities of all tested compounds are summarized and compared with affinities of racemic and enantiomeric RC-3322e as reference compounds. With the only exception of arylpyrrolidinol 5, which is a weak σ1 receptor binder, all compounds generally showed an interesting σ1 affinity, in accordance with our in silico predictions (Table 2). Most importantly, the (S)-configured enantiomers at the stereogenic center directly linked to the aromatic moiety exhibit a preferential interaction with the target protein, thus suggesting that the interaction with the receptor is stereoselective. This is particularly evident for (S)-1, which shows a eudismic ratio of about 8 and represents the compound with both the highest affinity and selectivity toward σ2 receptors among all molecules investigated (Kiσ1 = 4.7 ± 0.3 nM, Kiσ2/Kiσ1= 382, Table 3).
| Compound | K iσ1 (nM) ± SEM | K iσ2 (nM) ± SEM |
|---|---|---|
| (R,S)-RC-33 | 0.9 ± 0.3 | 103 ± 10 |
| (R)-RC-33 | 1.8 ± 0.1 | 45 ± 16 |
| (S)-RC-33 | 1.9 ± 0.2 | 98 ± 64 |
| (R,S)-1 | 6.57 ± 0.2 | 34.6 ± 47 |
| (R)-1 | 39 ± 8 | 4.3 μM ± 315 |
| (S)-1 | 4.7 ± 0.3 | 1.8 μM ± 288 |
| (R,S)-2 | 77 ± 23 | 66 ± 13 |
| (R)-2 | 205 ± 60 | 651 ± 67 |
| (S)-2 | 63 ± 39 | 75 ± 5 |
| (R,S)-3 | 41 ± 11 | 97 ± 18 |
| (R)-3 | 51 ± 14 | 133 ± 63 |
| (S)-3 | 25 ± 4 | 1.1 μM ± 223 |
| (2R,S/3S,R)-4 | 65 ± 18 | 366 ± 64 |
| (2R,3S)-4 | 86 ± 16 | 94 ± 23 |
| (2S,3R)-4 | 26 ± 2 | 432 ± 53 |
| (2R,S/3S,R)-5 | 1.9 μM ± 304 | 1.5 μM ± 219 |
| (2R,3S)-5 | 1.5 μM ± 226 | 1.2 μM ± 212 |
| (2S,3R)-5 | 1.2 μM ± 257 | 1.9 μM ± 293 |
Racemic and enantiomeric 1 were then selected for further investigation in our validated PC12 cell model of neuronal differentiation with the purpose of determining their agonistic/antagonistic profile and investigating the role of chirality and their effect on NGF-induced neurite outgrowth. The range of concentrations for racemic and enantiomeric 1 was chosen according to our previous assays22c performed on RC-33. Both (S)-1·d-tartrate and (R,S)-1·dl-tartrate displayed a σ1 agonistic profile, consistently and significantly potentiating NGF-induced neurite outgrowth at concentrations of 2.5 and 5 μM (p = 0.05 and p < 0.05, respectively, vs.NGF alone for (R,S)-1; p < 0.01 and p < 0.005, respectively, vs.NGF alone for (S)-1, Fig. 5). Consistently, the effect of these compounds was totally blocked by co-administration of the selective σ1antagonist NE-100. In contrast, (R)-1·l-tartrate did not affect the percentage of cells with neurite outgrowth with respect to NGF alone (Fig. 5). Importantly, (S)-1·dl-tartrate was more effective than the corresponding racemate in promoting NGF induced neurite outgrowth (p < 0.01 vs. (R,S)-1, Fig. 5).
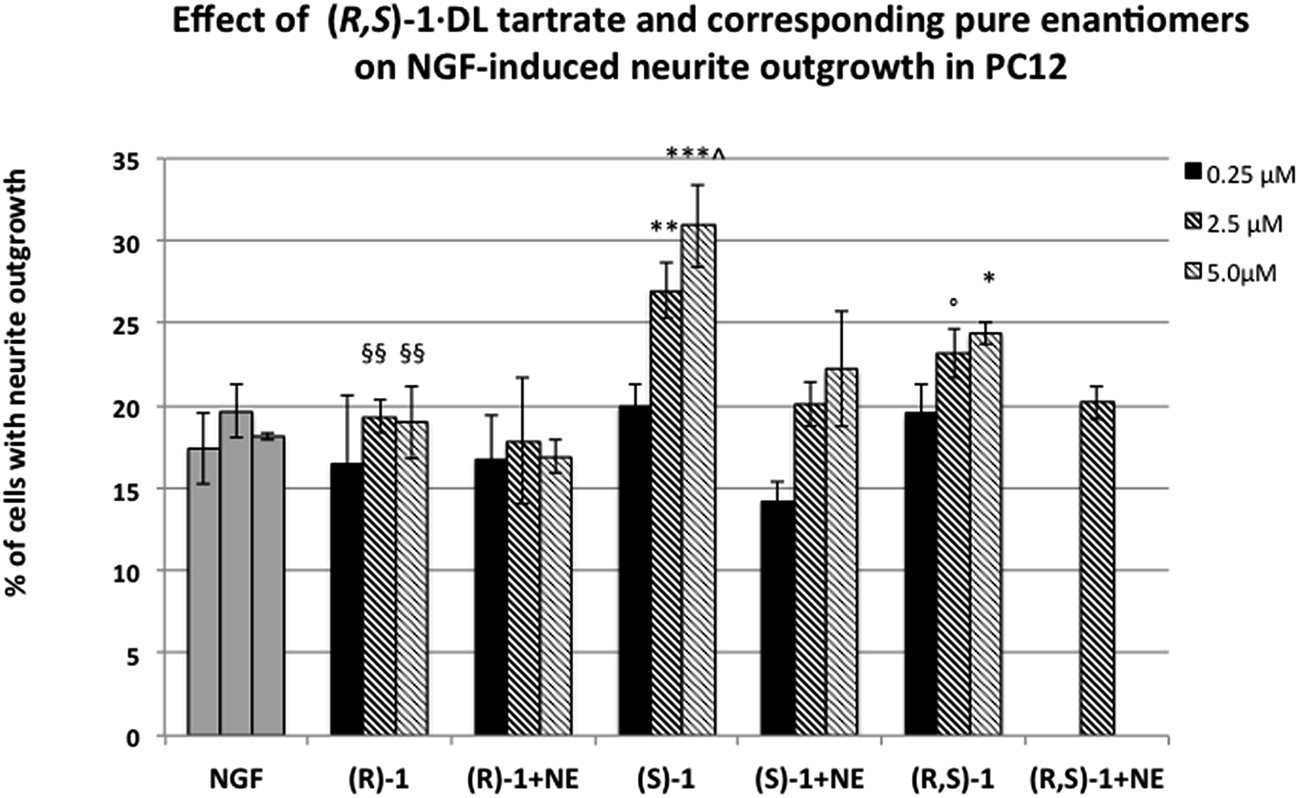 | ||
| Fig. 5 Effect of σ1 receptor ligands (R,S)-1·dl-tartrate and corresponding enantiomers on NGF-induced neurite outgrowth. Co-administration of NE-100 selective σ1 receptor antagonist totally blocked the potentiating effect of (R,S)-1 and (S)-1 compounds. Histograms represent the mean ± SEM of at least three different experiments performed in duplicate. *** = p < 0.005; ** = p < 0.01; * = p < 0.05; ° = p = 0.05 vs.NGF alone. ^ = p < 0.01 vs. (R,/S)-1; §§ = p < 0.01 vs. (S)-1. | ||
Taken together, these results show that (S)-1·d-tartrate is the eutomer. Indeed it enhances NGF-induced neurite outgrowth and its efficacy is greater than (R,S)-1·dl-tartrate, while (R)-1·l-tartrate is not effective in promoting NGF induced neurite outgrowth in PC12 cells at the same concentrations.
Conclusions
The stereoselectivity of the ligand binding to σ1 receptor remains one of the obscure yet intriguing aspects of the activity of this enigmatic transmembrane protein. In this paper, to enrich our knowledge of the structural origins of the enantioselective interaction of σ1 ligands we studied the enantiomeric compounds 1–5 structurally related to the potent σ1agonistRC-33. According to the latest and more specific σ1 receptor 3D pharmacophore models and with respect to the reference ligand RC-33 (for which the interaction with its target receptor is not dependent on the absolute configuration), compounds 1–5 present an additional pharmacophoric feature: a hydrogen bond center. Our in silico analysis of the binding modes and interactions between compounds 1–5 and the σ1 receptor revealed that, for these molecules, four major intermolecular interactions are involved in stabilizing ligand binding within the receptor binding pocket. Among those, the extra hydrogen bond interaction – missing in the RC-33/σ1 receptor complex – plays a role in mild enantiomeric binding discrimination. Interestingly, the mechanism of enantiomer recognition is typically described assuming that three31 or four32 key interactions are necessary to distinguish one enantiomer from the other. Thus, our results seem to be in line with this view. Accordingly, for all studied compounds, a weak (about two) to moderate (about eight) stereoselectivity in the interaction with the σ1 receptor was observed and (S)-1 was found to be the most active compound of the entire series (Kiσ1 = 4.7 nM, eudismic ratio = 8). In summary, we showed that the σ1 receptor exhibits enantiopreference toward compounds characterized by (S)-configuration at the stereogenic center bearing the aromatic moiety only when the alcoholic group is present at the chiral center. Although a more robust and populated dataset of compounds is undoubtedly needed to verify our hypothesis, we postulate that a heterogroup at the chiral center is required for a σ1–ligand interaction to be stereoselective. An effort to corroborate this claim is ongoing in our laboratories.Regarding the effect in promoting neurite outgrowth, the results of the functional assays related to 1 demonstrated that the chirality of the molecule affects the biological activity; indeed (S)-1 enhances NGF-induced neurite outgrowth and, also, its efficacy is greater than (R,S)-1. Most importantly (R)-1 is not effective in potentiating NGF-induced neurite outgrowth at the tested concentrations.
Altogether our observations provide further insights into the role of chirality in the σ1 receptor–ligand interaction and represent a step-forward in future development of more specific and effective σ1agonists.
Author contribution
Simona Collina, Sabrina Pricl and Daniela Curti conceived the work and contributed in reviewing the whole manuscript. S.C. was also responsible for the correctness of the whole studies. Daniela Rossi was responsible for the design of the experimental work and for data analysis of the whole study and wrote the manuscript. Annamaria Marra and Marta Rui performed the synthesis and chiral resolution of compounds (A.M. also contributed to the writing of the manuscript). Erik Laurini, Maurizio Fermeglia and Sabrina Pricl were responsible for the in silico studies (E.L also contributed the writing of molecular modeling section). Dirk Schepmann, Bernhard Wuensch Marco Peviani and Daniela Curti were responsible for biological investigations (D.S and B.W: binding assays; D.C. and M. P.: NGF-induced neurite outgrowth investigations) and contributed to the writing of the biological section.Acknowledgements
D.R., S.C., M.P., and D.C. gratefully acknowledge financial support from ARISLA (Grant SaNet-ALS). E.L., M.F., and S.P. gratefully acknowledge financial support from ESTECO s.r.l. (Gran DDOS). Access to the CINECA supercomputing facility was granted through the sponsored Italian Super Computing Resource Allocation (ISCRA), projects INSIDER and SIMBIOSY (to E.L. and S.P.).References
- W. R. Martin, C. E. Eades, J. A. Thompson, R. E. Huppler and P. E. Gilbert, J. Pharmacol. Exp. Ther., 1976, 197, 517–532 CAS.
- W. D. Bowen, Pharm. Acta Helv., 2000, 74, 211–218 CrossRef CAS.
- S. B. Hellewell and W. D. Bowen, Brain Res., 1990, 527, 235–236 CrossRef.
- Y. Itzhak and I. Stein, Brain Res., 1991, 566, 166–172 CrossRef CAS.
- R. Quirion, W. D. Bowen, Y. Itzhak, J. L. Junien, J. M. Mustacchio, R. B. Rothman, T. P. Su, S. W. Tam and D. P. Taylor, Trends Pharmacol. Sci., 1992, 13, 85–86 CrossRef CAS.
- S. McLean and E. Weber, Neuroscience, 1988, 25, 259–269 CrossRef CAS.
- R. R. Matsumoto, M. K. Hemstreet, N. L. Lai, A. Thurkauf, B. R. De Costa, K. C. Rice, S. B. Helleweel, W. D. Bowen and J. M. Walker, Pharmacol., Biochem. Behav., 1990, 36, 151–155 CrossRef CAS.
- G. Alonso, V. L. Phan, I. Guillemain, M. Saunier, A. Legrand, M. Anoal and T. Maurice, Neuroscience, 2000, 97, 155–170 CrossRef CAS.
- V. L. Phan, G. Alonso, F. Sandillon, A. Privat and T. Maurice, Soc. Neurosci. Abstr., 2000, 26, 2172 Search PubMed.
- S. Collina, R. Gaggeri, A. Marra, A. Bassi, S. Negrinotti, F. Negri and D. Rossi, Expert Opin. Ther. Pat., 2013, 23(5), 597–613 CrossRef CAS PubMed.
- T. Maurice and T. P. Su, Pharmacol. Ther., 2009, 124, 195–206 CrossRef CAS PubMed.
- S. K. Kulkarni and A. Dhir, Expert Rev. Neurother., 2009, 9, 1021–1034 CrossRef CAS PubMed.
- J. E. Bermack and G. J. Debonnel, Pharmacol. Sci., 2005, 97, 317–336 CrossRef CAS.
- S. H. Snyder and B. L. Largent, J. Neuropsychiatry Clin. Neurosci., 1989, 1(1), 7–15 CAS.
- A. A. Luty, J. B. Kwok, C. Dobson-Stone, C. T. Loy, K. G. Coupland, H. Karlstrom, T. Sobow, J. Tchorzewska, A. Maruszak, M. Barcikowska, P. K. Panegyres, C. Zekanowski, W. S. Brooks, K. L. Williams, I. P. Blair, K. A. Mather, P. S. Sachdev, G. M. Halliday and P. R. Schofield, Ann. Neurol., 2010, 68, 639–649 CrossRef CAS PubMed.
- M. Peviani, E. Salvaneschi, L. Bontempi, A. Petese, A. Manzo, D. Rossi, M. Salmona, S. Collina, P. Bigini and D. Curti, Neurobiol. Dis., 2014, 62, 218–232 CrossRef CAS PubMed.
- T. Hayashi, R. Rizzuto, G. Hajnoczky and T. P. Su, Trends Cell Biol., 2009, 19, 81–88 CrossRef CAS PubMed.
- T. Hayashi, Z. Justinova, E. Hayashi, G. Cormaci, T. Mori, S. Y. Tsai, C. Barnes, S. R. Goldberg and T. P. Su, J. Pharmacol. Exp. Ther., 2010, 332, 1054–1063 CrossRef CAS PubMed.
- T. Hayashi and T. P. Su, J. Pharmacol. Exp. Ther., 2003, 306, 726–733 CrossRef CAS PubMed.
- T. Hayashi, S. Y. Tsai, T. Mori, M. Fujimoto and T. P. Su, Expert Opin. Ther. Targets, 2011, 15, 557–577 CAS.
- J. M. Walker, W. D. Bowen, F. O. Walker, R. R. Matsumoto, B. De Costa and K. C. Rice, Pharmacol. Rev., 1990, 42, 355–402 CAS.
- (a) S. Collina, G. Loddo, M. Urbano, L. Linati, A. Callegari, F. Ortuso, S. Alcaro, C. Laggner, T. Langer, O. Prezzavento, G. Ronsisvalle and O. Azzolina, Bioorg. Med. Chem., 2007, 15, 771–783 CrossRef CAS PubMed; (b) D. Rossi, M. Urbano, A. Pedrali, M. Serra, D. Zampieri, M. G. Mamolo, C. Laggner, C. Zanette, C. Florio, D. Shepmann, B. Wünsch, O. Azzolina and S. Collina, Bioorg. Med. Chem., 2010, 18, 1204–1212 CrossRef CAS PubMed; (c) D. Rossi, A. Pedrali, M. Urbano, R. Gaggeri, M. Serra, L. Fernandez, M. Fernandez, J. Caballero, S. Rosinsvalle, O. Prezzavento, D. Shepmann, B. Wünsch, M. Peviani, D. Curti, O. Azzolina and S. Collina, Bioorg. Med. Chem., 2011, 19, 6210–6224 CrossRef CAS PubMed; (d) D. Rossi, A. Marra, P. Picconi, M. Serra, L. Catenacci, M. Sorrenti, E. Laurini, M. Fermeglia, S. Pricl, S. Brambilla, N. Almirante, M. Peviani, D. Curti and S. Collina, Bioorg. Med. Chem., 2013, 21, 2577–2586 CrossRef CAS PubMed; (e) D. Rossi, A. Pedrali, R. Gaggeri, A. Marra, L. Pignataro, E. Laurini, V. DalCol, M. Fermeglia, S. Pricl, D. Schepmann, B. Wünsch, M. Peviani, D. Curti and S. Collina, ChemMedChem, 2013, 8, 1514–1527 CrossRef CAS PubMed; (f) D. Rossi, A. Pedrali, A. Marra, L. Pignataro, D. Schepmann, B. Wünsch, L. Ye, K. Leuner, M. Peviani, D. Curti, O. Azzolina and S. Collina, Chirality, 2013, 25, 814–822 CrossRef CAS PubMed.
- D. Zampieri, M. G. Mamolo, E. Laurini, C. Zanette, C. Florio, S. Collina, D. Rossi, O. Azzolina and L. Vio, Eur. J. Med. Chem., 2009, 44, 124–130 CrossRef CAS PubMed.
- T. Schläger, D. Shepmann, K. Lehmkuhl, J. Holenz, J. M. Vela, H. Buschmann and B. Wünsch, J. Med. Chem., 2011, 54(19), 6704–6713 CrossRef PubMed.
- (a) R. A. Glennon, S. Y. Ablordeppey, A. M. Ismaiel, M. B. El-Ashmawy, J. B. Fischer and K. B. Howie, J. Med. Chem., 1994, 37, 1214–1219 CrossRef CAS; (b) T. M. Gund, J. Floyd and D. J. Jung, J. Mol. Graphics Modell., 2004, 22, 221–230 CrossRef CAS PubMed; (c) C. Laggner, C. Schieferer, B. Fiechtner, G. Poles, R. D. Hoffmann, H. Glossmann, T. Langer and F. F. Moebius, J. Med. Chem., 2005, 48, 4754–4764 CrossRef CAS PubMed; (d) D. Zampieri, M. G. Mamolo, E. Laurini, C. Florio, C. Zanette, M. Fermeglia, P. Posocco, M. S. Paneni, S. Pricl and L. Vio, J. Med. Chem., 2009, 52, 5380–5393 CrossRef CAS PubMed; (e) C. Oberdorf, T. J. Schmidt and B. Wünsch, Eur. J. Med. Chem., 2010, 45(7), 3116–3124 CrossRef CAS PubMed; (f) C. Meyer, D. Schepmann, S. Yanagisawa, J. Yamaguchi, V. Dal Col, E. Laurini, K. Itami, S. Pricl and B. Wuensch, J. Med. Chem., 2012, 55, 8047–8065 CrossRef CAS PubMed.
- R. Kekuda, P. D. Prasad, J. Y. Fei, F. H. Leibach and V. Ganapathy, Biochem. Biophys. Res. Commun., 1996, 229, 553–558 CrossRef CAS PubMed.
- R. A. Glennon, Mini-Rev. Med. Chem., 2005, 5, 927–940 CrossRef CAS.
- (a) O. Azzolina, S. Collina, G. Brusotti, D. Rossi, A. Callegari, L. Linati, A. Barbieri and V. Ghislandi, Tetrahedron: Asymmetry, 2002, 13, 1073–1081 CrossRef CAS; (b) S. Collina, O. Azzolina, D. Vercesi, M. Sbacchi, M. A. Scheideler, A. Barbieri, E. Lanzac and V. Ghislandi, Bioorg. Med. Chem., 2000, 8, 1925–1930 CrossRef CAS; (c) S. Collina, O. Azzolina, D. Vercesi, G. Brusotti, D. Rossi, A. Barbieri, E. Lanza, L. Mennuni, S. Alcaro, D. Battaglia, L. Linati and V. Ghislandi, Il Farmaco, 2003, 58, 939–946 CrossRef CAS; (d) S. Collina, D. Rossi, G. Loddo, A. Barbieri, E. Lanza, L. Linati, S. Alcaro, A. Gallelli and O. Azzolina, Bioorg. Med. Chem., 2005, 13, 3117–3126 CrossRef CAS PubMed.
- (a) J. Srinivasan, T. E. Cheatham, P. Cieplak, P. A. Kollman and D. A. Case, J. Am. Chem. Soc., 1998, 120, 9401–9409 CrossRef CAS; (b) P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- (a) E. Laurini, V. Dal Col, M. G. Mamolo, D. Zampieri, P. Posocco, M. Fermeglia, V. Vio and S. Pricl, ACS Med. Chem. Lett., 2011, 2, 834–839 CrossRef CAS PubMed; (b) E. Laurini, D. Marson, V. Dal Col, M. Fermeglia, M. G. Mamolo, D. Zampieri, L. Vio and S. Pricl, Mol. Pharmaceutics, 2012, 9, 3107–3126 CrossRef CAS PubMed.
- D. W. Armstrong, T. J. Ward, R. D. Armstrong and T. E. Beesley, Separation of drug stereoisomers by the formation of beta-cyclodextrin inclusion complexes, Science, 1986, 232(4754), 1132–1135 CAS.
- A. D. Mesecar and D. E. Koshland Jr, A new model for protein stereospecificity, Nature, 2000, 403(6770), 614–615 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of chemical synthesis, chiral resolution and absolute configuration assignment of compound 1, and all details of the molecular modeling study and biological investigation. See DOI: 10.1039/c4md00349g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |