Synthesis, SAR and selectivity of 2-acyl- and 2-cyano-1-hetarylalkyl-guanidines at the four histamine receptor subtypes: a bioisosteric approach†
Roland
Geyer‡
,
Patrick
Igel‡
,
Melanie
Kaske
,
Sigurd
Elz
and
Armin
Buschauer
*
Institute of Pharmacy, Pharmaceutical/Medicinal Chemistry, Faculty of Chemistry and Pharmacy, University of Regensburg, Universitätsstr. 31, D-93053 Regensburg, Germany. E-mail: armin.buschauer@ur.de
First published on 7th November 2013
Abstract
In the search for potential bioisosteres of the 4-imidazolyl ring in acylguanidines (e.g. UR-AK24), known to possess affinity to several histamine receptor subtypes (HxR, x = 1–4), and cyanoguanidine-type H4R agonists (e.g. UR-PI376), the contribution of various heterocycles to agonism, antagonism and HR subtype selectivity was studied (recombinant human H1,2,3,4Rs, isolated guinea pig organs (H1R, H2R)). While minor structural modifications of UR-PI376 analogues were not tolerated regarding H4R agonism, in the case of the acylguanidines, a 1,2,4-triazole ring shifted the selectivity toward the H2R.
Introduction
The physiological and pathophysiological effects of the biogenic amine histamine are mediated through four receptor subtypes, referred to as H1, H2, H3 and H4 receptors (H1R, H2R, H3R, and H4R), all belonging to class A of G protein coupled receptors.1–4 H1R and H2R antagonists have been used for decades in the treatment of allergic conditions and as antiulcer drugs, respectively. The H3R is mainly expressed in the brain and is considered a promising drug target, for instance, for the treatment of attention-deficit hyperactivity disorder, Alzheimer's disease, Parkinson's disease, sleep disorders or obesity.5 The H4R was discovered by several groups based on its high sequence homology with the H3R.6–12 The expression on hematopoietic cells such as mast cells, basophils, eosinophils, T-cells and dendritic cells suggests a role of the H4R in the regulation of immune responses and inflammation.13–15 Therefore, the H4R is considered a potential drug target for the treatment of diseases like allergic rhinitis, rheumatoid arthritis, bronchial asthma and pruritus.16–18 Recent reports on β-arrestin-mediated signalling19–21 of H4R ligands and partial agonistic effects of the standard H4R antagonist JNJ-7777120 (ref. 22) at certain H4R species orthologs suggest that the interpretation of ligand effects in vivo in terms of agonism or antagonism should be interpreted with caution.19,23 Hence, both selective antagonists and agonists are required as pharmacological tools to further explore the role of the H4R.15,24Guanidine-type compounds like arpromidine and analogues represent highly potent H2R agonists.25,26 Drawbacks due to the strongly basic guanidine, e.g. very low oral bioavailability and lack of penetration across the blood brain barrier, were eliminated by replacing the guanidine group with a considerably less basic acylguanidine moiety (Fig. 1, UR-AK24 (1)).27–29 However, these NG-acylated imidazolylpropylguanidines developed as H2R agonists lacked selectivity toward the hH3R and hH4R.30 By analogy with the H2R agonist amthamine (2),31 the replacement of the imidazole ring in acylguanidine-type ligands by a 2-amino-thiazole ring, resulted in selective H2R agonists (3 and 4).27,32,33 In contrast potent selective agonists for the hH4R were obtained by replacing the basic acylguanidine group with a cyanoguanidine moiety as in UR-PI376 (5).34 The highest potency resided in imidazolylalkylcyanoguanidines with a tetramethylene linker, connecting the imidazole and the cyanoguanidine moiety. Unlike hH4R agonists such as 5-methylhistamine,35 VUF-8430 (ref. 36) or OUP-16 (ref. 37) compound 5 is devoid of agonistic activities at hHR subtypes other than the hH4R.34
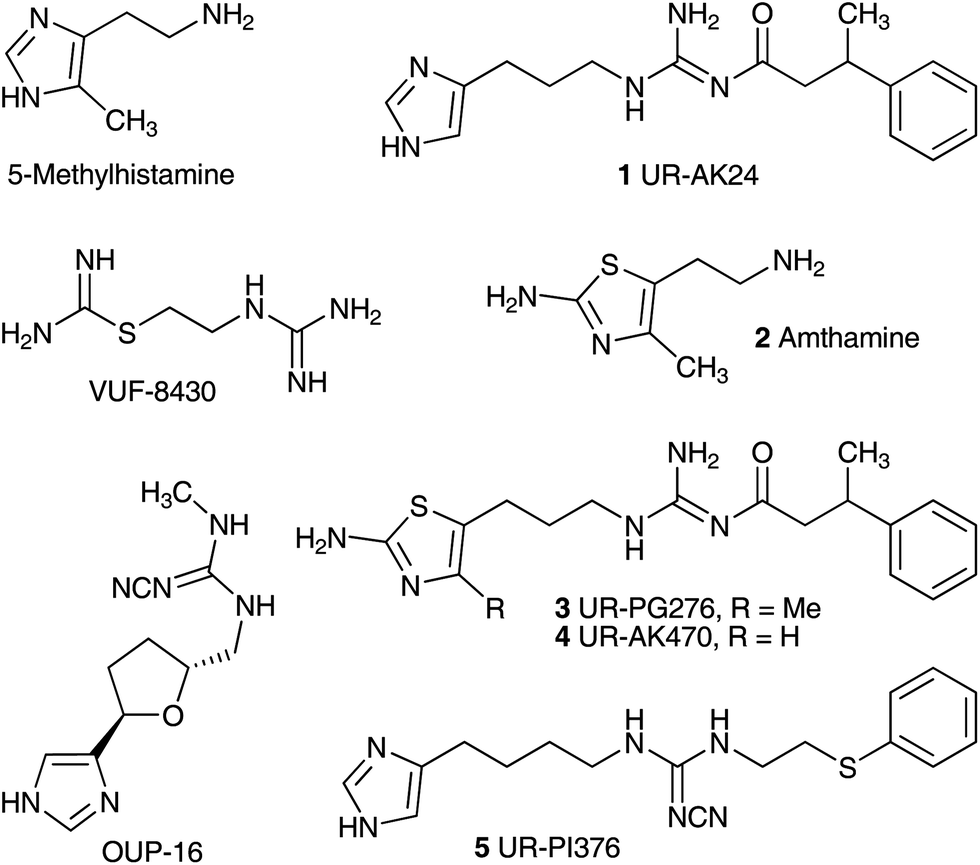 | ||
| Fig. 1 Structures of the selective H4R agonists 5-methylhistamine, VUF8430 and OUP-16, the NG-acylated imidazolylpropylguanidine UR-AK24 (1), which is active at the H2R, H3R and H4R, the H2R agonist amthamine (2), related potent and selective acylguanidine-type H2R agonists 3 and 4 and the potent and selective H4R agonist UR-PI376 (5). | ||
The previous results suggest that the bioisosteric approach harbours the potential of further increasing the preference or the selectivity of hetarylalkylguanidines for a certain HR subtype. In the present work various heterocycles were introduced to replace the 1H-imidazol-4-yl ring (Fig. 2). Special attention was paid to substructures known from early studies on hetaryl analogues of histamine.38,39 These structural modifications were combined with an acylguanidine or a cyanoguanidine moiety as less basic or non-basic guanidine replacements.
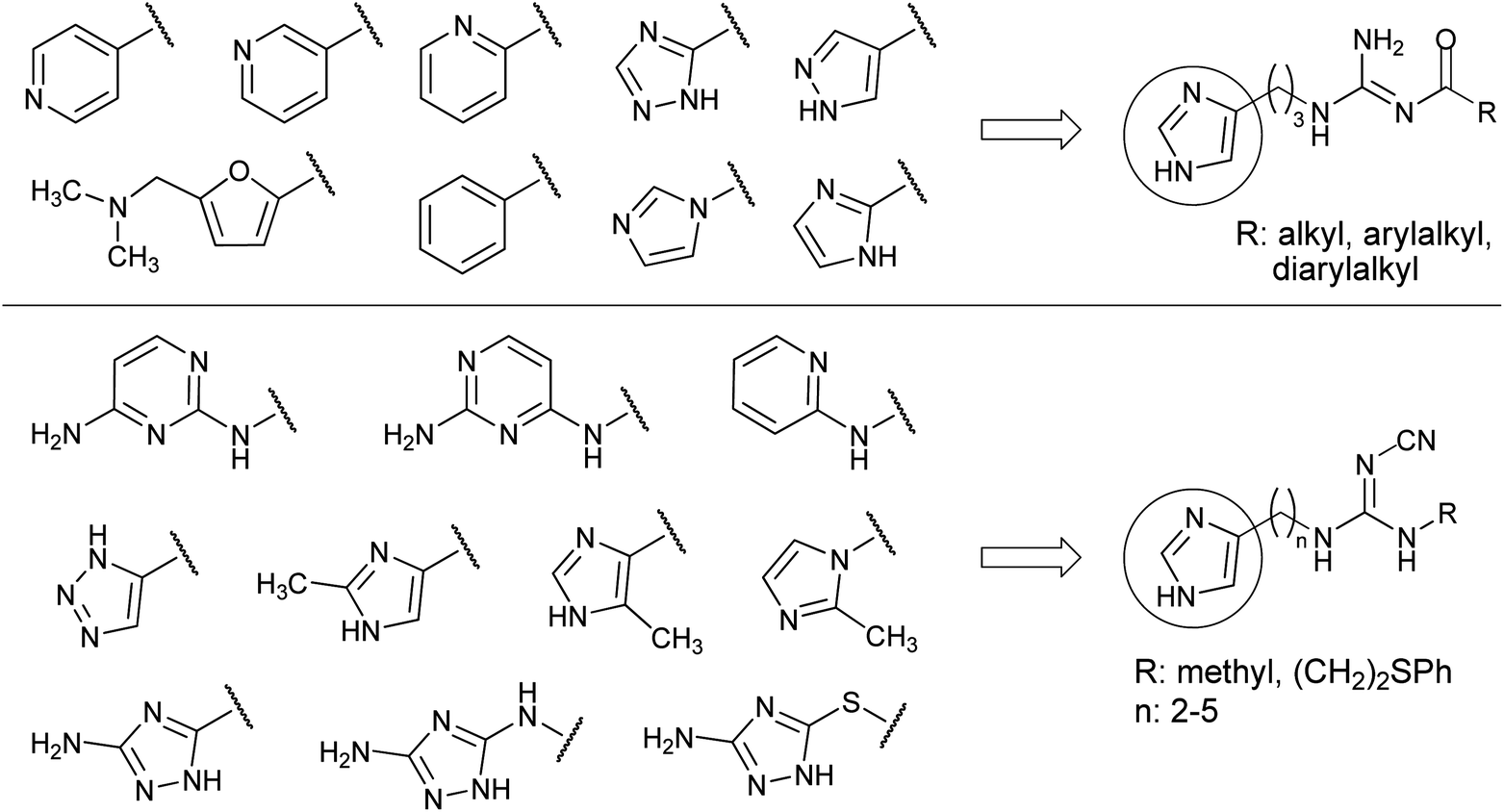 | ||
| Fig. 2 Imidazole replacement in acylguanidine-type non-selective H2R agonists and in cyanoguanidine-type H4R agonists. Overview of the introduced aromatic rings. | ||
Results and discussion
Chemistry
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and reduction of the ethyl ester yielded 20. Conversion of the pyridylpropanols 21–23 to the corresponding phthalimides 24–26 under Mitsunobu conditions47 followed by hydrazinolysis gave the pyridylpropylamines 27–29.48
C double bond and reduction of the ethyl ester yielded 20. Conversion of the pyridylpropanols 21–23 to the corresponding phthalimides 24–26 under Mitsunobu conditions47 followed by hydrazinolysis gave the pyridylpropylamines 27–29.48
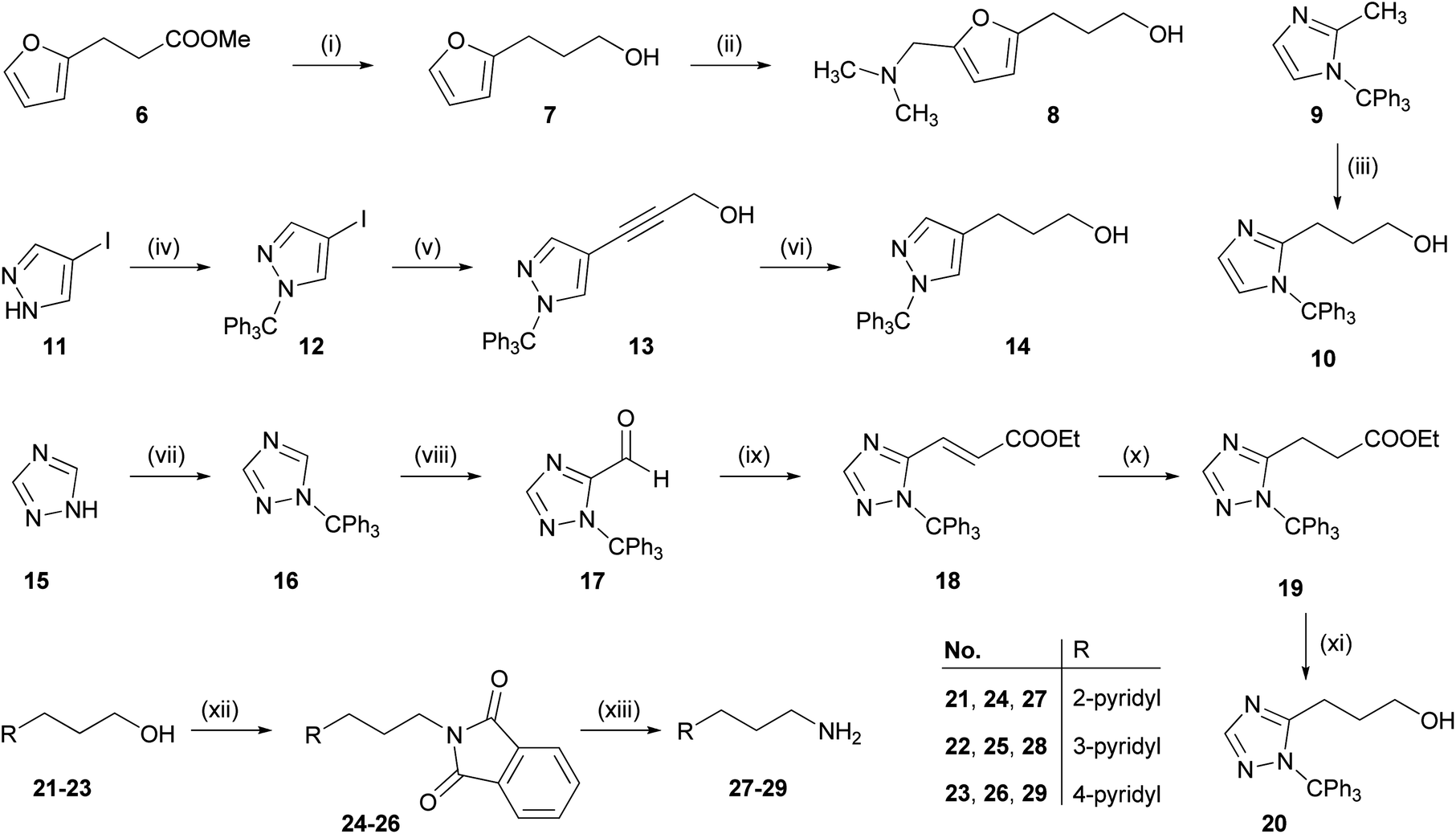 | ||
| Scheme 1 Synthesis of the arylpropyl alcohols 8, 10, 14 and 20, and the pyridylpropylamines 27–29. Reagents and conditions: (i) LiAlH4 (2 eq.), Et2O, overnight, 0 °C → rt; (ii) NH(CH3)2·HCl (1.6 eq.), (CH2O)n (1.6 eq.), EtOH, overnight, reflux; (iii) oxirane (5 eq.), n-BuLi (1.1 eq.), THF, overnight, −78 °C → rt; (iv) TrCl (1 eq.), NEt3 (1.2 eq.), DCM, 12 h, 0 °C → rt; (v) Pd(PPh3)2Cl2 (0.03 eq.), CuI (0.05 eq.), DIPA (4.5 eq.), propargyl alcohol (1.1 eq.), DMF, 48 h, −15 °C → rt; (vi) H2, Pd/C (10%) (cat.), MeOH, overnight, rt; (vii) TrCl (1 eq.), NEt3 (1 eq.), DCM, overnight, rt; (viii) TMEDA (1 eq.), n-BuLi (1.1 eq.), DMF (0.9 eq.), THF, 12 h, −78 °C; (ix) triethyl phosphonoacetate (1.2 eq.), NaH (60% dispersion in mineral oil) (1.2 eq.), THF, overnight, rt; (x) H2, Pd/C (10%) (cat.), EtOH/THF, overnight, rt; (xi) LiAlH4 (2 eq.), THF, 2 h, 0 °C → reflux; (xii) phthalimide (1.1 eq.), PPh3(1.1 eq.), DIAD (1.1 eq.), THF, overnight, 0 °C → rt. (xiii) N2H4·H2O (6 eq.), EtOH, overnight, rt. | ||
The arylpropylguanidines 44–52 were synthesized starting from the corresponding arylpropyl alcohols 8, 10, 14 and 20 or arylpropylamines 27–31 (Scheme 2). Conversion of the alcohol to the di-Cbz-protected guanidines 35–38 was accomplished under Mitsunobu conditions47 by analogy with the procedure described by Feichtinger et al.49 The arylpropylamines 27–31 were treated with the triflyl-di-Cbz-protected guanidine 34 (ref. 49) to give the di-Cbz protected arylpropylguanidines 39–43. Finally, the arylpropylguanidines 44–52 were obtained by hydrogenolytic cleavage of the Cbz groups. The NG-acylated arylpropylguanidines were prepared as outlined in Scheme 2. Coupling of the CDI-activated carboxylic acids50,5153–56 to the arylpropylguanidines 44–52 gave the acylguanidines 57–78.27 Tritylated heterocycles were deprotected under acidic conditions yielding 79–86.
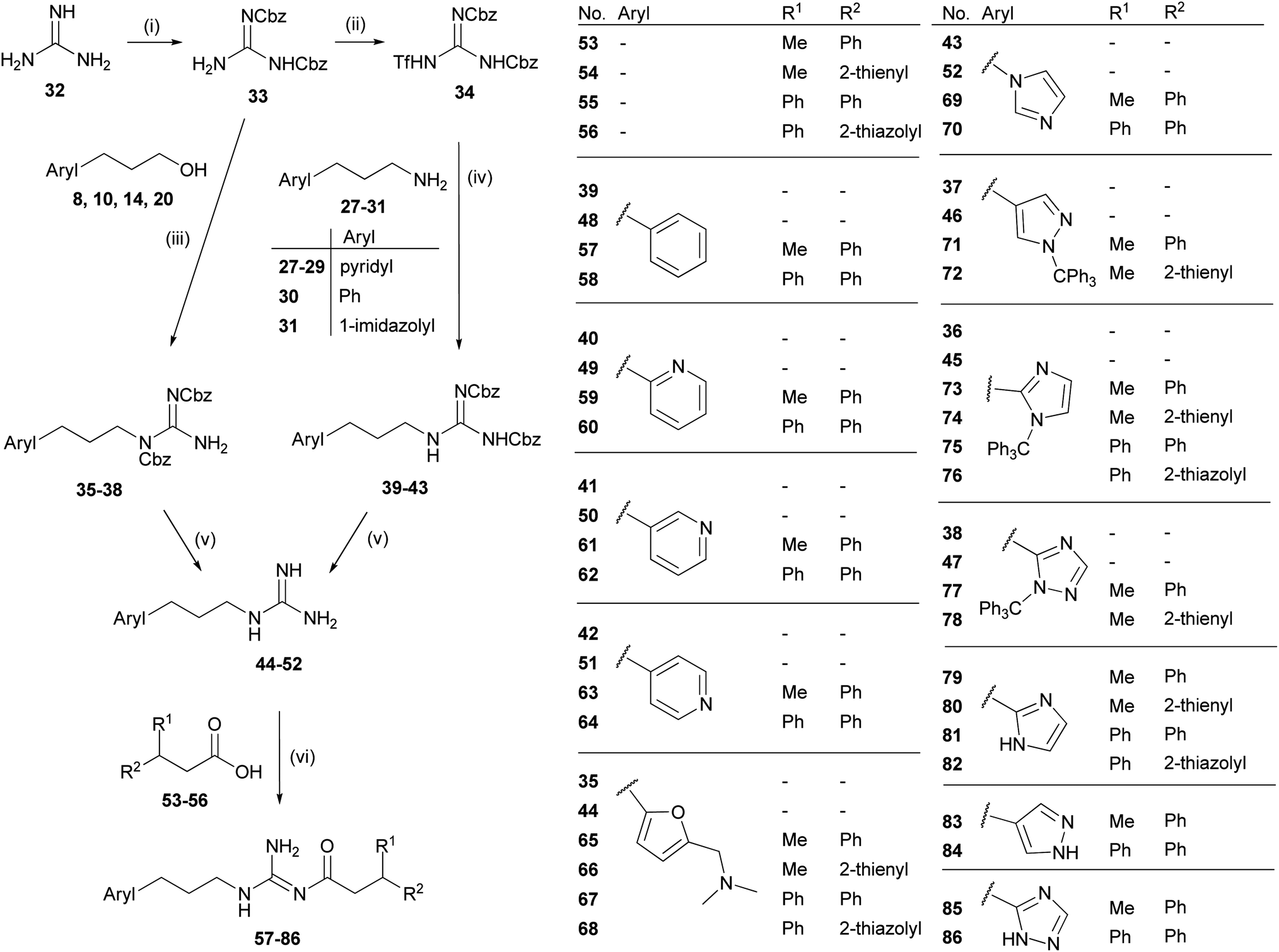 | ||
| Scheme 2 Synthesis of the NG-acylated arylpropylguanidines 57–86. Reagents and conditions: (i) benzyl chloroformate (3 eq.), NaOH (5 eq.), H2O/DCM, 20 h, 0 °C; (ii) Tf2O (1 eq.), NaH (60% dispersion in mineral oil) (2 eq.), chlorobenzene, overnight, −45 °C → rt; (iii) PPh3 (1.5 eq.), DIAD (1.5 eq.), THF, overnight, 0 °C → rt; (iv) NEt3 (1 eq.), DCM, overnight, rt; (v) H2, Pd/C (10%) (cat.), MeOH, 3 h, rt; (vi) (a) CDI (1.2 eq.), NaH (60% dispersion in mineral oil) (2 eq.), THF, 5 h, rt; (b) for trityl-protected intermediates 71–78: TFA (20%), DCM, 5 h, rt. | ||
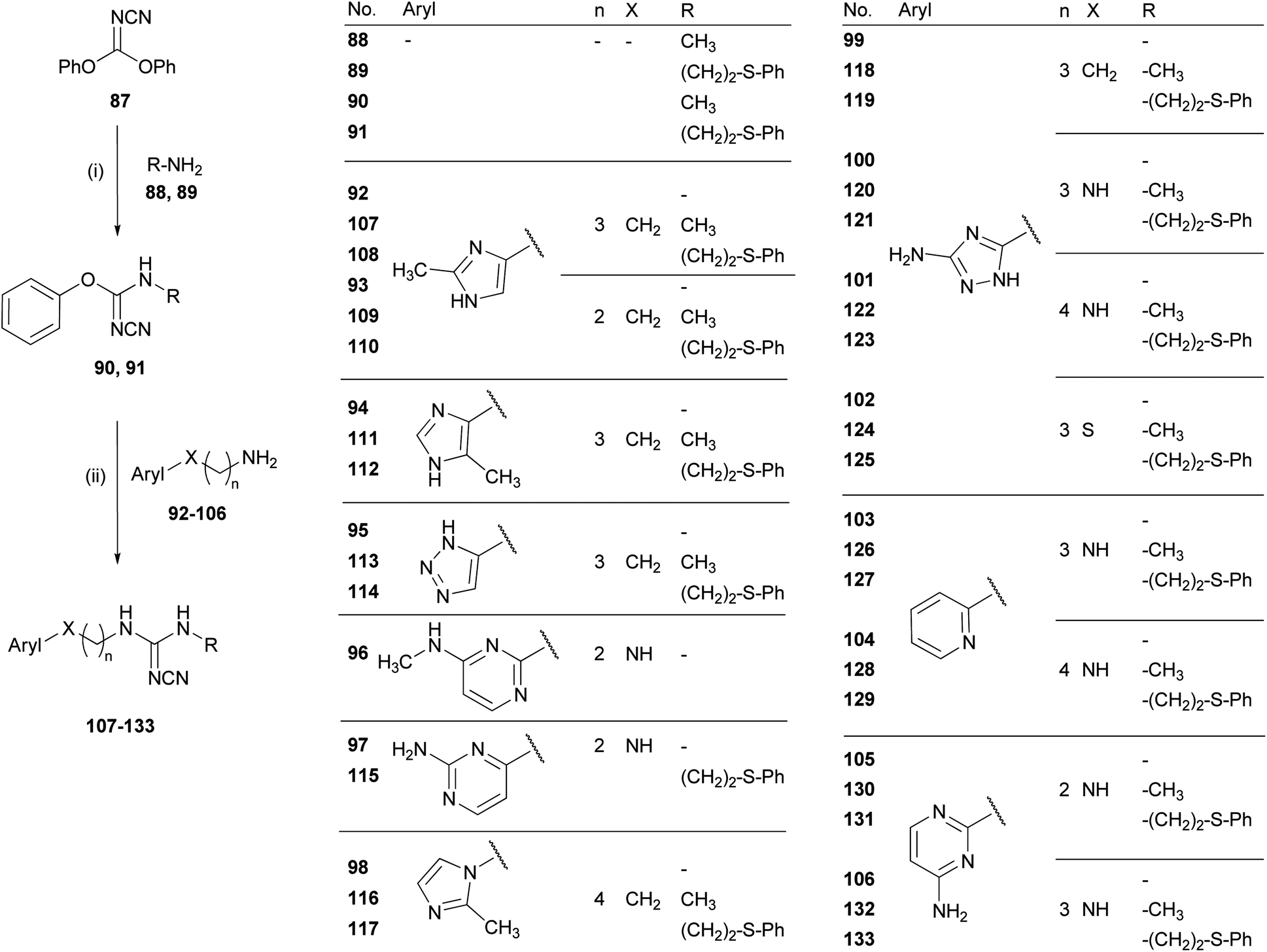 | ||
| Scheme 3 Synthesis of the cyanoguanidines 107–133. Reagents and conditions: (i) 2-propanol, 1 h, rt; (ii) MeCN, microwave 150 °C, 15 min. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) EC50) values. Intrinsic activities (α) refer to the maximal response induced by the standard agonist histamine. Compounds identified to be inactive as agonists (α < 0.1 or negative values, respectively, determined in the agonist mode) were investigated in the antagonist mode. The pKB values of neutral antagonists and inverse agonists were determined from the concentration-dependent inhibition of the histamine-induced increase in [35S]GTPγS binding or [γ32P]GTP ([γ33P]GTP) hydrolysis, respectively.
EC50) values. Intrinsic activities (α) refer to the maximal response induced by the standard agonist histamine. Compounds identified to be inactive as agonists (α < 0.1 or negative values, respectively, determined in the agonist mode) were investigated in the antagonist mode. The pKB values of neutral antagonists and inverse agonists were determined from the concentration-dependent inhibition of the histamine-induced increase in [35S]GTPγS binding or [γ32P]GTP ([γ33P]GTP) hydrolysis, respectively.
| Compound | hH1R | hH2R | hH3R | hH4R | ||||
|---|---|---|---|---|---|---|---|---|
| pEC50 or (pKB) | N | pEC50 or (pKB) | N | pEC50 or (pKB) | N | pEC50 or (pKB) | N | |
| a Steady-state GTPase activity in Sf9 insect cell membranes expressing the hH1R + RGS4, hH2R-GsαS fusion protein, hH3R + Gαi2 + Gβ1γ2 + RGS4 or hH4R-RGS19 fusion protein + Gαi2 + Gβ1γ2 was determined as described in the ESI. N gives the number of independent experiments performed in duplicate. b n.d.: not determined. | ||||||||
| Histamine | 6.72 ± 0.02 (ref. 28) | 5.92 ± 0.11 (ref. 28) | 7.60 ± 0.05 | 3 | 7.92 ± 0.11 | 8 | ||
| α: 1.00 | a: 1.00 | a: 1.00 | a: 1.00 | |||||
| UR-AK24 (1) | (<5)27 | 7.17 (ref. 29) | 8.60 ± 0.11 | 2 | 7.82 ± 0.01 | 2 | ||
| — | α: 0.87 | a: 0.24 ± 0.02 | a: 0.84 ± 0.06 | |||||
| Thioperamide | n.d. | n.d. | (7.01 ± 0.08) | 5 | (6.96 ± 0.06) | 6 | ||
| a: −0.71 ± 0.6 | a: −0.95 ± 0.07 | |||||||
| 57 | (5.05 ± 0.06) | 2 | (5.89 ± 0.04) | 2 | (<5) | 2 | (<5) | 2 |
| a: −0.02 ± 0.05 | a: −0.10 ± 0.00 | a: −0.19 ± 0.03 | n.d. | |||||
| 58 | (5.26 ± 0.03) | 2 | (5.89 ± 0.08) | 2 | (5.10 ± 0.0) | 2 | (<5) | 2 |
| a: −0.01 ± 0.01 | a: −0.12 ± 0.01 | a: −0.24 ± 0.01 | n.d. | |||||
| 59 | (5.34 ± 0.01) | 2 | 6.08 ± 0.11 | 3 | (<5) | 2 | (<5) | 2 |
| a: 0.21 ± 0.03 | a: 0.30 ± 0.01 | a: −0.17 ± 0.01 | n.d. | |||||
| 60 | (5.41 ± 0.02) | 2 | (5.68 ± 0.01) | 2 | (5.35 ± 0.04) | 2 | (<5) | 2 |
| a: 0.10 ± 0.02 | a: −0.00 ± 0.03 | a: −0.44 ± 0.01 | n.d. | |||||
| 61 | (<5) | 2 | (5.27 ± 0.0) | 2 | (<5) | 2 | (<5) | 2 |
| n.d. | a: −0.11 ± 0.05 | a: −0.01 ± 0.01 | n.d. | |||||
| 62 | (5.04 ± 0.04) | 2 | (6.14 ± 0.08) | 2 | (<5) | 2 | (<5) | 2 |
| a: 0.06 ± 0.04 | a: −0.16 ± 0.05 | a: −0.14 ± 0.01 | n.d. | |||||
| 63 | (<5) | 2 | (5.54 ± 0.01) | 2 | (<5) | 2 | (<5) | 2 |
| a: 0.12 ± 0.06 | a: 0.00 ± 0.09 | a: −0.07 ± 0.06 | n.d. | |||||
| 64 | (<5) | 2 | (6.14 ± 0.01) | 2 | (5.06 ± 0.01) | 2 | (<5) | 2 |
| a: 0.08 ± 0.07 | a: −0.12 ± 0.06 | a: −0.08 ± 0.07 | n.d. | |||||
| 65 | (<5) | 2 | (6.52 ± 0.03) | 3 | (5.92 ± 0.01) | 2 | (<5) | 2 |
| a: 0.04 ± 0.01 | a: −0.12 ± 0.02 | a: −0.64 ± 0.00 | n.d. | |||||
| 66 | n.d. | (6.51 ± 0.12) | 2 | (5.77 ± 0.04) | 2 | (<5) | 2 | |
| a: −0.12 ± 0.02 | a: −0.73 ± 0.01 | a: −0.14 ± 0.05 | ||||||
| 67 | (5.11 ± 0.01) | 2 | (7.03 ± 0.13 | 2 | (5.89 ± 0.07) | 2 | (<5) | 2 |
| a: −0.02 ± 0.00 | a: −0.16 ± 0.02 | a: −0.65 ± 0.01 | a: −0.16 ± 0.09 | |||||
| 68 | n.d. | (6.13 ± 0.12) | 2 | (5.51 ± 0.03) | 2 | (<5) | 2 | |
| a: −0.13 ± 0.02 | a: −0.62 ± 0.04 | n.d. | ||||||
| 69 | (<5) | 2 | (<5) | 2 | (<5) | 2 | 6.00 ± 0.13 | 2 |
| n.d. | n.d. | a: −0.05 ± 0.03 | a: 0.77 ± 0.01 | |||||
| 70 | (<5) | 2 | (5.82 ± 0.03) | 2 | (<5) | 2 | (<5) | 2 |
| a: 0.01 ± 0.01 | a: −0.07 ± 0.02 | a: −0.07 ± 0.02 | n.d. | |||||
| 79 | (<5) | 2 | (5.30 ± 0.02) | 2 | (5.41 ± 0.11) | 2 | 5.54 ± 0.19 | 2 |
| a: 0.09 ± 0.06 | a: 0.03 ± 0.03 | a: −0.32 ± 0.02 | a: 0.35 ± 0.01 | |||||
| 80 | n.d. | (5.26 ± 0.14) | 2 | (5.35 ± 0.14) | 2 | 6.11 ± 0.12 | 3 | |
| a: 0.04 ± 0.00 | a: −0.46 ± 0.02 | a: 0.50 ± 0.04 | ||||||
| 81 | (5.07 ± 0.09) | 2 | (6.13 ± 0.12) | 2 | (5.47 ± 0.04) | 2 | (5.85 ± 0.15) | 2 |
| a: 0.04 ± 0.01 | a: −0.04 ± 0.06 | a: −0.33 ± 0.00 | a: −0.08 ± 0.19 | |||||
| 82 | n.d. | (5.19 ± 0.11) | 2 | (5.28 ± 0.04) | 2 | (<5) | 3 | |
| a: −0.00 ± 0.00 | a: −0.46 ± 0.01 | n.d. | ||||||
| 83 | (<5) | 4 | 6.62 ± 0.11 | 2 | (<5) | 2 | (<5) | 2 |
| n.d. | a: 0.44 ± 0.01 | a: 0.04 ± 0.01 | n.d. | |||||
| 84 | (5.06 ± 0.03) | 2 | (5.42 ± 0.09) | 2 | (<5) | 2 | (<5) | 2 |
| a: 0.06 ± 0.02 | a: 0.08 ± 0.04 | a: −0.02 ± 0.01 | n.d. | |||||
| 85 | (<5) | 2 | 6.13 ± 0.03 | 3 | (<5) | 2 | Inactive | 2 |
| n.d. | a: 0.66 ± 0.02 | a: −0.03 ± 0.00 | ||||||
| 86 | (<5) | 2 | 6.39 ± 0.01 | 2 | (<5) | 2 | Inactive | 3 |
| a: 0.06 ± 0.02 | a: 0.42 ± 0.01 | a: −0.03 ± 0.02 | ||||||
| Compound | gpH1R | gpH2R | ||
|---|---|---|---|---|
| pA2 | N | pEC50b/(pA2)/[pD'2]c/αd | N | |
| a Number of experiments. b pEC50 was calculated from the mean shift ΔpEC50 of the agonist curve relative to the histamine reference curve by equation: pEC50 = 6.00 + 0.13 + ΔpEC50. Summand 0.13 represents the mean desensitization observed for control organs when two successive curves for histamine were performed (0.13 ± 0.02, N = 16). The SEM given for pEC50 is the SEM calculated for ΔpEC50. c pD'2 values given in brackets for compounds producing a significant, concentration-dependent reduction of the maximal response of histamine. d Efficacy, maximal response, relative to the maximal increase in heart rate induced by the reference compound histamine. e E max (histamine) at 10 μM: 0.68 ± 0.01. f E max (histamine) at 30 μM: 0.69 ± 0.03. g E max (histamine) at 30 μM: 0.54 ± 0.09. h pA2 of cimetidine (10 μM, N = 2): 6.32 ± 0.06. For experimental details, cf. ESI. | ||||
| Histamine | — | — | 6.00 ± 0.10 | >50 |
| a: 1.00 ± 0.02 | ||||
| UR-AK24 (1)27 | 5.87 ± 0.14 | 4 | 7.80 ± 0.07 | 4 |
| a: 0.99 ± 0.02 | ||||
| UR-PG276 (3) | n.d. | (<4.5) | 2 | |
| [< 4.5] | ||||
| 59 | n.d. | 6.71 ± 0.04 | 3 | |
| a: 0.26 ± 0.03 | ||||
| 61 | n.d. | (<4.5) | 2 | |
| [4.22 ± 0.04] | ||||
| 63 | n.d. | (4.72 ± 0.34) | 2 | |
| [4.54 ± 0.03] | ||||
| 65 | 5.52 ± 0.06 | 18 | (6.28 ± 0.13) | 2 |
| a: 0e | ||||
| 69 | 5.95 ± 0.05 | 18 | (<4.5) | 2 |
| [4.16 ± 0.05] | ||||
| a: 0f | ||||
| 79 | 5.63 ± 0.04 | 18 | (4.90 ± 0.16) | 2 |
| [4.44 ± 0.15] | ||||
| a: <10g | ||||
| 83 | 5.42 ± 0.10 | 15 | 6.33 ± 0.07 | 3 |
| a: 0.54 ± 0.03 | ||||
| 84 | 5.59 ± 0.09 | 17 | 6.44 ± 0.11 | 3 |
| a: 0.41 ± 0.05 | ||||
| 85 | 5.83 ± 0.07 | 16 | 7.00 ± 0.07h | 3 |
| a: 1.00 ± 0.02 | ||||
| 86 | 5.79 ± 0.04 | 16 | 6.69 ± 0.02 | 3 |
| a: 0.83 ± 0.06 | ||||
| Compound | hH1R | hH2R | hH3R | hH4R | ||||
|---|---|---|---|---|---|---|---|---|
| pEC50 or (pKB) | N | pEC50 or (pKB) | N | EC50 or (pKB) | N | EC50 or (pKB) | N | |
| a Functional [35S]GTPγS binding assay with membrane preparations of Sf9 cells expressing the hH3R + Gαi2 + Gβ1γ2 or the hH4R + Gαi2 + Gβ1γ2 or the hH2R-Gsαs fusion protein were performed as described in the ESI. b Steady-state GTPase activity in Sf9 cell membranes expressing the hH1R + RGS4 was determined as described under Pharmacological methods. c Reaction mixtures contained ligands at a concentration range from 1 nM to 1 mM as appropriate to generate saturated concentration/response curves. N gives the number of independent experiments performed in duplicate. The intrinsic activity (α) of histamine was set to 1.00 and α values of other compounds were referred to this value. The α values of neutral antagonists and inverse agonists were determined at a concentration of 10 μM. The pKB values of neutral antagonists and inverse agonists were determined in the antagonist mode versus histamine as the agonist. | ||||||||
| Histamine | 6.72 ± 0.02 (ref. 28) | 5.92 ± 0.11 (ref. 28) | 7.89 ± 0.07 | 3 | 7.96 ± 0.12 | 5 | ||
| α: 1.00 | a: 1.00 | a: 1.00 | a: 1.00 | |||||
| UR-PI376 (5) | (<5)34 | (<5)34 | (6.14 ± 0.02) | 2 | 7.43 ± 0.04 | 3 | ||
| α: 0.07 | α: 0.08 | a: −0.52 ± 0.05 | a: 0.88 ± 0.08 | |||||
| Cimetidine | n.d. | (5.77 ± 0.11)62 | n.d. | (<5)35 | ||||
| a: −0.08 ± 0.01 | ||||||||
| 107 | (<5) | 2 | (<5) | 2 | (<5) | 2 | (<5) | 2 |
| α: 0.01 ± 0.03 | a: 0.04 ± 0.02 | a: −0.13 ± 0.08 | a: −0.23 ± 0.03 | |||||
| 108 | (<5.30) | 2 | 4.79 ± 0.01 | 2 | (<5) | 2 | (6.21 ± 0.04) | 2 |
| a: −0.01 ± 0.03 | a: −0.05 ± 0.02 | a: −0.47 ± 0.04 | a: −0.24 ± 0.11 | |||||
| 109 | Inactive | 2 | (<5) | 2 | (<5) | 2 | 6.26 ± 0.02 | 2 |
| a: −0.02 ± 0.01 | a: −0.52 ± 0.04 | a: 0.74 ± 0.02 | ||||||
| 110 | (<5.30) | 2 | (<5) | 2 | (<5) | 2 | 6.33 ± 0.03 | 2 |
| a: 0.03 ± 0.05 | a: −0.05 ± 0.02 | a: −1.11 ± 0.03 | a: 0.40 ± 0.07 | |||||
| 111 | (<5) | 2 | (<5) | 2 | 5.97 ± 0.04 | 2 | 5.44 ± 0.04 | 2 |
| a: −0.03 ± 0.02 | a: 0.04 ± 0.01 | a: 0.23 ± 0.03 | a: 0.60 ± 0.17 | |||||
| 112 | (<5.30) | 2 | (5.36 ± 0.04) | 2 | (5.50 ± 0.02) | 2 | 6.61 ± 0.0 | 2 |
| a: −0.03 ± 0.01 | a: −0.06 ± 0.02 | a: −0.65 ± 0.02 | a: 0.37 ± 0.1 | |||||
| 113 | Inactive | 2 | Inactive | 2 | Inactive | 2 | Inactive | 2 |
| 114 | (<5) | 2 | (<5) | 2 | Inactive | 2 | Inactive | 2 |
| a: −0.02 ± 0.01 | a: −0.04 ± 0.0 | |||||||
| 115 | (<5) | 2 | (<5) | 2 | (5.54 ± 0.01) | 2 | (5.43 ± 0.09) | 2 |
| a: −0.02 ± 0.04 | a: −0.07 ± 0.01 | a: −1.03 ± 0.01 | a: −0.83 ± 0.06 | |||||
| 116 | (<5) | 2 | (<5) | 2 | (<5) | 2 | Inactive | 2 |
| a: −0.01 ± 0.03 | a: −0.07 ± 0.01 | a: −0.26 ± 0.03 | a: 0.06 ± 0.03 | |||||
| 117 | (<5) | 2 | (<5) | 2 | (<5) | 2 | (<5) | 2 |
| a: 0.03 ± 0.01 | a: −0.08 ± 0.00 | a: −0.42 ± 0.06 | a: −0.05 ± 0.04 | |||||
| 118 | Inactive | 2 | <5 | 2 | Inactive | 2 | Inactive | 2 |
| a: 0.31 ± 0.02 | ||||||||
| 119 | (<5) | 2 | (<5) | 2 | Inactive | 2 | Inactive | 2 |
| a: 0.01 ± 0.04 | a: 0.03 ± 0.02 | |||||||
| 120 | Inactive | 2 | (<5) | 2 | Inactive | 2 | Inactive | 2 |
| a: −0.03 ± 0.0 | ||||||||
| 121 | Inactive | 2 | (<5) | 2 | Inactive | 2 | Inactive | 2 |
| a: −0.01 ± 0.04 | ||||||||
| 122 | Inactive | 2 | Inactive | 2 | Inactive | 2 | Inactive | 2 |
| 123 | Inactive | 2 | (<5) | 2 | Inactive | 2 | Inactive | 2 |
| a: −0.09 ± 0.01 | ||||||||
| 124 | Inactive | 2 | (<5) | 2 | Inactive | 2 | Inactive | 2 |
| a: −0.02 ± 0.0 | ||||||||
| 125 | Inactive | 2 | (<5) | 2 | Inactive | 2 | Inactive | 2 |
| a: −0.05 ± 0.00 | ||||||||
| 126 | Inactive | 2 | Inactive | 2 | (<5) | 2 | (<5) | 2 |
| a: −0.03 ± 0.04 | a: −0.13 ± 0.05 | |||||||
| 127 | (<5) | 2 | (<5) | 2 | (<5) | 2 | (<5) | 2 |
| a: 0.02 ± 0.03 | a: −0.12 ± 0.02 | a: −0.21 ± 0.02 | a: −0.10 ± 0.01 | |||||
| 128 | (<5) | 2 | Inactive | 2 | (<5) | 2 | (<5) | 2 |
| a: 0.03 ± 0.03 | a: −0.10 ± 0.03 | a: −0.11 ± 0.03 | ||||||
| 129 | (<5) | 2 | (<5) | 2 | (<5) | 2 | (<5) | 2 |
| a: −0.01 ± 0.01 | a: −0.13 ± 0.01 | a: −0.36 ± 0.04 | a: −0.34 ± 0.01 | |||||
| 130 | Inactive | 2 | Inactive | 2 | (<5) | 2 | (<5) | 2 |
| a: −0.16 ± 0.02 | a: −0.11 ± 0.03 | |||||||
| 131 | (<5) | 2 | (<5) | 2 | (<5) | 2 | Inactive | 2 |
| a: 0.01 ± 0.03 | a: −0.08 ± 0.02 | a: −1.43 ± 0.14 | ||||||
| 132 | (<5) | 2 | Inactive | 2 | (<5) | 2 | Inactive | 2 |
| a: 0.03 ± 0.02 | a: −0.24 ± 0.05 | |||||||
| 133 | (<5) | 2 | (<5) | 2 | (5.14 ± 0.01) | 2 | (<5) | 2 |
| a: 0.01 ± 0.02 | a: −0.11 ± 0.01 | a: −0.71 ± 0.03 | a: −0.09 ± 0.05 | |||||
Replacing the imidazole ring in 1 with a 2-pyridyl ring resulted in an hH2R partial agonist (59) with a potency comparable to that of the endogenous ligand histamine (pEC50 = 6.08, Emax = 0.30). At the hH1R, this compound also behaved as a weak partial agonist. This is in agreement with data for the 2-pyridyl analogue of histamine, betahistine, which is a weak agonist at the hH1R and hH2R.56,59 The bulky diphenylpropanoyl residue in 60 was not tolerated in terms of agonistic potency. At the hH3R and hH4R, 59 and 60 were almost inactive. The 3- and 4-pyridyl analogues 61–64 displayed moderate antagonism at the hH2R and negligible activities at the other HR subtypes.
Replacement of the imidazole ring by the 5-[(dimethylamino)methyl]furan-2-yl group, reminiscent of the H2R antagonist ranitidine, afforded hH2R antagonists (65–68): all prepared compounds turned out to be superior to ranitidine (pKB ∼ 6.10)59 at the hH2R, with the highest antagonist activity exhibited by the diphenylpropanoylguanidine 67 (pKB = 7.03). 65–68 were weak inverse agonists at the hH3R and almost inactive at the hH1R and hH4R.
Apart from other heterocycles, isomers of the 1H-imidazol-4-yl ring were investigated. The 1H-imidazol-1-yl isomer (69) was comparable with UR-AK24 (1) regarding intrinsic activity (Emax: 0.77 vs. 0.84) at the hH4R, but the potency was 65-fold lower (pEC50: 6 vs. 7.82). The activities (69) at the other HRs were negligible (pKB < 5). However, the results for 69 suggest that, in contrast to other HR subtypes, an imidazole–NH group is obviously dispensible, though it is crucial to obtain highly potent hH4R agonists. As obvious from the diphenylpropanoyl analogue 70, which is almost inactive at the hH4R, the hH4R agonism strongly depends on the constitution of the acyl residue.
For the 1H-imidazol-2-yl isomers 79–82 similar pharmacological activities at the hH4R were observed as for the 1H-imidazol-1-yl isomers 69 and 70. Both 3-arylbutanoylguanidines (79 and 80) exhibited moderate partial agonistic potencies and low intrinsic activities. Similar to the isomer 70, diarylpropanoyl residues (81 and 82) abolished agonism at the hH4R. At the other HR subtypes, 79–82 were very weak antagonists or inverse agonists.
The results for the imidazole isomers suggest that, regarding agonism and compared to the other HR subtypes, the hH4R tolerates some modifications in the arrangement of side chains and nitrogen atoms in the heterocycle.
Exchange of the imidazole ring in UR-AK24 by a 1H-pyrazol-4-yl ring (83) resulted in a moderate decrease in potency and efficacy at the hH2R (pEC50 = 6.62, Emax = 0.44). Interestingly, this compound was virtually inactive at all other HRs, suggesting the pyrazole ring to be an appropriate bioisostere of the imidazole ring to shift the receptor subtype selectivity toward the hH2R. However, the bulky diphenylpropanoyl residue in 84 was deleterious for agonistic activity at the hH2R.
Compared to the pyrazole 83, the 1H-1,2,4-triazol-3-yl analogue 85 was more efficacious, but slightly less potent at the hH2R (pEC50 = 6.13, Emax = 0.66). In contrast to the pyrazole 84, the triazole analog 86 with a diphenylpropanoyl moiety was an hH2R partial agonist with slightly higher potency than 85 (pEC50 = 6.39, Emax = 0.42). This suggests that the acylguanidines containing a triazole or a pyrazole ring can adopt different binding modes at the hH2R. Like the pyrazoles 83 and 84, the triazoles 85 and 86 were almost inactive at the other HRs.
The basicity of pyrazole (pKa ≈ 3)60 and triazole (pKa ≈ 3)60 is considerably lower than that of imidazole (pKa ≈ 7).60 This may be interpreted as a hint that the presence of a heterocycle, which is positively charged at physiological pH value, is not required for hH2R activation. Moreover, the modification of the acyl residue in triazolylalkylguanidines obviously harbours the potential of increasing H2R selectivity.
The results from isolated guinea pig organs were essentially in line with the data gained from recombinant human H1 and H2 receptors, but, in general, the guinea pig receptors proved to be more sensitive than the human orthologs. At the guinea pig ileum the investigated NG-acylated arylpropylguanidines (Table 2) were moderate H1R antagonists. Like at the hH2R, introduction of a phenyl (58), 3-pyridyl (61), 4-pyridyl (63), 5-[(dimethylamino)-methyl]furan-2-yl (65), 1H-imidazol-1-yl (69) and 1H-imidazol-2-yl moiety (79) resulted in a loss of agonistic efficacy at the gpH2R relative to compound 1 and yielded weak gpH2R antagonists. Remarkably, the H2R antagonist activity of compound 65 was comparable to that of cimetidine at the guinea pig right atrium.27
In accordance with the results at the hH2R, the exchange of the 1H-imidazol-4-yl ring in UR-AK24 (1) by a 2-pyridyl ring (59) resulted in a gpH2R partial agonist with lower potency and efficacy than 1 (Emax = 0.26). This tendency is reminiscent of the close histamine analogues 2-(2-pyridyl)ethanamine and betahistine, which likewise display weak partial agonism at the guinea pig right atrium.61 Replacing the 1H-imidazol-4-yl by a 1H-pyrazol-4-yl ring (83 and 84) resulted in compounds with retained gpH2R partial agonistic activity. However, relative to UR-AK24 (1), the potency decreased by about one order of magnitude. In contrast to 83 and 84, the analogues bearing a 1H-1,2,4-triazol-3-yl ring (85, 86) and UR-AK24 were equiefficacious, though 6-fold less potent at the guinea pig right atrium. These findings support the hypothesis that the 1H-1,2,4-triazol-3-yl ring is a potential bioisostere of the 1H-imidazol-4-yl moiety with respect to gpH2R affinity.
As expected, the compounds 107–112, bearing a methyl substituted imidazole ring, showed some activity at the H4R. The 2-methylimidazole derivatives 107 and 108 with a tetramethylene chain connecting imidazole and cyanoguanidine were weak inverse agonists at the H4R, devoid of noteworthy activity at the other HR subtypes. As reported previously, the phenylthioethyl residue confers higher potency at the H4R compared to a methyl substitution. Reducing the spacer chain length to three carbon atoms provides the hH4R partial agonists 109 and 110 with pEC50 values around 6.3 and no agonistic activity at the other three histamine receptors. This is in agreement with the results for the amines 92 and 93.52 Compound 111, the carba analogue of cimetidine,61 was a weak partial agonist at the hH4R (pEC50 = 5.44) and the hH3R (pEC50 = 5.97) and showed only very weak antagonistic properties at the hH1R and at the hH2R. This is in accordance with data reported for H2R antagonism at guinea pig right atrium and the rat uterus, where 111 was inferior to cimetidine by a factor of 6 to 10.61 The phenylthioethyl cyanoguanidine 112 was 15 times more potent as an hH4R agonist than the methyl cyanoguanidine 111. With a pEC50 value of 6.61 at the H4R, the 5-methyl analogue of UR-PI376, compound 112, showed a more than 10-fold selectivity for the H4R over the H3R and the other HR subtypes. Nevertheless, none of the investigated hetarylalkylcyanoguanidines was superior to UR-PI376 in terms of H4R agonistic potency or receptor subtype selectivity.
Conclusions
In summary, for most of the investigated acylguanidines the replacement of the 1H-imidazol-4-yl ring with isomers or other heterocycles resulted in considerably reduced potency and efficacy at the hH2R, hH3R and hH4R. This underlines the substantial contribution of an appropriate arrangement of the nitrogen atoms in the heterocycle for binding to the H2R, H3R and H4R and for stabilizing an active conformation of the H2R and H4R. Strikingly, the imidazol-1-yl-propylguanidine derivative 69 displayed a comparable maximal response as its isomer, reference compound UR-AK24 (1), at the H4R subtype, although, the potency was about 50-fold lower. As these acylguanidines were poorly active at the other HR subtypes and the hH4R activity largely depended on the type of acyl residue, further modifications in this moiety appear promising with respect to the development of more potent and selective hH4R agonists.Introduction of a 2-pyridyl (59 and 60), a 1H-pyrazol-4-yl (83 and 84) or a 1H-1,2,4-triazol-3-yl (85 and 86) ring provided compounds exhibiting partial to full agonist activity at the hH2R and gpH2R. Except for the 2-pyridyl analogues, these compounds had negligible activities at the other hHR subtypes. In particular, the triazole ring was identified as a promising bioisostere for the imidazole ring with respect to H2R activity. At the gpH2R, the NG-acylated triazolylpropylguanidines (85 and 86) were equiefficacious to UR-AK24, but devoid of activities at the other hHR subtypes, suggesting the 1H-1,2,4-triazol-3-yl ring as a potential bioisostere for the design of H2R selective ligands. 2-Aminothiazole analogues of acylguanidine-type H2R ligands are described33,63 as highly selective agonists with higher potencies compared to the triazole analogs. However, compared to the aminothiazoles, the triazole ring is considered as relatively stable against enzymatic oxidation.64 This may offer an alternative to improve the drug-like properties.
The cyanoguanidines derived from OUP-16 and UR-PI376 revealed high sensitivity against both, replacement of the heterocycle and minor structural modifications such as methyl-substitution of the imidazole ring. None of the analogues showed improved potency and/or H4R selectivity compared to UR-PI376. Except for the heterocycle, the cyanoguanidines are devoid of basic groups. Since previous studies revealed higher potency of H4R agonists with retained basicity in the central structural motif, e.g. acylguanidines, a combination of bioisosteric replacement of both, imidazole ring and cyanoguanidine moiety, should be considered in future ligand design.
In conclusion, the presented data suggest alternative bioisosteric approaches, including the synthesis and pharmacological evaluation of additional heterocyclic analogues of known histamine receptor ligands, with respect to retained/increased potency, improved receptor subtype selectivity and drug-like properties.
Acknowledgements
The authors are grateful to Maria Beer-Kroen, Kerstin Fisch, Karin Schadendorf, Gertraud Wilberg and Astrid Seefeld for expert technical assistance. This work was supported by the Graduate Training Program (Graduiertenkolleg) GRK 760 of the Deutsche Forschungsgemeinschaft and the COST program BM0806 (H4R network) of the European Union.Notes and references
- S. J. Hill, C. R. Ganellin, H. Timmerman, J.-C. Schwartz, N. P. Shankley, J. M. Young, W. Schunack, R. Levi and H. L. Haas, Pharmacol. Rev., 1997, 49, 253–278 CAS.
- L. B. Hough, Mol. Pharmacol., 2001, 59, 415–419 CAS.
- R. Seifert, A. Strasser, E. H. Schneider, D. Neumann, S. Dove and A. Buschauer, Trends Pharmacol. Sci., 2013, 34, 33–58 CrossRef CAS PubMed.
- A. Strasser, H.-J. Wittmann, A. Buschauer, E. H. Schneider and R. Seifert, Trends Pharmacol. Sci., 2013, 34, 13–32 CrossRef CAS PubMed.
- K. Sander, T. Kottke and H. Stark, Biol. Pharm. Bull., 2008, 31, 2163–2181 CAS.
- T. Oda, N. Morikawa, Y. Saito, Y. Masuho and S. Matsumoto, J. Biol. Chem., 2000, 275, 36781–36786 CrossRef CAS PubMed.
- T. Nakamura, H. Itadani, Y. Hidaka, M. Ohta and K. Tanaka, Biochem. Biophys. Res. Commun., 2000, 279, 615–620 CrossRef CAS PubMed.
- Y. Zhu, D. Michalovich, H.-L. Wu, K. B. Tan, G. M. Dytko, I. J. Mannan, R. Boyce, J. Alston, L. A. Tierney, X. Li, N. C. Herrity, L. Vawter, H. M. Sarau, R. S. Ames, C. M. Davenport, J. P. Hieble, S. Wilson, D. J. Bergsma and L. R. Fitzgerald, Mol. Pharmacol., 2001, 59, 434–441 CAS.
- K. L. Morse, J. Behan, T. M. Laz, R. E. West, Jr, S. A. Greenfeder, J. C. Anthes, S. Umland, Y. Wan, R. W. Hipkin, W. Gonsiorek, N. Shin, E. L. Gustafson, X. Qiao, S. Wang, J. A. Hedrick, J. Greene, M. Bayne and F. J. Monsma, Jr, J. Pharmacol. Exp. Ther., 2001, 296, 1058–1066 CAS.
- F. Coge, S.-P. Guenin, H. Rique, J. A. Boutin and J.-P. Galizzi, Biochem. Biophys. Res. Commun., 2001, 284, 301–309 CrossRef CAS PubMed.
- C. Liu, X.-J. Ma, X. Jiang, S. J. Wilson, C. L. Hofstra, J. Blevitt, J. Pyati, X. Li, W. Chai, N. Carruthers and T. W. Lovenberg, Mol. Pharmacol., 2001, 59, 420–426 CAS.
- T. Nguyen, D. A. Shapiro, S. R. George, V. Setola, D. K. Lee, R. Cheng, L. Rauser, S. P. Lee, K. R. Lynch, B. L. Roth and B. F. O'Dowd, Mol. Pharmacol., 2001, 59, 427–433 CAS.
- H. Engelhardt, R. A. Smits, R. Leurs, E. Haaksma and I. J. de Esch, Curr. Opin. Drug Discovery Dev., 2009, 12, 628–643 CAS.
- E. Tiligada, E. Zampeli, K. Sander and H. Stark, Expert Opin. Invest. Drugs, 2009, 18, 1519–1531 CrossRef CAS PubMed.
- R. Kiss and G. M. Keseru, Expert Opin. Ther. Pat., 2009, 19, 119–135 CrossRef CAS PubMed.
- R. A. Smits, R. Leurs and I. J. P. de Esch, Drug Discovery Today, 2009, 14, 745–753 CrossRef CAS PubMed.
- J. M. Cowden, J. P. Riley, J. Y. Ma, R. L. Thurmond and P. J. Dunford, Respir. Res., 2010, 11, 86 CrossRef PubMed.
- J. M. Cowden, M. Zhang, P. J. Dunford and R. L. Thurmond, J. Invest. Dermatol., 2010, 130, 1023–1033 CrossRef CAS PubMed.
- R. Seifert, E. H. Schneider, S. Dove, I. Brunskole, D. Neumann, A. Strasser and A. Buschauer, Mol. Pharmacol., 2011, 79, 631–638 CrossRef CAS PubMed.
- S. Nijmeijer, H. F. Vischer, E. M. Rosethorne, S. J. Charlton and R. Leurs, Mol. Pharmacol., 2012, 82, 1174–1182 CrossRef CAS PubMed.
- E. M. Rosethorne and S. J. Charlton, Mol. Pharmacol., 2011, 79, 749–757 CrossRef CAS PubMed.
- J. A. Jablonowski, C. A. Grice, W. Chai, C. A. Dvorak, J. D. Venable, A. K. Kwok, K. S. Ly, J. Wei, S. M. Baker, P. J. Desai, W. Jiang, S. J. Wilson, R. L. Thurmond, L. Karlsson, J. P. Edwards, T. W. Lovenberg and N. I. Carruthers, J. Med. Chem., 2003, 46, 3957–3960 CrossRef CAS PubMed.
- D. Neumann, S. Beermann and R. Seifert, Pharmacology, 2010, 85, 217–223 CrossRef CAS PubMed.
- P. Igel, S. Dove and A. Buschauer, Bioorg. Med. Chem. Lett., 2010, 20, 7191–7199 CrossRef CAS PubMed.
- A. Buschauer, J. Med. Chem., 1989, 32, 1963–1970 CrossRef CAS PubMed.
- A. Buschauer, A. Friese-Kimmel, G. Baumann and W. Schunack, Eur. J. Med. Chem., 1992, 27, 321–330 CrossRef CAS.
- P. Ghorai, A. Kraus, M. Keller, C. Goette, P. Igel, E. Schneider, D. Schnell, G. Bernhardt, S. Dove, M. Zabel, S. Elz, R. Seifert and A. Buschauer, J. Med. Chem., 2008, 51, 7193–7204 CrossRef CAS PubMed.
- S.-X. Xie, P. Ghorai, Q.-Z. Ye, A. Buschauer and R. Seifert, J. Pharmacol. Exp. Ther., 2006, 317, 139–146 CrossRef CAS PubMed.
- S.-X. Xie, A. Kraus, P. Ghorai, Q.-Z. Ye, S. Elz, A. Buschauer and R. Seifert, J. Pharmacol. Exp. Ther., 2006, 317, 1262–1268 CrossRef CAS PubMed.
- P. Igel, E. Schneider, D. Schnell, S. Elz, R. Seifert and A. Buschauer, J. Med. Chem., 2009, 52, 2623–2627 CrossRef CAS PubMed.
- G. Coruzzi, H. Timmerman, M. Adami and G. Bertaccini, Naunyn-Schmiedeberg's Arch. Pharmacol., 1993, 348, 77–81 CrossRef CAS PubMed.
- P. Ghorai, A. Kraus, T. Birnkammer, R. Geyer, G. Bernhardt, S. Dove, R. Seifert, S. Elz and A. Buschauer, Bioorg. Med. Chem. Lett., 2010, 20, 3173–3176 CrossRef CAS PubMed.
- A. Kraus, P. Ghorai, T. Birnkammer, D. Schnell, S. Elz, R. Seifert, S. Dove, G. Bernhardt and A. Buschauer, ChemMedChem, 2009, 4, 232–240 CrossRef CAS PubMed.
- P. Igel, R. Geyer, A. Strasser, S. Dove, R. Seifert and A. Buschauer, J. Med. Chem., 2009, 52, 6297–6313 CrossRef CAS PubMed.
- H. D. Lim, R. M. van Rijn, P. Ling, R. A. Bakker, R. L. Thurmond and R. Leurs, J. Pharmacol. Exp. Ther., 2005, 314, 1310–1321 CrossRef CAS PubMed.
- H. D. Lim, R. A. Smits, R. A. Bakker, C. M. E. vanDam, I. J. P. deEsch and R. Leurs, J. Med. Chem., 2006, 49, 6650–6651 CrossRef CAS PubMed.
- T. Hashimoto, S. Harusawa, L. Araki, O. P. Zuiderveld, M. J. Smit, T. Imazu, S. Takashima, Y. Yamamoto, Y. Sakamoto, T. Kurihara, R. Leurs, R. A. Bakker and A. Yamatodani, J. Med. Chem., 2003, 46, 3162–3165 CrossRef CAS PubMed.
- Chemistry and Structure–Activity Relationships of Drugs Acting at Histamine Receptors, ed. C. R. Ganellin, Wright PSG, Bristol, London, Boston, 1982 Search PubMed.
- C. R. Ganellin, Pharmacochemistry of H1 and H2 Receptors, Wiley-Liss, Inc., 1992 Search PubMed.
- J. Navarro, Pharmazie, 1995, 50, 12–15 CAS.
- B. Striegl, Doctoral thesis, University of Regensburg, Germany, 2006.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- P. G. Bulger, I. F. Cottrell, C. J. Cowden, A. J. Davies and U.-H. Dolling, Tetrahedron Lett., 2000, 41, 1297–1301 CrossRef CAS.
- M. Kunze, Doctoral thesis, University of Regensburg, Germany, 2006.
- Application: J. M. Cox, P. Bellini, R. Barrett, R. M. Ellis and T. R. Hawkes (Zeneca Ltd.), WO Pat., WO 9315610, 1993, Chem. Abstr., 1993, 120, 134813.
- W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS.
- O. Mitsunobu, M. Yamada and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1967, 40, 935–939 CrossRef CAS.
- Application: J. R. Allen, S. A. Hitchcock, B. Liu and W. W. Turner, Jr. (Eli Lilly Co.), WO Pat., WO 2005009941, 2005, Chem. Abstr., 2005, 142, 197700.
- K. Feichtinger, H. L. Sings, T. J. Baker, K. Matthews and M. Goodman, J. Org. Chem., 1998, 63, 8432–8439 CrossRef CAS.
- R. Paul and G. W. Anderson, J. Am. Chem. Soc., 1960, 82, 4596–4600 CrossRef CAS.
- R. Paul and G. W. Anderson, J. Org. Chem., 1962, 27, 2094–2099 CrossRef CAS.
- R. Geyer, M. Kaske, P. Baumeister and A. Buschauer, Arch. Pharm., 2014, 347 DOI:10.1002/ardp.201300316 , in press.
- R. L. Webb and C. S. Labaw, J. Heterocycl. Chem., 1982, 19, 1205–1206 CrossRef CAS.
- R. Geyer and A. Buschauer, Arch. Pharm., 2011, 344, 775–785 CrossRef CAS PubMed.
- M. T. Kelley, T. Bürckstümmer, K. Wenzel-Seifert, S. Dove, A. Buschauer and R. Seifert, Mol. Pharmacol., 2001, 60, 1210–1225 CAS.
- R. Seifert, K. Wenzel-Seifert, T. Bürckstümmer, H. H. Pertz, W. Schunack, S. Dove, A. Buschauer and S. Elz, J. Pharmacol. Exp. Ther., 2003, 305, 1104–1115 CrossRef CAS PubMed.
- T. Asano, S. E. Pedersen, C. W. Scott and E. Ross, Biochemistry, 1984, 23, 5460–5467 CrossRef CAS PubMed.
- G. Hilf, P. Gierschik and K. H. Jakobs, Eur. J. Biochem., 1989, 186, 725–731 CrossRef CAS PubMed.
- H. Preuss, P. Ghorai, A. Kraus, S. Dove, A. Buschauer and R. Seifert, J. Pharmacol. Exp. Ther., 2007, 321, 983–995 CrossRef CAS PubMed.
- pKa-value was calculated with ACD Labs 9.0, Advanced Chemistry Development, Toronto (Canada).
- G. J. Durant, J. C. Emmett, C. R. Ganellin, P. D. Miles, M. E. Parsons, H. D. Prain and G. R. White, J. Med. Chem., 1977, 20, 901–906 CrossRef CAS PubMed.
- H. Preuss, Doctoral thesis, University of Regensburg, 2007.
- T. Birnkammer, A. Spickenreither, I. Brunskole, M. Lopuch, N. Kagermeier, G. Bernhardt, S. Dove, R. Seifert, S. Elz and A. Buschauer, J. Med. Chem., 2012, 55, 1147–1160 CrossRef CAS PubMed.
- D. K. Dalvie, A. S. Kalgutkar, S. C. Khojasteh-Bakht, R. S. Obach and J. P. O'Donnell, Chem. Res. Toxicol., 2002, 15, 269–299 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis procedures, analytical data and pharmacological methods. See DOI: 10.1039/c3md00245d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |