Improved synthesis of 4-/6-substituted 2-carboxy-1H-indole-3-propionic acid derivatives and structure–activity relationships as GPR17 agonists†
Younis
Baqi
*a,
Samer
Alshaibani
b,
Kirsten
Ritter
b,
Aliaa
Abdelrahman
b,
Andreas
Spinrath
c,
Evi
Kostenis
c and
Christa E.
Müller
*b
aDepartment of Chemistry, Faculty of Science, Sultan Qaboos University, PO Box 36, Postal Code 123, Muscat, Oman. E-mail: baqi@squ.edu.om; Fax: +968 2414 1469; Tel: +968 2414 2347
bPharma-Zentrum Bonn, Pharmazeutisches Institut, Pharmazeutische Chemie I, Universität Bonn, Bonn, Germany. E-mail: christa.mueller@uni-bonn.de; Fax: +49 228 73 2567; Tel: +49 228 73 2301
cInstitute of Pharmaceutical Biology, Section Molecular-, Cellular-, and Pharmacobiology, University of Bonn, Bonn, Germany
First published on 22nd November 2013
Abstract
The orphan G protein-coupled receptor GPR17 was shown to be involved in myelin repair and has been proposed as a novel drug target for the treatment of brain and spinal cord injury and for multiple sclerosis. Recently, 3-(2-carboxy-4,6-dichloro-indol-3-yl)propionic acid (MDL29,951, 1a) was discovered and characterized as a potent synthetic GPR17 agonist. In the present study we substantially optimized the preparation of 1a, which is carried out via Japp–Klingemann condensation of 3,5-dichlorophenyldiazonium chloride and deprotonated 2-(ethoxycarbonyl)cyclopentanone yielding phenylhydrazone derivative 5a followed by Fischer indole (diaza-Cope) rearrangement. A robust synthesis of 1a (75% yield) was developed to allow upscaling of the procedure. The developed method was applied to the synthesis of a series of 10 derivatives, eight of which represent new compounds. Biological evaluation in calcium mobilization assays using 1321N1-astrocytoma cells recombinantly expressing the human GPR17 provided first insights into their structure–activity relationships. 3-(2-Carboxy-4,6-dibromo-indol-3-yl)propionic acid (1b) showed similar potency to 1a and represents the most potent synthetic GPR17 agonist described to date with an EC50 value of 202 nM.
Introduction
The orphan G protein-coupled receptor 17 (GPR17) belongs to the large family of rhodopsin-like class A G protein-coupled receptors (GPCRs). It is coupled to inhibition of adenylate cyclase via Gi proteins resulting in decreased intracellular cAMP levels, and to Gq proteins which activate phospholipase C leading to IP3-mediated intracellular calcium release.1,2 GPR17 was recently shown to be involved in myelin repair and has therefore been proposed as a novel drug target for the treatment of multiple sclerosis, brain and spinal cord injury, and neurodegenerative diseases.2–6 Thus, the development of GPR17 modulators is of high pharmacological relevance. Several compounds have been postulated as physiological agonists of GPR17, including cysteinylleukotrienes (CysLTs) C4 and D4, UDPglucose, UDPgalactose, and UDP.1 However, several groups, including ours, were unable to confirm the described effects.2,4,7,8 Recently, Hennen et al. identified 3-(2-carboxy-4,6-dichloro-indol-3-yl)propionic acid (MDL29,951, 1a, Fig. 1) as a synthetic agonist for GPR17 and characterized it broadly in recombinant and native cells.2 Compound 1a showed high potency at GPR17 in the nanomolar range; the determined EC50 value was found to be dependent on the assay system and the receptor expression level.2 | ||
| Fig. 1 The first reported synthetic GPR17 agonist.2 | ||
The described synthesis of 1a provides only moderate overall yields. For extended studies of GPR17 using 1a as a tool compound, and for setting up a high-throughput (HTS) screening assay to identify GPR17 antagonists gram amounts of the agonist are required. Therefore the goal of the present study was to develop a significantly improved synthetic procedure for 1a by carefully studying and optimizing the critical reaction steps. Furthermore, the new method was to be applied to the preparation of analogs to study their structure–activity relationships (SARs).
Results and discussion
Syntheses
The synthetic pathway to indole derivative 1a and its analogs starts with a Japp–Klingemann reaction of aniline derivatives 2 with 2-(ethoxycarbonyl)cyclopentanone (4) to obtain the corresponding phenylhydrazone derivatives 5. Subsequent heating in the presence of a strong acid (Fischer indole synthesis, a special type of the (diaza)-Cope rearrangement) leads to ring closure producing the indole mono-ethyl esters 6, which can be saponified to yield the desired products 1 (Scheme 1).2,9 The published procedures for the preparation of 1a suffer from low yields.2,9 Therefore, we systematically studied the low-yielding Fischer indole cyclization of dichlorophenylhydrazone derivative 5a using four different protocols (Scheme 2). At first we applied the procedure published by Salituro et al.9 using concentrated sulfuric acid in refluxing ethanol, which in our hands resulted in a low yield of 10.5% (Scheme 2).2 By applying polyphosphoric acid in refluxing toluene, or in acetic acid, respectively, formation of the expected product was not observed at all. Finally we applied para-toluenesulfonic acid in refluxing ethanol,9 and again only a low yield of 6a was obtained (9%, Scheme 2).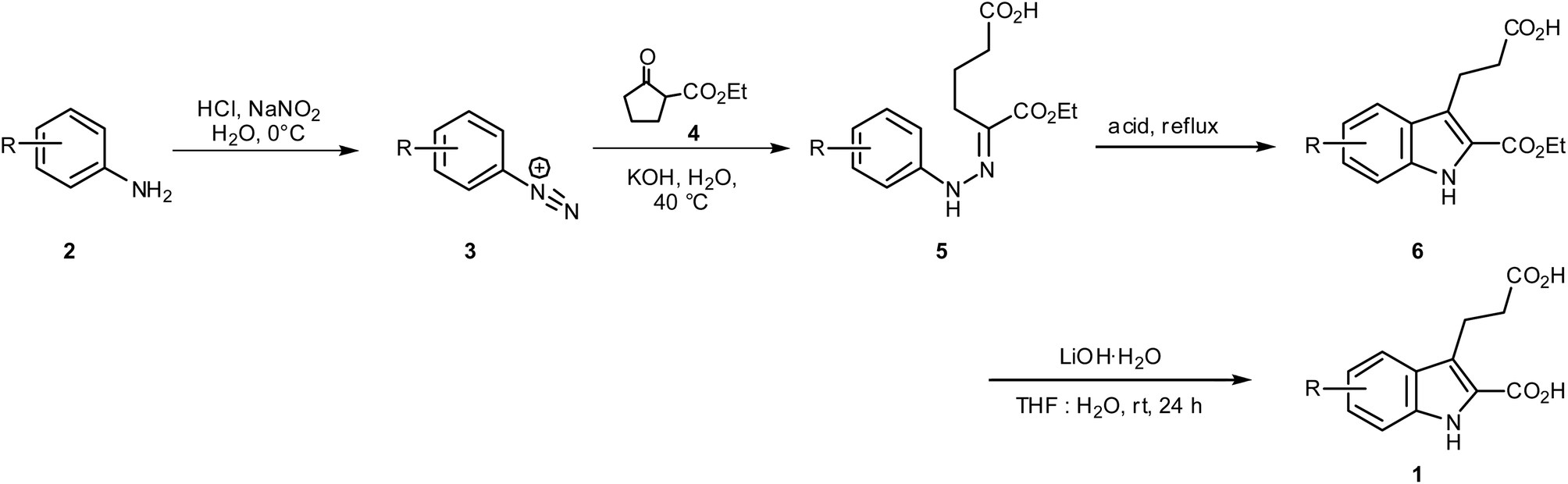 | ||
| Scheme 1 General Japp–Klingemann/Fischer indole synthesis pathway for the preparation of indole derivatives 1. | ||
 | ||
| Scheme 2 Different cyclization methods of compound 5a. | ||
Since we suspected that the free carboxylate function of 5a might be involved in side-reactions we decided to convert it to an ester group prior to Fischer indole cyclization. This simple intermediate reaction step, combined with optimized cyclization reaction conditions, led to a dramatically increased yield of indole 6a. Thus, mono-ester 5a was converted to its di-ethyl ester 8a, which was subsequently cyclized to the indole derivative 9a according to the following conditions (Scheme 3): compound 8a was added to a p-toluenesulfonic acid (p-TSA) solution in toluene, which had previously been dried by means of a Dean–Stark apparatus in refluxing toluene for ca. 1 h (for more details see the Experimental part). The solution was heated for 5 h under the same conditions. The ester functions of indole 9a were subsequently hydrolyzed. With these modifications, an overall yield of the GPR17 agonist 1a of 75% was obtained starting from the corresponding aniline derivative (3,5-dichloroaniline), as compared to only 10.5% in our previous study,2 or 40% reported by Salituro et al.,9 a yield that had not been reproducible by three different experienced chemists in our laboratory using the previously reported method.
 | ||
| Scheme 3 Improved procedure for the synthesis of 3-(2-carboxy-indol-3-yl)propionic acid derivatives. | ||
The complete optimized synthetic procedure for indole derivative 1a as well as its analogs 1b–j is depicted in Scheme 2 and Scheme 3. In the first step, the substituted anilines (2) are diazotized by adding sodium nitrite in the presence of hydrogen chloride at low temperature (0–5 °C) to yield diazonium salts 3, followed by the addition of an aqueous mixture of 2-(ethoxycarbonyl)cyclopentanone (4) and KOH. The resulting mixture is treated with ice and stirred at 40 °C until the phenylhydrazine mono-ethyl ester 5a–j is formed (the reaction is monitored by TLC). Carboxylic acids 5a–j are then refluxed in ethanol at 100 °C in the presence of sulfuric acid to obtain the di-ethyl esters of phenylhydrazones 8a–j, which are subsequently cyclized to the corresponding indole di-ethyl ester derivatives 9a–j. Finally, compounds 9a–j are saponified to yield the desired indoles 1a–j containing two carboxylic acid functions (Scheme 3). This improved method was applied to the synthesis of 10 analogous indole derivatives, of which eight were new, not previously described compounds (1b, c, e, f–j, Table 1). Besides 4,6-disubstituted indole derivatives (1a–e), which were obtained starting from symmetrically 3,5-disubstituted anilines, 4- and 6-monosubstituted derivatives (1g–j), were prepared starting from meta-mono-substituted anilines. Symmetrically substituted aniline derivatives produced a single product (1a–f, Table 1), while meta-mono-substituted anilines gave two isomers: 4- and 6-substituted indole derivatives (1g–j, Table 1). The separation of the two formed isomers was achieved on the di-ester stage by silica gel column chromatography of indoles 9g/9h, and 9i/9j, respectively, using 20% ethyl acetate in cyclohexane as an eluent. All final products were analyzed by 1H- and 13C-NMR spectroscopy, elemental analysis and high-performance liquid chromatography (HPLC) coupled with electrospray ionization (ESI) mass spectrometry (MS). Purity as determined by HPLC-ESI-MS was in all cases greater than 95%.
| Product | Aniline derivative | Product (1a–j) | m.p. (°C) | Yielda (%) | EC50 ± SEMb (μM) |
|---|---|---|---|---|---|
| a Total isolated yield. b Potency to induce calcium mobilization in 1321N1 astrocytoma cells transfected with the human GPR17. c Also no antagonistic activity was observed. | |||||
| 1a |

|
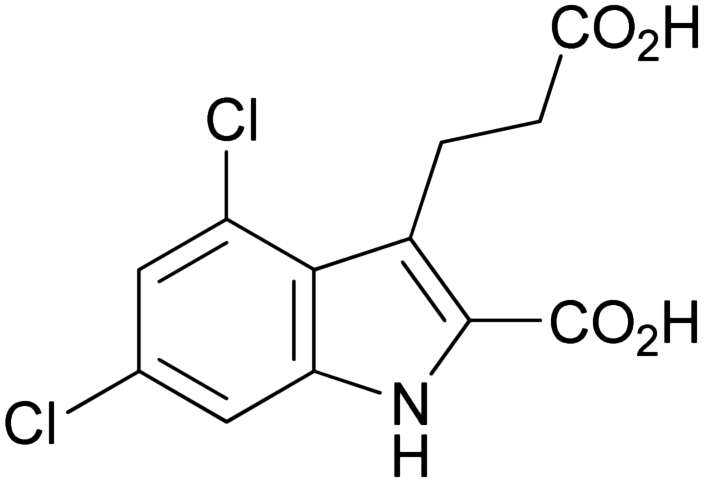
|
275–277 | 75 | 0.331 ± 0.087 |
| 1b |

|

|
259–260 | 63 | 0.202 ± 0.063 |
| 1c |

|

|
280–282 | 35 | 4.79 ± 0.877 |
| 1d |
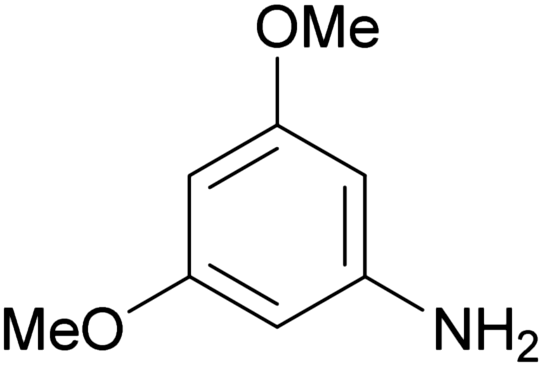
|
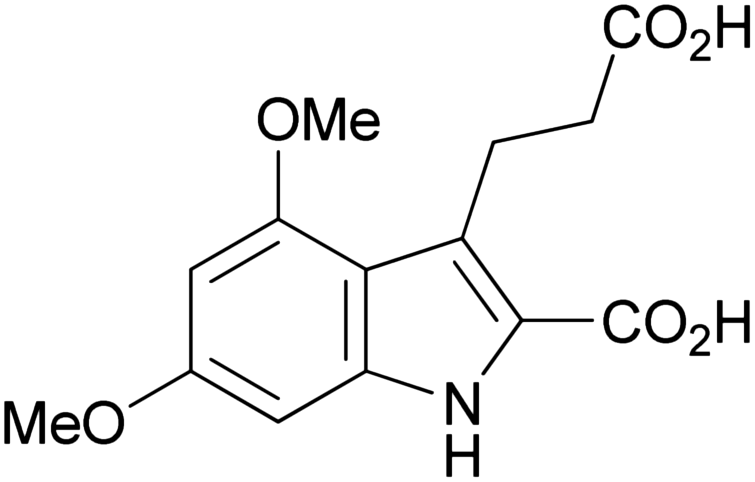
|
239–241 | 36 | ≫100c |
| 1e |

|

|
249–251 | 45 | 17.1 ± 4.12 |
| 1f |
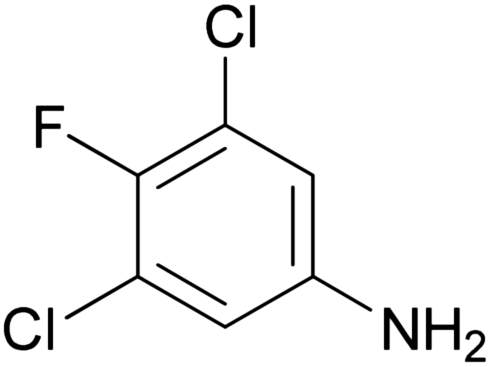
|

|
292–294 | 65 | 52.0 ± 5.50 |
| 1g |
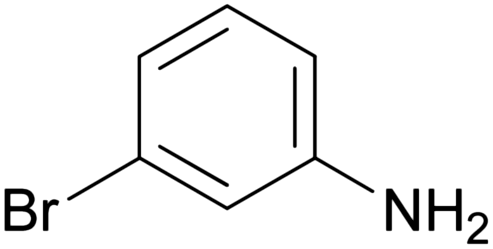
|

|
235–237 | 23 (69% total) | 45.5 ± 8.66 |
| 1h |
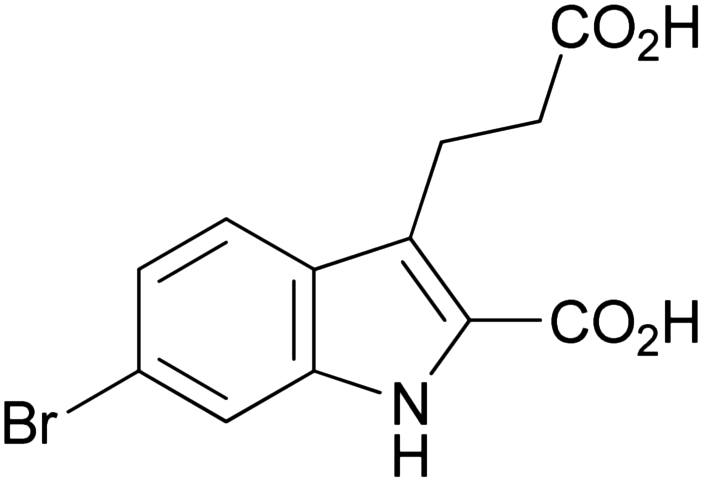
|
234–235 | 46 (69% total) | 2.24 ± 0.68 | |
| 1i |

|

|
246–248 | 30 (90% total) | 36.8 ± 1.81 |
| 1j |

|
230–232 | 60 (90% total) | 0.715 ± 0.128 | |
Biological evaluation
The compounds were investigated for their potential to induce calcium mobilization in 1321N1 astrocytoma cells stably transfected with the human GPR17 using the calcium-chelating fluorescent dye Oregon Green®.Structure–activity relationships
GPR17 has been shown to be coupled to different G proteins,1,2 including Gq, which mediates the release of inositol trisphosphate (IP3). IP3 binds to ryanodine receptors on the endoplasmic reticulum and triggers the opening of calcium channels resulting in an increase in intracellular calcium levels. In 1321N1 astrocytoma cells recombinantly expressing moderate levels of the human GPR17 an EC50 value of 330 nM was determined for 1a in calcium mobilization assays (see Table 1). The Hill slope of the curve for 1a as well as for all other investigated indole derivatives was not significantly different from 1.In the present study initial SARs were obtained for a set of 10 compounds. Different tri- and tetra-substituted indole derivatives were investigated (see Table 1). Analogs of the 4,6-dichloro-substituted indole derivative 1a, in which the chlorine atoms were replaced by bromine or iodine atoms, or by methoxy or trifluoromethoxy groups, showed the following rank order of potency: dibromo (1b) > dichloro (1a) > diiodo (1c) > di-(trifluoromethoxy) (1e) ≫ dimethoxy (1d). These results indicate that the size of the substituents is important for receptor interaction. The dibromo-substituted indole showed a somewhat higher potency (EC50 = 202 nM) than the dichloro-substituted lead structure 1a, although the difference was not statistically significant. However, larger substituents were less well tolerated and led to reduced potency. Besides sterical effects, electronic effects are also important: high electronegativity of the 4,6-substituents appears to be required for potency (compare e.g.1d and 1e).
Introducing a small substituent (fluoro) in position 5 of lead structure 1a significantly decreased potency by >150-fold (compare 1a with 1f, Table 1). This means that a substituent in position 5, at least one with a high electronegativity, is hardly tolerated by the receptor. Finally, derivatives substituted either in position 4 or 6 of the indole moiety with a halogen atom (Br, I) were investigated. It was found that a bulky halogen atom in position 6 was very important for high potency. The larger iodine atom was superior to a bromine atom in that position (compare 1h and 1j). In contrast, indole derivatives 1g and 1i, which were only substituted in position 4 but not in position 6, were only weak agonists at GPR17. Based on a series of 10 indole derivatives it can be recognized that their SARs as GPR17 agonists are steep, and small modifications can modulate potency and even abolish their activity.
Experimental part
General
All materials were used without prior purification. Thin-layer chromatography was performed using TLC aluminum sheets silica gel 60 F254. Synthesized compounds were visualized under UV light (254 nm). 1H- and 13C-NMR data were measured in DMSO-d6 as a solvent. Chemical shifts are reported in parts per million (ppm) relative to the deuterated solvent (DMSO-d6), δ1H: 2.49 ppm, 13C: 39.7 ppm, coupling constants J are given in Hertz and spin multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet), sext (sextet), m (multiplet), br (broad). The purities of isolated products were determined by ESI-mass spectra obtained on an HPLC-MS instrument (LC-MS) using the same procedure as previously published.10 The purity of the compounds was determined at 254 nm. For further proof of purity of the final compounds (1a–j), elemental analysis and 1H-NMR were determined. Melting points were measured on a melting point apparatus and are uncorrected.General procedures for the synthesis of the products 1a–j
(i) Preparation of benzenediazonium salt derivatives (mixture I). To a well stirred suspension of aniline derivatives (10 mmol) in 16.6 ml aq. HCl (5 M) at 0–5 °C was dropwise added a solution of sodium nitrite (1.38 g, 20 mmol, 2 equiv.) in 8 ml water, previously cooled to 0–5 °C in an ice bath. The addition of sodium nitrite solution was slow, in order to keep the temperature of the mixture below 8 °C. The resulting orange-red mixture was stirred at 0–5 °C for additional 20 min in an ice bath.
(ii) Preparation of 2-(ethoxycarbonyl)cyclopentanone anion (mixture II). 2-(Ethoxycarbonyl)cyclopentanone (2.512 ml, 1.344 g, 15 mmol) was dissolved in ethanol (4.2 ml) and cooled to 0–5 °C. Then, a potassium hydroxide solution (5.040 g, 90 mmol, 6 equiv.) in water (5 ml) previously cooled to 0–5 °C was added dropwise within ca. 30 min in order to keep the temperature below 8 °C. The mixture turned to a white-milky appearance, and the final mixture was stirred at 0–5 °C for further 30 min.
(iii) Synthesis of compounds 5a–j. Ice (50 g) was added to mixture II with stirring at 0–5 °C in an ice bath, followed by the addition of mixture I, and stirring continued for 1 h at 40 °C. The combined mixtures were then let to cool to rt and the pH was subsequently adjusted to 4–5 by adding 1 M aq. HCl. The desired product was extracted with diethyl ether (3 × 50 ml). The combined organic layers were collected, dried over magnesium sulfate, filtered, and the filtrate was evaporated to dryness yielding a gummy material (95–100%). This material was used without further purification for the next step.
Synthesis of compounds 8a–j. Compound 5a–j (10 mmol), obtained from general procedure A, was dissolved in absolute ethanol (100 ml) followed by the addition of concentrated sulfuric acid (2.7 ml, 50.5 mmol, 5.1 equiv.). The mixture was then allowed to reflux for 1 h at 100 °C. Then the ethanol was evaporated and the residue was treated with 100 ml of ice water. The aqueous solution was extracted with dichloromethane (3 × 50 ml); the organic layer was dried over magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography using 20% of ethyl acetate in cyclohexane yielding a white solid in 85–100% yield.
Synthesis of indole di-ethyl esters (9a–j). A mixture of p-toluenesulfonic acid (2.954 g, 15 mmol, 1.5 equiv.) and 100 ml of dry toluene was refluxed for 1 h at 140 °C; water was continuously removed by means of a Dean–Stark trap. Subsequently, 10 mmol of the starting material 8a–j dissolved in a minimum amount of dry toluene (ca. 15 ml) was added and the mixture was refluxed for 5 h. Then it was allowed to cool down to rt, and toluene was removed under reduced pressure and the residue was dissolved in dichloromethane and washed with water. The organic layer was dried over magnesium sulfate, filtered, and evaporated to dryness. The residue was purified by silica gel column chromatography using 20% of ethyl acetate/cyclohexane as eluent.
Saponification of indole diethyl esters 9a–j yielding the final products 1a–j. Compound 9a–j (10 mmol) was dissolved in 25 ml tetrahydrofuran (THF) with stirring at rt. Then a solution of 1.26 g of lithium hydroxide trihydrate (3 equiv.) in 25 ml water was added and the resulting mixture was left to stir at rt for 24 h. After completion of the reaction, THF was removed under reduced pressure, the pH was adjusted to 4–5, and the product was extracted with diethyl ether (3 × 30 ml). The organic layers were dried over magnesium sulfate, filtered, and evaporated to dryness to yield the final products (1a–j) as solids in excellent isolated yield (95–100%).
Analytical data of the synthesized products 1a–j
Biological evaluation
1321N1 astrocytoma cells stably transfected with the human GPR17 were used for fluorimetric measurement of intracellular calcium release induced by the test compounds in analogy to previously described procedures.2,11Conclusions
In conclusion, we have developed a fast, efficient, and high-yielding procedure for the synthesis of 3-(2-carboxy-4,6-dichloro-indol-3-yl)propionic acid (1a, MDL29,951), which will allow the preparation of multi-gram amounts of this recently discovered potent GPR17 agonist. The developed method was used for the total synthesis of 10 indole derivatives, eight of which are new compounds (1b, c, e, f–j), while two derivatives (1a, d) had been previously reported.9 Compound 1b (3-(2-carboxy-4,6-dibromo-indol-3-yl)propionic acid) showed the highest potency of the tested compound series at human GPR17 expressed in 1321N1 astrocytoma cells with an EC50 value of 202 nM. Steep SARs have been discovered for this class of compounds and further, more extensive exploration is warranted. New ligands for GPR17 are of great interest as pharmacological tools and as potential drug candidates.Acknowledgements
Y. B. is grateful for an SQU grant (IG/SCI/CHEM/13/02). This study was supported by the German Federal Ministry of Education and Research (BMBF) within the BioPharma – Neuroallianz consortium and by UCB Pharma GmbH, Monheim, Germany.Notes and references
- P. Ciana, M. Fumagalli, M. L. Trincavelli, C. Verderio, P. Rosa, D. Lecca, S. Ferrario, C. Parravicini, V. Capra, P. Gelosa, U. Guerrini, S. Belcredito, M. Cimino, L. Sironi, E. Tremoli, G. E. Rovati, C. Martini and M. P. Abbracchio, EMBO J., 2006, 25, 4615 CrossRef CAS PubMed.
- S. Hennen, L. Peters, H. Wang, A. Spinrath, N. Merten, S. Blättermann, R. Akkari, R. Schrage, K. Simon, R. Schröder, D. Schulz, C. Vermeiren, K. Zimmermann, S. Kehraus, C. Drewke, A. Pfeifer, G. M. König, K. Mohr, M. Gillard, C. E. Müller, Q. R. Lu, J. Gomeza and E. Kostenis, Sci. Signaling, 2013, 6, ra93 CrossRef PubMed.
- D. Lecca, M. L. Trincavelli, P. Gelosa, L. Sironi, P. Ciana, M. Fumagalli, G. Villa, C. Verderio, C. Grumelli, U. Guerrini, E. Tremoli, P. Rosa, S. Cuboni, C. Martini, A. Buffo, M. Cimino and M. P. Abbracchio, PLoS ONE, 2008, 3, e3579 Search PubMed.
- Y. Chen, H. Wu, S. Wang, H. Koito, J. Li, F. Ye, J. Hoang, S. S. Escobar, A. Gow, H. A. Arnett, B. D. Trapp, N. J. Karandikar, J. Hsieh and Q. R. Lu, Nat. Neurosci., 2009, 11, 1398 CrossRef PubMed.
- M. Fumagalli, S. Daniele, D. Lecca, P. R. Lee, C. Parravicini, R. D. Fields, P. Rosa, F. Antonucci, C. Verderio, M. L. Trincavelli, P. Bramanti, C. Martini and M. P. Abbracchio, J. Biol. Chem., 2011, 286, 10593 CrossRef CAS PubMed.
- H. Franke, C. Parravicini, D. Lecca, E. R. Zanier, C. Heine, K. Bremicker, M. Fumagalli, P. Rosa, L. Longhi, N. Stocchetti, M. G. De Simoni, M. Weber and M. P. Abbracchio, Purinergic Signalling, 2013, 9, 451–462 CrossRef CAS PubMed.
- A. Maekawa, B. Balestrieri and Y. Kanaoka, Proc. Natl. Acad. Sci. USA, 2009, 106, 11685–11690 CrossRef CAS PubMed.
- A.-D. Qi, K. Harden and R. A. Nicholas, J. Pharmacol. Exp. Ther., 2013, 347, 38–46 CrossRef CAS PubMed.
- F. G. Salituro, B. L. Harrison, B. M. Baron, P. L. Nyce, K. T. Stewart, J. H. Kehne, H. S. White and I. A. McDonald, J. Med. Chem., 1992, 35, 1791 CrossRef CAS.
- Y. Baqi and C. E. Müller, Nat. Protoc., 2010, 5, 945–953 CrossRef CAS PubMed.
- P. Hillmann, G. Y. Ko, A. Spinrath, A. Raulf, I. von Kügelgen, S. C. Wolff, R. A. Nicholas, E. Kostenis, H. D. Höltje and C. E. Müller, J. Med. Chem., 2009, 52, 2762–2775 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data of the di-ethyl ester indole derivatives (9a–j).See DOI: 10.1039/c3md00309d |
| This journal is © The Royal Society of Chemistry 2014 |