Novel protein–protein interaction inhibitor of Nrf2–Keap1 discovered by structure-based virtual screening†
Hao-Peng
Sun‡
abc,
Zheng-Yu
Jiang‡
ab,
Ming-Ye
Zhang
ab,
Meng-Chen
Lu
ab,
Ting-Ting
Yang
ab,
Yang
Pan
ab,
Hao-Ze
Huang
ab,
Xiao-Jin
Zhang
abd and
Qi-dong
You
*ab
aJiang Su Key Laboratory of Drug Design and Optimization, China Pharmaceutical University, Nanjing 210009, China. E-mail: youqidong@gmail.com; Fax: +86 025 83271351; Tel: +86 025 83271351
bState Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, China
cDepartment of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China
dDepartment of Organic Chemistry, School of Science, China Pharmaceutical University, Nanjing 210009, China
First published on 30th October 2013
Abstract
Herein we first reported hierarchical structure-based virtual screening utilizing the receptor–ligand binding model of Nrf2–Keap1. The most promising compound, 15, which is one of the most potent direct PPI inhibitors of Nrf2–Keap1 reported so far, can effectively disrupt the Nrf2–Keap1 interaction with the in vitro EC50 of 9.80 μM in the fluorescence polarization (FP) assay. 15 can also activate the Nrf2 transcription activity in the cell-based ARE–luciferase reporter assays in a dose-dependent manner. The compound can serve as a promising starting point for the discovery of potent inhibitors of Nrf2–Keap1 interaction.
Introduction
The human body is surrounded by various oxidative and electrophilic chemicals from both endogenous and exogenous sources.1 Sustained oxidative stress and elevated redox state are the major causes of chronic inflammation which could be closely related with cancer, neurodegenerative and cardiovascular diseases, and aging.2 The Keap1–Nrf2–ARE signaling pathway plays a central role in the antioxidant defense mechanism.3 This pathway has three main components, namely Kelch-like ECH-associated protein 1 (Keap1), nuclear factor erythroid 2-related factor 2 (Nrf2), and antioxidant response elements (ARE). The transcriptional factor Nrf2 is a cap‘n’collar (CNC) basic-region leucine zipper (bZIP) protein which mediates the expression of more than 100 oxidative stress related genes containing the enhancer sequence ARE (5′-GTGACnnnGC-3′) in their promoter regulatory regions.4 Keap1, a cysteine-rich protein (27 cysteines in 626 amino acids of human Keap1), is the endogenic negative modulator of Nrf2. Under normal physiological conditions, Nrf2 is sequestered in the cytosol and maintained at a low level through Keap1-dependent ubiquitination and proteasomal degradation. Upon oxidative stress, such as reactive oxygen species (ROS) and electrophilic chemicals, Keap1 serves as a redox sensor through its reactive cysteines. Covalent modifications of the cysteines result in the conformational changes of Keap1 which finally lead to relieving Nrf2 from Keap1-directed degradation. In brief, the Keap1–Nrf2–ARE signaling pathway can modulate the redox state of the cells to keep cellular homeostasis, as a result, it has become an attractive target for the prevention and treatment of oxidative stress and cellular homeostasis related diseases and conditions including cancer, and neurodegenerative, cardiovascular, metabolic and inflammatory diseases.5 The most well-known modulators are the triterpenoid CDDO-methyl esters (bardoxolone methyl) developed by Reata pharmaceuticals6 and Tecfidera™, the dimethyl, monomethyl fumarates developed by Biogen Idec, Inc.7However, most known Nrf2 activators are electrophilic species or metabolically activated to become electrophilic, and subsequently react with Keap1 cysteine residues which ultimately leads to the dissociation of the Nrf2–Keap1 complex.8 The molecular mechanism of these activators is the covalent binding to the thiol of the cysteine, which does not possess selectivity and specificity for the ubiquitous cysteines in cells. Thus, the targets of current known Nrf2 activators could be promiscuous, which may lead to side effects. Nevertheless, protein–protein interaction (PPI) inhibitors of Nrf2–Keap1 can overcome this disadvantage of current known Nrf2 activators. Recently, two high throughput screenings for small molecular PPI inhibitors of Nrf2–Keap1 have been reported by different groups. Fluorescence polarization (FP) based screening of the MLPCN library containing 330![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds gave the hit with an IC50 of 3 μM.9 The homogeneous confocal fluorescence anisotropy assay (two-dimensional fluorescence intensity distribution analysis, 2D-FIDA) based screening of Evotec Lead Discovery library screened out Cpd15 and Cpd16 with the EC50 of 118 μM and 2.7 μM, respectively.10 In this paper, we report our version of discovering a small molecular PPI inhibitor of Nrf2–Keap1 through virtual screening of the Specs database. Compound 15 (Specs no. AN-465/14458038) can disrupt the PPI of Nrf2–Keap1 in the in vitro FP experiments with an EC50 of 9.80 μM. It also dose-dependently induces the expression of ARE genes in a cell-based luciferase reporter assay, which further indicates the potential in vivo efficacy.
000 compounds gave the hit with an IC50 of 3 μM.9 The homogeneous confocal fluorescence anisotropy assay (two-dimensional fluorescence intensity distribution analysis, 2D-FIDA) based screening of Evotec Lead Discovery library screened out Cpd15 and Cpd16 with the EC50 of 118 μM and 2.7 μM, respectively.10 In this paper, we report our version of discovering a small molecular PPI inhibitor of Nrf2–Keap1 through virtual screening of the Specs database. Compound 15 (Specs no. AN-465/14458038) can disrupt the PPI of Nrf2–Keap1 in the in vitro FP experiments with an EC50 of 9.80 μM. It also dose-dependently induces the expression of ARE genes in a cell-based luciferase reporter assay, which further indicates the potential in vivo efficacy.
Results and discussions
To understand the molecular mechanisms underlying the interaction of Keap1 with Nrf2 is the key to develop the PPI inhibitors of Nrf2–Keap1. Neh2 domain of Nrf2 contains two binding motifs ETGE and DLG that bind to the DC domain of Keap1 with different affinities.11 Both motifs are indispensable for the ubiquitination of the poly-lysine region between the two motifs, which are known as the ‘hinge and latch’ model.11,12 Among the model, the high-affinity binding of ETGE motif functions as a ‘hinge’ to fix Nrf2 to Keap1 and the low-affinity binding of DLG motif functions as a ‘latch’ to lock down the Neh2 domain that facilitates the ubiquitination by the Cul3-based E3 ligase complex.13 The crystal structure of Keap1 DC domain with ETGE14 or DLG peptides13 provides more detailed information about the Nrf2–Keap1 interaction (Fig. 1). | ||
| Fig. 1 Molecular interactions of Nrf2 ETGE motif and DLG motif binding to Keap1 DC domain. | ||
In the crystal structure of 16mer Nrf2 ETGE peptide and DC domain of Keap1 (PDB code: 2FLU), only the side chains of Glu79 and Glu82 in the ETGE peptide make specific interactions with the DC domain (as shown in Fig. 2A). The carboxylate oxygen atoms of Glu79 contact with the side chains of Arg415, Arg483, and Ser508, whereas the carboxylate oxygen atoms of Glu82 make hydrogen bonds with the side chains of Ser363, Asn382, and Arg380. It indicates that two Glutamate residues are crucial to the recognition between Nrf2 and Keap1. The peptide backbone makes five contacts with the DC domain, four from the carbonyl oxygen atoms of Glu78, Glu79, Thr80, and Phe83, and one from a backbone amide group of Phe83.14b In the case of 6mer Nrf2 DLG peptide and Kelch domain of Keap1 (PDB code: 2DYH), the side chains of Gln26 and Asp27 make specific interactions with the DC domain. The amide nitrogen atom of Gln26 takes part in the electrostatic interaction with the side chain atom of Arg483 and the amide oxygen atom makes hydrogen bonds with Ser508 and Arg415, whereas the carboxylate oxygen atoms of Asp27 make multiple interactions with Ser602, Gly603, and Arg415.13
 | ||
| Fig. 2 The pharmacophore based on receptor–ligand interactions. (A) & (B) Two pharmacophores derived from the crystal structures of Nrf2 ETGE peptide and Kelch domain of Keap1 (PDB code 1X2R and 2FLU), (C) superimposition of the two pharmacophores, (D) the final pharmacophore used for screening. Hypothesis features are color-coded as follows: hydrogen bond donor, violet; hydrogen bond acceptor, green; negative ionizable center, dark blue. | ||
Despite the huge difference of the binding affinity,11b ETGE and DLG peptides all possess electrostatic interaction with Arg415 and Arg483, indicating their significant contribution to the recognition of Nrf2. On the other hand, it manifests that the PPI inhibitors of Nrf2–Keap1 should have a negative charged group, especially the carboxyl group, to recognize Keap1. It has also been confirmed by the alanine mutation experiment.13 Furthermore, the research of the peptide also proved that carboxyl group that interacted with the key arginines is indispensable to Keap1 binding.15
These results taken together indicate that the small molecular PPI inhibitors of Nrf2–Keap1 should mimic the side chains of the acidic residues, which means a negative ionizable center should be included in the PPI inhibitors. Depending on this deduction, the formal charge of each compound in the Specs database was calculated at pH = 7.4. All the compounds with formal charge less than or equal to −1 were kept to construct the focus library of Nrf2–Keap1. Compared to 251774 compounds in the Specs database, the focus library of Nrf2–Keap1 only contains 21199 compounds, more than 90% of the compounds have been excluded before the time-consuming screening procedure. A large amount of useless computation can be saved though the construction of a focus library of Nrf2–Keap1 relying on the informatics analysis of the Nrf2–Keap1 complex. This analysis provides some lessons for virtual screening of a large-scale database, especially for those targets without known small molecular modulators as templates.
Since more and more protein structures have been and are being identified, the structure-based pharmacophore method, especially receptor–ligand complex based pharmacophore method, becomes more and more efficient.16 However, the traditional receptor–ligand complex based pharmacophore method mainly applies to the small molecular ligand. Here, the range of the ligand is extended to the small peptide. Pharmacophore models were generated from two crystal structures of Nrf2 ETGE peptide and Kelch domain of Keap1 (PDB code 1X2R and 2FLU). The Receptor–Ligand Pharmacophore Generation Protocol in Discovery Studio 3.0 was applied to detect and interpret the crucial interaction patterns between Keap1 and its binding partners. The pharmacophores (as shown in Fig. 2A and B), generated from 1X2R and 2FLU, were superimposed and the overlapping pharmacophore features were retained to generate the final pharmacophore model (Fig. 2D) for screening. The final model contains one hydrogen bond donor (HBD), two hydrogen bond acceptors (HBA) and three negative ionizable centers. The three negative ionizable centers represent the carboxylic groups of three acidic residues (Asp77, Glu79, and Glu82), the hydrogen bond donor feature points to the amide NH group of Phe83 and hydrogen bond acceptors characterize the carbonyl group of Glu79's side chain and Glu78's backbone. However, considering this model is too complex for the recognition of small molecules, especially three negative ionizable centers, two features can be omitted in the ligand–pharmacophore mapping procedure. Only 2325 of the 21199 molecules were mapped to the pharmacophore which were retained for docking screening.
In the Ligandfit docking screening procedure, the docking site was derived from the position of the peptide ligand co-crystallized in the binding site of Keap1. The consensus score of seven scoring functions, including the DockScore, LigScore1, LigScore2, -PLP1, -PLP2, Dockscore and -PMF, was calculated to filter the compounds. 40% of top molecules were included in the consensus. Ligands in the top percentile are assigned “1”. The remaining entries in the ranking lists are set to “0”. The consensus score for each molecule is obtained by summing its list entries overall. The remaining 225 compounds whose consensus scores were equal or greater than 4 were then examined by the binding energy calculation. The Implicit Solvent Model used in this procedure was PBSA. Considering the amount of computations needed, the minimized structure was used to carry out the MM–PBSA binding energy calculation. The top 10% of compounds were selected for visual inspection. In the visual inspection procedure, we mainly focus on the examination of the electrostatic interaction, especially the salt bridge, between the small molecule and the key arginines of Keap1 (Arg380, Arg415 and Arg483). These docking poses, without possessing any electrostatic interaction with the key arginines, were removed directly. 17 compounds were finally added to purchasing list.
The VS hits were experimentally tested in the FP assay to identify the small-molecule inhibitors of the Keap1–Nrf2 interaction. The experiment procedure was conducted as previously reported with slight modifications (detailed information can be found in the ESI†).17 The 9-mer Nrf2 peptide, Ac–LDEETGEFL–NH2, could effectively inhibit the PPI of the Nrf2–Keap1 with the EC50 of 638 nM (as shown in Fig. 3C). The reported PPI inhibitor, LH601,9 was also evaluated as the positive control, resulting in the EC50 of 13.7 μM (as shown in Fig. 3D). After screening, compound 15 was found to possess the desired activity in binding to the Keap1 DC domain and inducing the dissociation of the Nrf2 peptide with the EC50 of 9.80 μM (as shown in Fig. 3B), indicating that 15 can effectively inhibit the PPI of Nrf2–Keap1 in vitro.
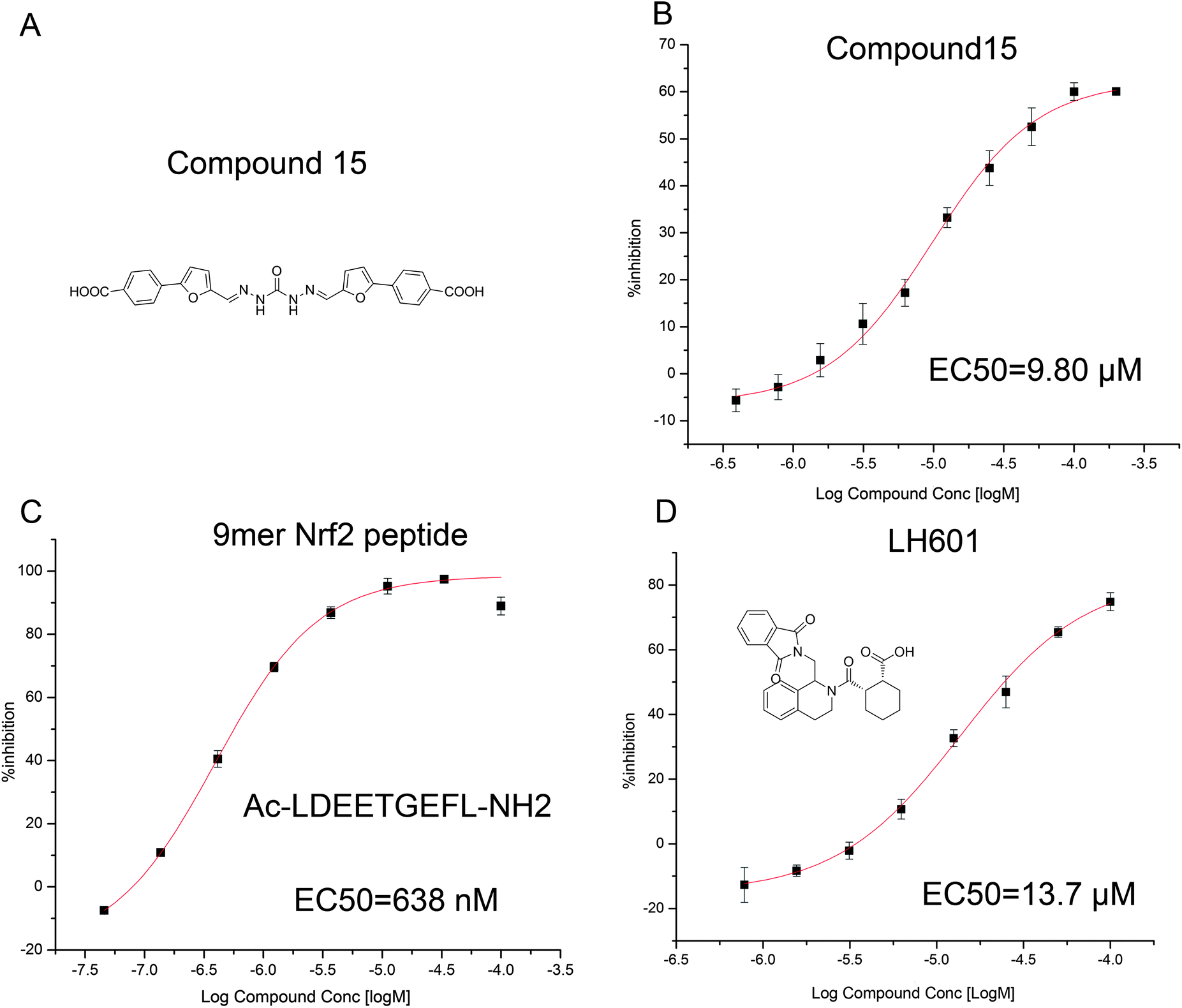 | ||
| Fig. 3 (A) The structure of compound 15 identified through FP assay, (B) dose–response curve of compound 15 in the FP assay, (C) dose–response curve of the 9mer Nrf2 peptide in the FP assay, (D) dose–response curve of LH601 in the FP assay. | ||
To investigate the effects of 15 on the Nrf2 transcription activity, we did the dose–response experiments in the HepG2–ARE–C8 cells, which contain a stable expressed ARE–luciferase reporter, with t-BHQ serving as a positive control, DMSO as a negative control and the luciferase cell culture lysis reagent as a blank. The effect of 15 was determined at 5, 25, 50, 100 and 200 μM and showed a clear dose response curve. The compound produced about 10-fold induction at 200 μM without any cytotoxic effects. The ARE-dependent luciferase activity of traditional ARE inducers targeting the cysteine residues was short acting for the metabolic instability and high reactivity. The ARE induction activity of t-BHQ, for example, falls rapidly from 12 hours to 24 hours, as shown in Fig. 4. Meanwhile, in the case of the PPI inhibitor, the induction activity only shows a slight decline indicating that the resident time of the PPI inhibitors is much longer than the traditional Nrf2 activators. Nevertheless, restricted by the carboxyl groups and the hydrazide scaffold, the cellular level activity of the compound was not very potent for its poor cell permeability. It should be optimized in future research.
 | ||
| Fig. 4 Comparison of ARE-inducing activity between compound 15 and t-BHQ. (A) The dose response curve of compound 15 at 12 h and 24 h, (B) the dose response curve of t-BHQ at 12 h and 24 h. The y axis is the average value of the induction fold and the x axis is the concentration of the compound. | ||
Compared to the ETGE motif of Nrf2, the docking result study shows a distinct binding mode of compound 15 (as shown in Fig. 5A). It covers a large binding surface depending on the four aromatic rings in its structure. The carboxyl groups at the edge of 15 make multiple hydrogen bonds and electrostatic interactions with Arg415 and Arg483 separately. The cation–Pi and Pi–Pi interactions also contribute to the binding. The phenyl ring A makes a Pi–Pi interaction with Tyr572; the phenyl ring D possesses cation–Pi interaction with Arg380. The two furan rings also occupy the hydrophobic cavity. The polar hydrogen in the carbohydrazide fragment takes part in the hydrogen bond with the phenolic group of Tyr334. The proposed binding mode indicates that the hydrophobic interaction can be used to improve the affinity between the small molecular and Keap1.
 | ||
| Fig. 5 Proposed binding mode of compound 15 to the Nrf2 binding site of Keap1 Kelch domain. (A) 15 mapped to the pharmacophore model. (B) Proposed binding mode of 15 from docking studies. The binding surface of Keap1 is colored depending on the partial charge. Compound 15 is shown as sticks and the residues is shown as lines. The hydrogen bonds and Pi interactions are labeled in the figure. | ||
Conclusions
In summary, we have discovered, through virtual screening of the Specs database, a direct inhibitor of Nrf2–Keap1 PPI. These results above demonstrate that compound 15 can potently disrupt the PPI of the Nrf2–Keap1 in both target-based and cell-based level. The compound may offer a promising starting point for the discovery of potent inhibitors of Keap1–Nrf2. The docking study indicated that hydrophobic interactions can contribute to the binding of Keap1. Importantly, this study also demonstrates that the activation of Nrf2 through direct inhibition of Keap1–Nrf2 could be more long-acting and less toxic compared to the traditional covalent binding based Nrf2 activators. With the development of small molecular direct inhibitors of the Keap1–Nrf2 interaction, the disruption of the Keap1–Nrf2 complex is believed to be an effective strategy in many human disorders, including cancer, Alzheimer's disease (AD), Parkinson's disease (PD) and diabetes.Acknowledgements
This work is supported by the project 81230078 (key program), 81202463 (youth foundation) and 91129732 of National Natural Science Foundation of China, Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (no. JKGQ201103), 2012AA020301 of 863 program, 2010ZX09401-401 and 2009ZX09501-003 of the National Major Science and Technology Project of China (Innovation and Development of New Drugs). The authors declare no other conflicts of interest.Notes and references
- T. Finkel and N. J. Holbrook, Oxidants, oxidative stress and the biology of ageing, Nature, 2000, 408(6809), 239–247 CrossRef CAS PubMed.
- (a) K. J. Barnham, C. L. Masters and A. I. Bush, Neurodegenerative diseases and oxidative stress, Nat. Rev. Drug Discovery, 2004, 3(3), 205–214 CrossRef CAS PubMed; (b) C. C. Benz and C. Yau, Ageing, oxidative stress and cancer: paradigms in parallax, Nat. Rev. Cancer, 2008, 8(11), 875–879 CrossRef CAS PubMed; (c) C. Leeuwenburgh and J. W. Heinecke, Oxidative stress and antioxidants in exercise, Curr. Med. Chem., 2001, 8(7), 829–838 CrossRef CAS.
- R. Brigelius-Flohe and L. Flohe, Basic principles and emerging concepts in the redox control of transcription factors, Antioxid. Redox Signaling, 2011, 15(8), 2335–2381 CrossRef CAS PubMed.
- (a) P. Moi, K. Chan, I. Asunis, A. Cao and Y. W. Kan, Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(21), 9926–9930 CrossRef CAS; (b) K. Itoh, K. Igarashi, N. Hayashi, M. Nishizawa and M. Yamamoto, Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins, Mol. Cell. Biol., 1995, 15(8), 4184–4193 CAS; (c) A. Martin-Montalvo, J. M. Villalba, P. Navas and R. de Cabo, NRF2, cancer and calorie restriction, Oncogene, 2011, 30(5), 505–520 CrossRef CAS PubMed.
- (a) T. W. Kensler, N. Wakabayashi and S. Biswal, Cell survival responses to environmental stresses via the Keap1–Nrf2–ARE pathway, Annu. Rev. Pharmacol., 2007, 47, 89–116 CrossRef CAS PubMed; (b) S. Magesh, Y. Chen and L. Hu, Small Molecule Modulators of Keap1–Nrf2–ARE Pathway as Potential Preventive and Therapeutic Agents, Med. Res. Rev., 2012, 32(4), 687–726 CrossRef CAS PubMed.
- S. A. Reisman, G. M. Chertow, S. Hebbar, N. D. Vaziri, K. W. Ward and C. J. Meyer, Bardoxolone Methyl Decreases Megalin and Activates Nrf2 in the Kidney, J. Am. Soc. Nephrol., 2012, 23(10), 1663–1673 CrossRef CAS PubMed.
- R. A. Linker, D. H. Lee, S. Ryan, A. M. van Dam, R. Conrad, P. Bista, W. Zeng, X. Hronowsky, A. Buko, S. Chollate, G. Ellrichmann, W. Bruck, K. Dawson, S. Goelz, S. Wiese, R. H. Scannevin, M. Lukashev and R. Gold, Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway, Brain, 2011, 134(3), 678–692 CrossRef PubMed.
- W. Hur and N. S. Gray, Small molecule modulators of antioxidant response pathway, Curr. Opin. Chem. Biol., 2011, 15(1), 162–173 CrossRef CAS PubMed.
- L. Hu, S. Magesh, L. Chen, L. Wang, T. A. Lewis, Y. Chen, C. Khodier, D. Inoyama, L. J. Beamer, T. J. Emge, J. Shen, J. E. Kerrigan, A. N. Kong, S. Dandapani, M. Palmer, S. L. Schreiber and B. Munoz, Discovery of a small-molecule inhibitor and cellular probe of Keap1–Nrf2 protein–protein interaction, Bioorg. Med. Chem. Lett., 2013, 23(10), 3039–3043 CrossRef CAS PubMed.
- D. Marcotte, W. Zeng, J.-C. Hus, A. McKenzie, C. Hession, P. Jin, C. Bergeron, A. Lugovskoy, I. Enyedy, H. Cuervo, D. Wang, C. Atmanene, D. Roecklin, M. Vecchi, V. Vivat, J. Kraemer, D. Winkler, V. Hong, J. Chao, M. Lukashev and L. Silvian, Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism, Bioorg. Med. Chem., 2013, 21(14), 4011–4019 CrossRef CAS PubMed.
- (a) M. McMahon, N. Thomas, K. Itoh, M. Yamamoto and J. D. Hayes, Dimerization of Substrate Adaptors Can Facilitate Cullin-Mediated Ubiquitylation of Proteins by a Tethering, Mechanism, J. Biol. Chem., 2006, 281(34), 24756 CrossRef CAS PubMed; (b) K. I. Tong, Y. Katoh, H. Kusunoki, K. Itoh, T. Tanaka and M. Yamamoto, Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model, Mol. Cell. Biol., 2006, 26(8), 2887–2900 CrossRef CAS PubMed.
- K. I. Tong, A. Kobayashi, F. Katsuoka and M. Yamamoto, Two-site substrate recognition model for the Keap1–Nrf2 system: a hinge and latch mechanism, Biol. Chem., 2006, 387(10–11), 1311–1320 CAS.
- K. I. Tong, B. Padmanabhan, A. Kobayashi, C. Shang, Y. Hirotsu, S. Yokoyama and M. Yamamoto, Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response, Mol. Cell. Biol., 2007, 27(21), 7511–7521 CrossRef CAS PubMed.
- (a) B. Padmanabhan, K. I. Tong, T. Ohta, Y. Nakamura, M. Scharlock, M. Ohtsuji, M.-I. Kang, A. Kobayashi, S. Yokoyama and M. Yamamoto, Structural Basis for Defects of Keap1 Activity Provoked by Its Point Mutations in Lung Cancer, Mol. Cell, 2006, 21(5), 689–700 CrossRef CAS PubMed; (b) S.-C. Lo, X. Li, M. T. Henzl, L. J. Beamer and M. Hannink, Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling, EMBO J., 2006, 25(15), 3605–3617 CrossRef CAS PubMed.
- (a) R. Hancock, H. C. Bertrand, T. Tsujita, S. Naz, A. El-Bakry, J. Laoruchupong, J. D. Hayes and G. Wells, Peptide inhibitors of the Keap1–Nrf2 protein–protein interaction, Free Radical Biol. Med., 2012, 52(2), 444–451 CrossRef CAS PubMed; (b) R. Steel, J. Cowan, E. Payerne, M. A. O'Connell and M. Searcey, Anti-Inflammatory Effect of a Cell-Penetrating Peptide Targeting the Nrf2–Keap1 Interaction, ACS Med. Chem. Lett., 2012, 3(5), 407–410 CrossRef CAS PubMed; (c) R. Hancock, M. Schaap, H. Pfister and G. Wells, Peptide inhibitors of the Keap1–Nrf2 protein–protein interaction with improved binding and cellular activity, Org. Biomol. Chem., 2013, 11(21), 3553–3557 RSC; (d) Z.-Y. Jiang, H.-X. Chu, M.-Y. Xi, T.-T. Yang, J.-M. Jia, J.-J. Huang, X.-K. Guo, X.-J. Zhang, Q.-D. You and H.-P. Sun, Insight into the Intermolecular Recognition Mechanism between Keap1 and IKKβ Combining Homology Modelling, Protein–Protein Docking, Molecular Dynamics Simulations and Virtual Alanine Mutation, PLoS One, 2013, 8(9), e75076 CAS.
- (a) A. R. Leach, V. J. Gillet, R. A. Lewis and R. Taylor, Three-dimensional pharmacophore methods in drug discovery, J. Med. Chem., 2010, 53(2), 539–558 CrossRef CAS PubMed; (b) F. Caporuscio and A. Tafi, Pharmacophore Modelling: A Forty Year Old Approach and its Modern Synergies, Curr. Med. Chem., 2011, 18(17), 2543–2553 CrossRef CAS.
- D. Inoyama, Y. Chen, X. Huang, L. J. Beamer, A. N. Kong and L. Hu, Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1–Nrf2 interaction, J. Biomol. Screening, 2012, 17(4), 435–447 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Assay for fluorescence polarization (FP) to identify small-molecule inhibitors of the Keap1–Nrf2 interaction, assay for ARE luciferase reporter activity, validation of the docking method, list of chosen compounds and the overall workflow scheme. See DOI: 10.1039/c3md00240c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |