Identification and optimisation of 3,3-dimethyl-azetidin-2-ones as potent and selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1)†
William
McCoull
*a,
Martin
Augustin
c,
Caroline
Blake
a,
Anne
Ertan
b,
Elaine
Kilgour
a,
Stephan
Krapp
c,
Jane E.
Moore
a,
Nicholas J.
Newcombe
a,
Martin J.
Packer
a,
Amanda
Rees
a,
John
Revill
a,
James S.
Scott
a,
Nidhal
Selmi
a,
Stefan
Gerhardt
a,
Derek J.
Ogg
a,
Stefan
Steinbacher
c and
Paul R. O.
Whittamore
a
aCardiovascular and Gastrointestinal Innovative Medicines Unit, AstraZeneca, Alderley Park, Macclesfield, Cheshire SK10 4TG, UK. E-mail: william.mccoull@astrazeneca.com; Tel: +44 (0)1625 519444
bMedicines Evaluation Pharmaceutical Development, AstraZeneca R&D, 151 85 Södertälje, Sweden
cProteros biostructures GmbH, Am Klopferspitz 19, Martinsried, Germany
First published on 6th November 2013
Abstract
3,3-Di-methyl-azetidin-2-ones were identified as potent and selective 11β-HSD1 inhibitors against the human and mouse forms of the enzyme. Structure guided optimisation of LLE was conducted, utilising a key polar interaction and identifying stereochemical preference for the 4S isomer. Metabolic stability was improved to afford oral exposure, providing tool compounds suitable for pre-clinical evaluation.
Introduction
The principal strategy in the management or prevention of type II diabetes and cardiovascular disease involves treatment of a collection of risk factors. When a patient exhibits central obesity in combination with two of the following risk factors; elevated fasting plasma glucose, dyslipidemia, hypertension, reduced high density lipoprotein (HDL) cholesterol they are defined as having the metabolic syndrome.1 The underlying causes of the metabolic syndrome are complex and the synthesis and metabolism of glucocorticoids have been proposed to play a key role.2,3 Human patients with Cushing's syndrome, who exhibit elevated circulating glucocorticoid levels, display a phenotype similar to the metabolic syndrome.4 Although patients with the metabolic syndrome do not exhibit elevated plasma glucocorticoid levels,5 it has been hypothesised that elevated intracellular concentrations may play a crucial role. Therefore, the ability to reduce intracellular glucocorticoid concentrations has been proposed as an attractive therapeutic paradigm for the metabolic syndrome.6–911β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) is a NADPH dependent enzyme that is widely expressed, notably in liver, adipose tissue, and brain.10 It catalyses the interconversion of the inactive glucocorticoid hormone cortisone 1 to the active glucocorticoid hormone cortisol 2 and therefore plays a key role in the regulation of intracellular cortisol concentrations (Fig. 1).11 11β-Hydroxysteroid dehydrogenase type 2 (11β-HSD2) is an NAD dependent oxidase expressed mainly in kidney and colon tissue that catalyses the reverse reaction and prevents cortisol activation of mineralocorticoid receptors.12 Inhibition of this enzyme has been associated with hypertension and other complications and therefore selectivity over this isoform is a key requirement for any potential therapeutic agent.13 Carbenoxolone (CBX) 3, originally developed as a treatment for oesophageal ulceration and inflammation, is a potent but unselective inhibitor of both 11β-HSD1 & 11β-HSD2.14 It has been shown to improve insulin sensitivity and reduce glucose production in patients with type II diabetes but is not used clinically for the treatment of the metabolic syndrome due to the associated hypertension risk.15
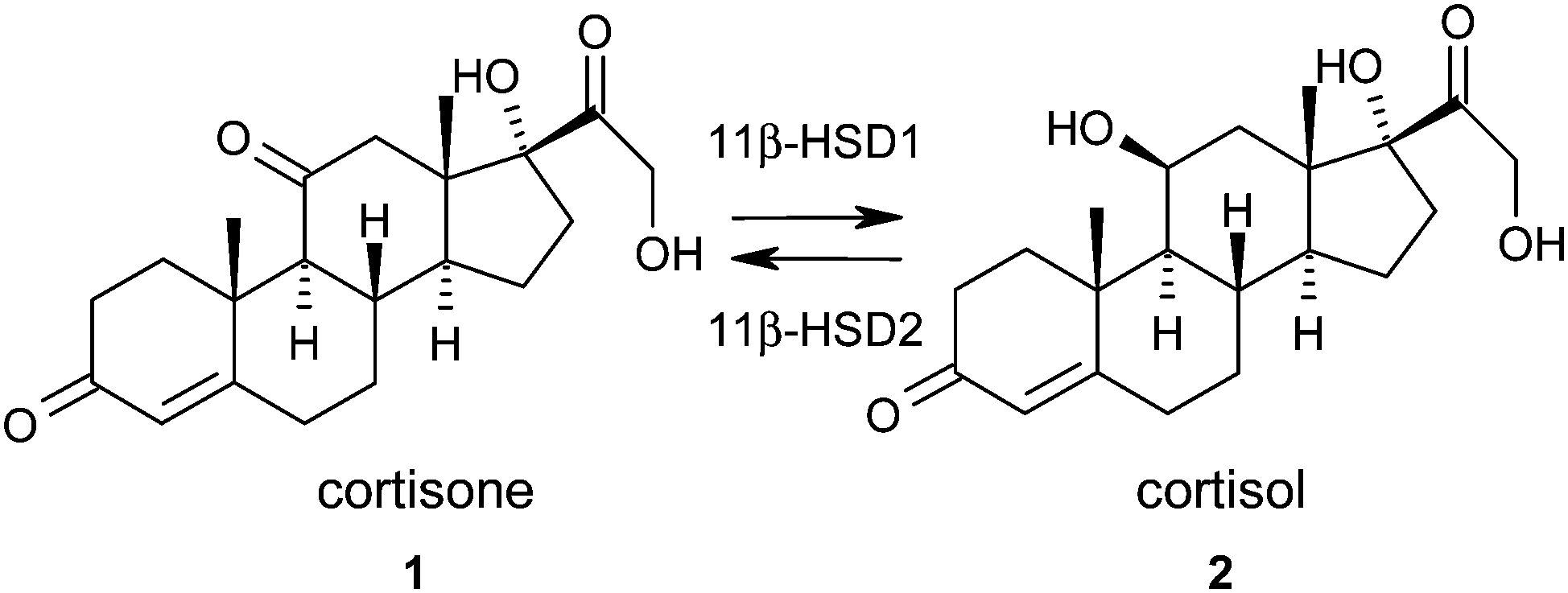 | ||
| Fig. 1 Interconversion of cortisone 1 to cortisol 2 by 11β-HSD1 and 2. | ||
There has been significant interest in the therapeutic potential of selective inhibitors of 11β-HSD1. Over 250 patent applications from over 25 pharmaceutical companies and academic groups have now been published and comprehensively reviewed.16–18 Our approach to identifying 11β-HSD1 inhibitors was to run a high throughput screen (HTS) and progress multiple chemical series in parallel. A 2-thioalkyl nicotinamide chemotype was progressed but initially exhibited modest rodent potency as recently reported elsewhere.19–23 We wished to progress another novel, structurally distinct series that, while selective for 11β-HSD1 over 11β-HSD2, was also potent against a rodent species to allow pre-clinical evaluation. In this communication we describe the optimisation of an azetidin-2-one series to deliver human and mouse potent 11β-HSD1 inhibitors that are selective over 11β-HSD2 with sufficient oral exposure to be used as preclinical tool compounds.
Results and discussion
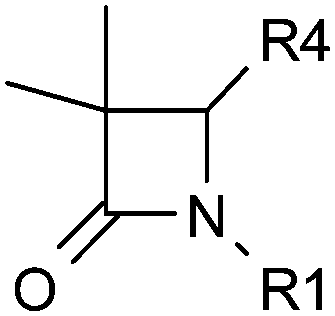
The lead compound 4 was obtained direct from HTS and had several attractive features. It was potent against human and murine 11β-HSD1 (although not rat), selective against 11β-HSD2 and hERG and displayed sub-optimal physicochemical properties (Table 1). High log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D7.4 and very high in vitro metabolism translated into zero oral exposure in rat and this was a key focus of the optimisation campaign. The low molecular weight and synthetic accessibility of the azetidin-2-one core allowed for SAR exploration around all three positions.
D7.4 and very high in vitro metabolism translated into zero oral exposure in rat and this was a key focus of the optimisation campaign. The low molecular weight and synthetic accessibility of the azetidin-2-one core allowed for SAR exploration around all three positions.
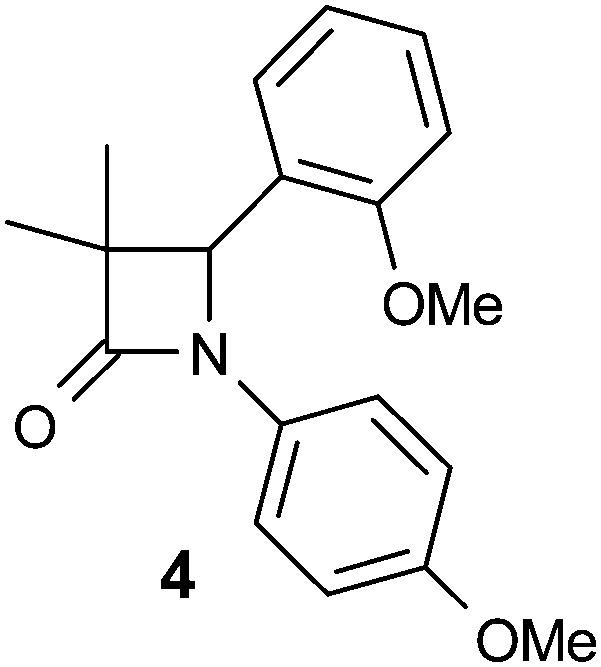
Our initial focus was to establish SAR to achieve reduction of lipophilicity while maintaining potency. In doing so we expected that the two electron-rich aryl rings (likely metabolic soft spots) would be replaced leading to modifications that would suggest a way to reduce metabolism as well. Our tactic was to keep at least one of these aryl groups and use ligand lipophilicity efficiency (LLE)24 as a key optimisation parameter. At this time, a crystal structure with carbenoxolone (CBX) bound to human 11β-HSD1 was available (PDB code 2bel)25 and compound design was guided by docking virtual ligands into this structure.
Our analysis of the CBX/11β-HSD1 structure identified that the active site Tyr183 and Ser170 formed polar interactions with the tertiary acid and ketone groups, spaced about 6 Å apart (Fig. 2); there is also a hydrogen bond interaction between the tertiary acid and a ribose hydroxyl group of the NAD co-factor. We believed that the azetidin-2-one carbonyl would mimic the ketone of CBX and postulated that adding a polar interaction about 6 Å distant from this into our ligands could realise an additional binding interaction. Docking azetidin-2-ones into this structure using Glide26 oriented the carbonyl as expected and the gem-di-Me group into shallow lipophilic pockets as shown in Fig. 3.27 The aryl ring on the azetidin-2-one nitrogen was orientated in the direction of Tyr183, which suggested the incorporation of a polar substituent, capable of hydrogen bonding, at the meta-position, as a way to increase LLE. Furthermore, only the (4S)-enantiomer docked well suggesting that it would be more potent than the (4R)-enantiomer.
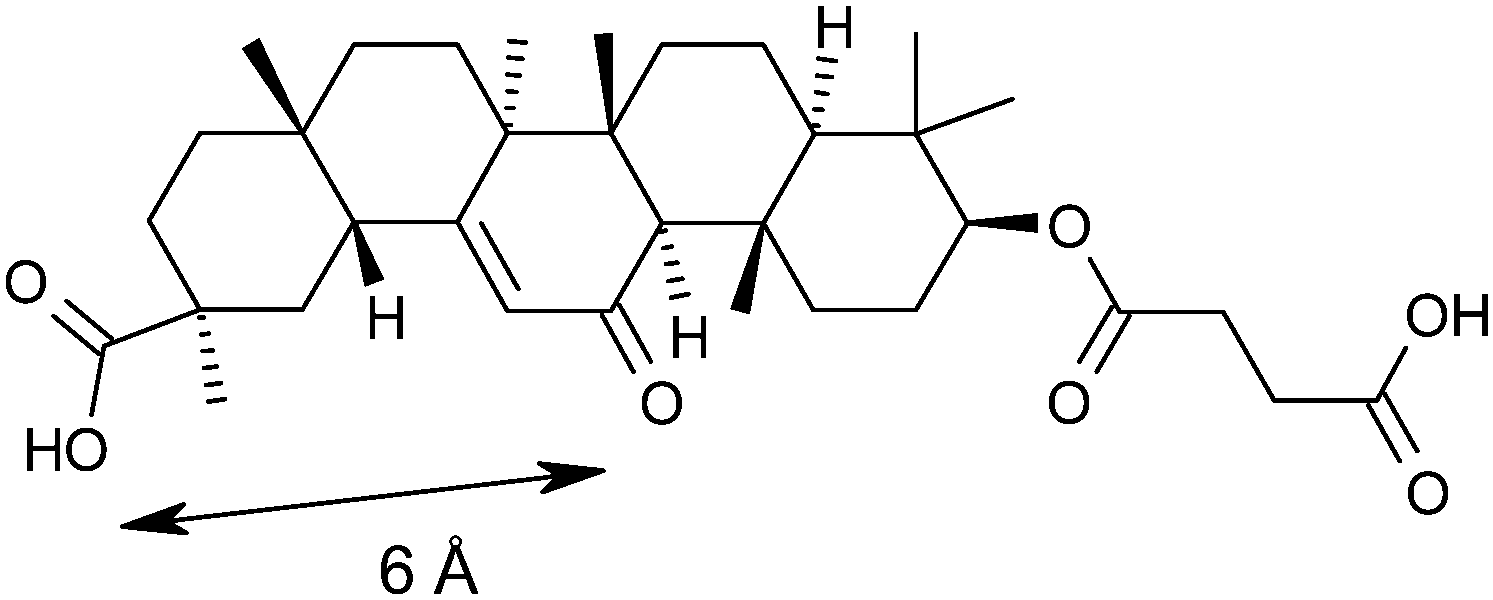 | ||
| Fig. 2 Key interaction pharmacophore defined by the crystal structure of carbenoxolone 3 bound to human 11β-HSD1, showing a 6 Å separation between acceptor groups bound in the active site. See Fig. 5 for a detailed view of the active site as compared with the 2-azetidinone series. | ||
 | ||
| Fig. 3 Docking of 43 into 11β-HSD1 binding site, which was modelled from the 2bel crystal structure. | ||
3,3-Dimethyl-azetidin-2-ones were synthesised through routes summarised in Scheme 1. The Staudinger cycloaddition28 was used to directly prepare di-aryl azetidin-2-ones 6 from imines 5 usually in good yield. Azetidin-2-ones with other 3-alkyl substituents were prepared similarly. To prepare N-alkyl analogues 9, the para-methoxyphenyl (PMP) protecting group was oxidatively cleaved by treatment with ceric ammonium nitrate,29 followed by alkylation of 8 at low temperature.30 Alternatively, N-arylation could be achieved from 8via Buchwald–Hartwig amination.31,32 Alkyl groups were incorporated into the C4 position by conversion of the easily accessible aldehyde 11 into the corresponding vinylazetidine, which was hydrogenated in excellent yield to give 12, or reduction and methylation to give ether 13.
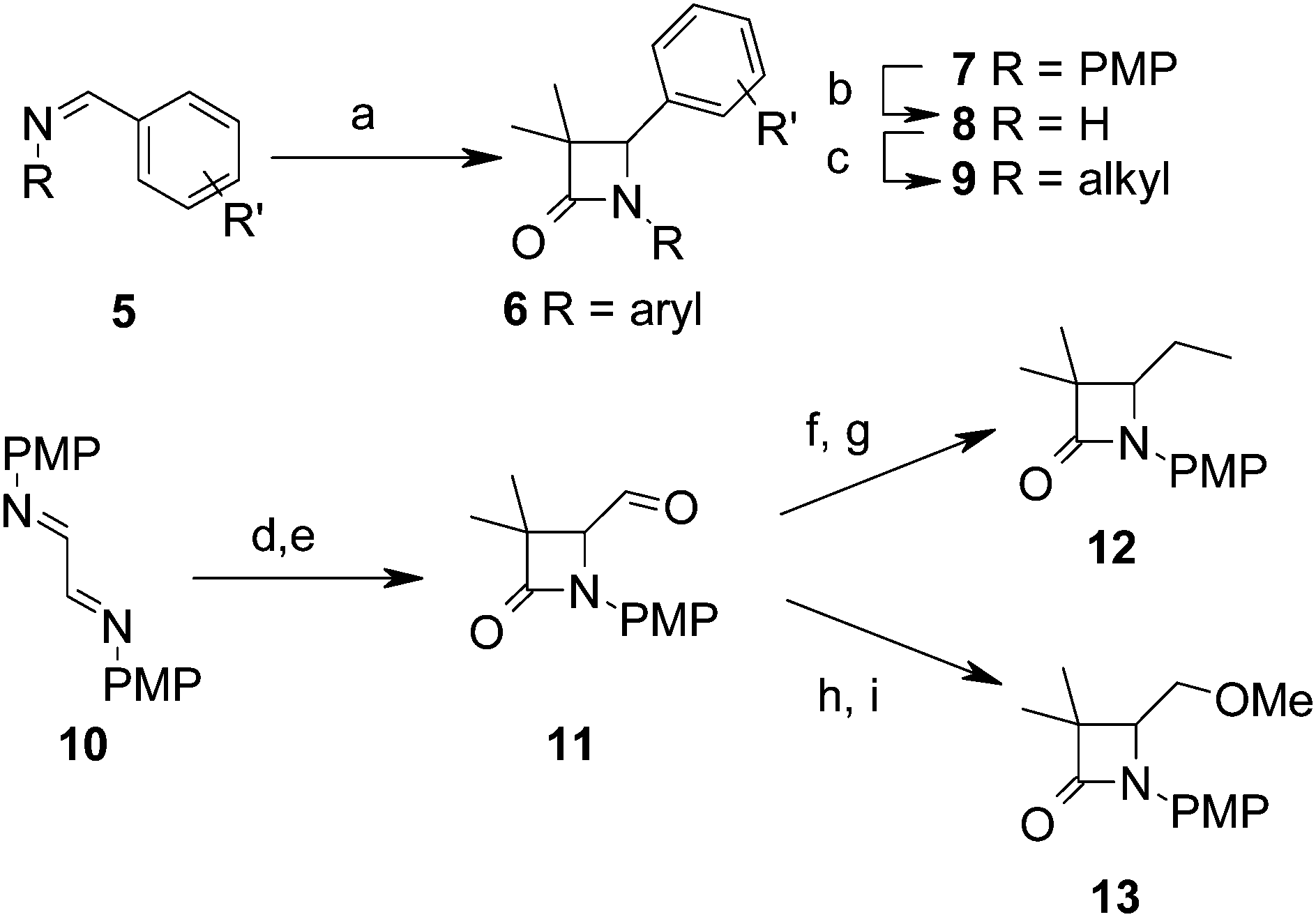 | ||
| Scheme 1 Representative synthetic route to 3,3-dimethylazetidine-2-ones. Reagents and conditions: PMP = para-MeO-phenyl. (a) iPrCO2Et, LiHMDS, PhMe–THF, 0 °C – RT, 8–90%; (b) CeIV(NH4)2(NO3)6, MeCN–H2O, 0 °C, 50%; (c) LiHMDS, R–X, −78 °C, THF, 29–45% or Pd2(dba)3, xantphos, Cs2CO3, dioxane, PhBr, 150 °C 30%; (d) iPrCO2Et, LDA, THF, 0 °C – RT, 55%; (e) HCl, MeOH, 65%; (f) tBuOK, THF, Ph3P+MeI−, 86%. (g) H2, 10% Pd/C, EtOAc, RT, 3 h, 98%; (h) NaBH4, MeOH, 75%; (i) NaH, MeI, THF, 55%. | ||
Firstly, substitution was investigated at the 3-position with gem-di-Me found to be optimal for potency (Table 2). According to our postulated docking pose, both methyl groups fit snugly into lipophilic pockets and even small changes were not tolerated thus we accepted that this was not a position where lipophilicity could be reduced. It is notable that a butyrolactam series of 11β-HSD1 inhibitors33 has been reported where gem-di-methyl substitution next to the lactam carbonyl was found to be optimal for human and murine potency, consistent with our results. All compounds in Table 2 had logD7.4 that were too high to measure accurately (>3.8) and rat hepatocytes gave high Clint values (>300 μL per min per 106 cells).
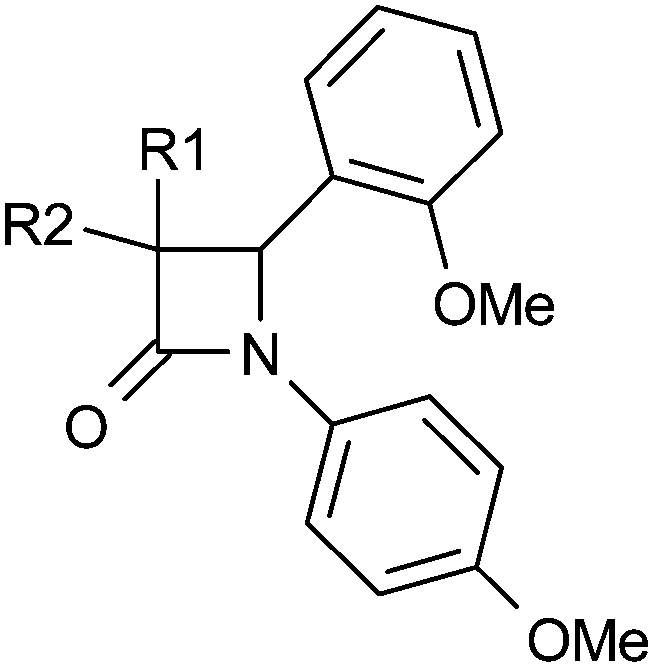
The N1 position of the azetidin-2-one was more tolerant to structural modification and allowed lipophilicity to be lowered (Table 3). Alkyl substituents such as ethyl (19) were less potent, although benzyl (20) did maintain potency. Introduction of nitrogen in pyridyl (22) did lower lipophilicity but lost potency relative to phenyl (21). A scan of fluorine and methyl groups at all three positions was conducted (23–28) which suggested that substitution at all positions could be tolerated. We envisaged that a sulfone at the meta-position, about 6 Å distant from the carbonyl would satisfy the favourable polar interaction with Tyr183 as suggested by our docking studies. This was confirmed when all three sulfones were tested, the meta-sulfone 30 being 9-fold more potent than the para-sulfone 31, and 170-fold more potent than ortho-sulfone 29. Furthermore, the meta-sulfide 32 lacking the hydrogen bonding acceptor was less potent while the meta-sulfoxide 33 which does provide an acceptor was equipotent. Consequently, 30 and 33 represent significant progress in terms of reducing logD7.4 and improving LLE relative to the initial lead 4.
D7.4 and LLE of selected 1-substituted azetidin-2-ones
|
|
||||
|---|---|---|---|---|
| R | h 11β-HSD1 IC50a (μM) | logD7.4 |
LLE | |
| a Mean value of at least 3 experiments. b Diastereomers unresolved. | ||||
| 4 | 4-MeO-Ph | 0.098 | >4.2 | — |
| 19 | Et | 2.8 | 2.7 | 2.9 |
| 20 | PhCH2 | 0.20 | 3.7 | 3.0 |
| 21 | Ph | 0.089 | >4.3 | — |
| 22 | 2-Pyridyl | 2.5 | 3.2 | 2.4 |
| 23 | 2-F-Ph | 0.20 | 4.2 | 2.5 |
| 24 | 3-F-Ph | 0.30 | >4 | — |
| 25 | 4-F-Ph | 0.15 | >4.3 | — |
| 26 | 2-Me-Ph | 0.11 | >4.3 | — |
| 27 | 3-Me-Ph | 0.029 | >4.3 | — |
| 28 | 4-Me-Ph | 0.057 | >4.1 | — |
| 29 | 2-MeSO2-Ph | 13 | 2.8 | 2.1 |
| 30 | 3-MeSO2-Ph | 0.076 | 2.9 | 4.2 |
| 31 | 4-MeSO2-Ph | 0.66 | 3.2 | 3 |
| 32 | 3-MeS-Ph | 0.40 | 3.8 | 2.6 |
| 33 | 3-MeSO-Ph | 0.057 | 2.8 | 4.5 |
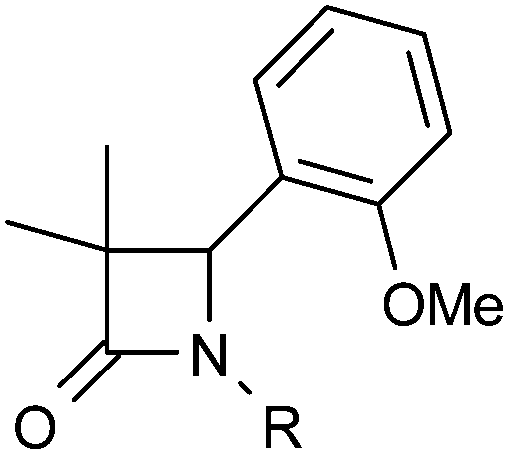
In parallel, we explored the 4-position of the azetidin-2-one with the N1 position fixed as 4-methoxyphenyl (Table 4). Although non-polar alkyl groups were tolerated, ethyl (12) resulted in lower potency relative to phenyl (34). Attempts to introduce polarity such as pyridine (35) resulted in lower potency albeit with similar LLE. Aryl substitution was tolerated at all positions (4, 36–39) and it was confirmed that the (4S)-enantiomer 41 was more potent than (4R)-enantiomer 40 in accord with our docking hypothesis. The absolute stereochemistry of (4R)-40 was unambiguously determined by X-ray crystallographic analysis (Fig. 4).34 Naphthyl 42 was potent and was evolved into quinoxaline 43 which, in line with our docking studies, realised increased potency and the highest LLE from this set.
D7.4 and LLE of selected 4-substituted azetidin-2-ones
|
|
||||
|---|---|---|---|---|
| R | h 11β-HSD1 IC50a (μM) | logD7.4 |
LLE | |
| a Mean value of at least 3 experiments. | ||||
| 12 | Et | 6.3 | 3.0 | 2.2 |
| 13 | MeOCH2 | >30 | 2.2 | — |
| 34 | Ph | 0.24 | 3.7 | 2.9 |
| 35 | 2-Pyridyl | 6.0 | 2.6 | 2.6 |
| 4 | 2-MeO-Ph | 0.098 | >4.2 | — |
| 36 | 3-MeO-Ph | 0.28 | 3.9 | 2.7 |
| 37 | 4-MeO-Ph | 0.13 | 3.8 | 3.1 |
| 38 | 2-HO-Ph | 0.086 | 3.9 | 3.1 |
| 39 | 4-F-Ph | 0.18 | 3.4 | 3.3 |
| 40 | (R)-4-Br-Ph | 3.0 | >4.3 | — |
| 41 | (S)-4-Br-Ph | 0.11 | >4.1 | — |
| 42 | 1-Naphthyl | 0.079 | >4.3 | — |
| 43 | 5-Quinoxalyl | 0.033 | 3.7 | 3.8 |
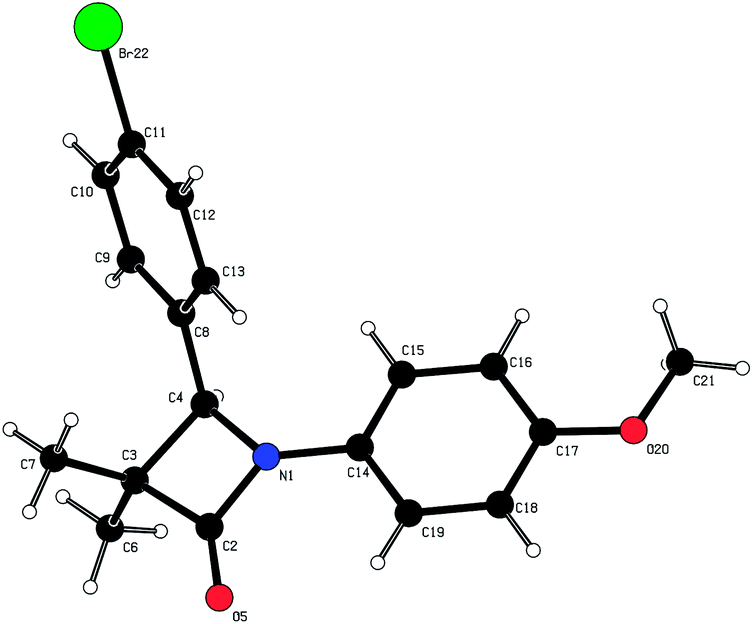 | ||
| Fig. 4 X-ray crystallographic structure of (4R)-40. | ||
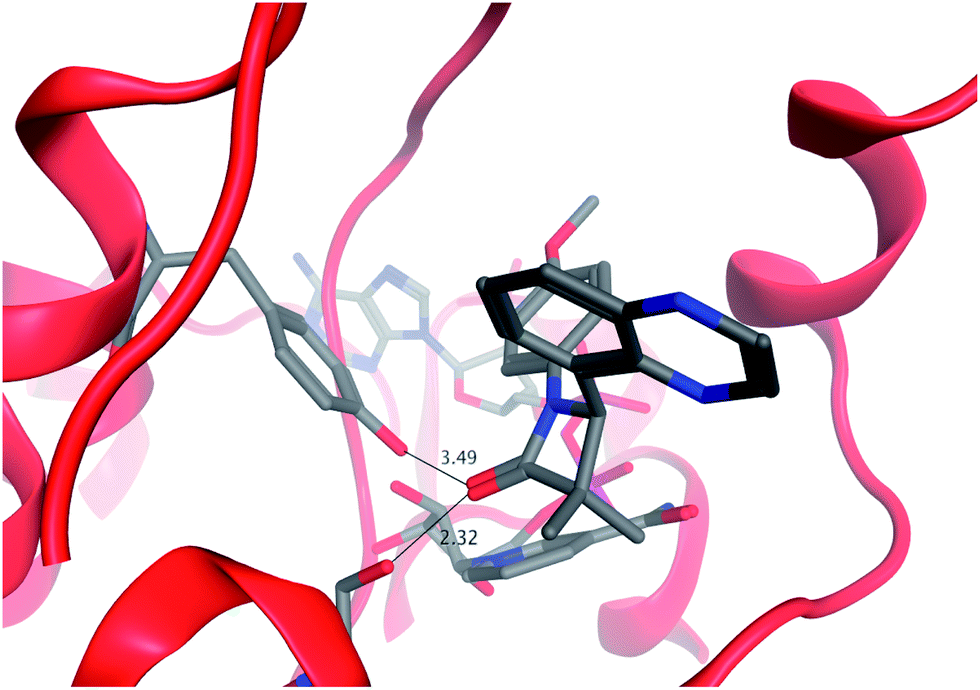
The crystal structure of sulfone 30 bound to murine 11β-HSD1 was obtained to explore our binding pharmacophore hypothesis of two polar interactions spaced about 6 Å apart (Fig. 5). The potency of 30 against the murine form of 11β-HSD1 (IC50 = 0.15 μM) is similar to that in human, thus we expected similar binding characteristics in both species. The tertiary acid of CBX makes a hydrogen bond contact to a ribose hydroxyl group and to Tyr183; we therefore expected the sulfone group of 30 to mimic this interaction. However, it appears from the contact network that Tyr183 is forced to act as proton donor to both the sulfone and to the azetidin-2-one carbonyl for ligand 30. The hydroxyl proton could be located between the two acceptor oxygen atoms to create a favourable polar environment, which is however not optimal for either interaction. This motif is not present for CBX, where the ketone group is deployed towards Ser170 only, leaving Tyr183 to act as donor for the acid. The azetidin-2-one carbonyl, is oriented between Ser170 and Tyr183, rather than only towards Ser170 as for the CBX ketone group. There is also a short contact to the serine, suggesting a strong hydrogen bond. This defines a favourable network of contacts, with the azetidin-2-one carbonyl positioned more optimally than the ketone of CBX. It is a feature of inhibitors of this target that contacts are observed to one or both of the active site residues.23
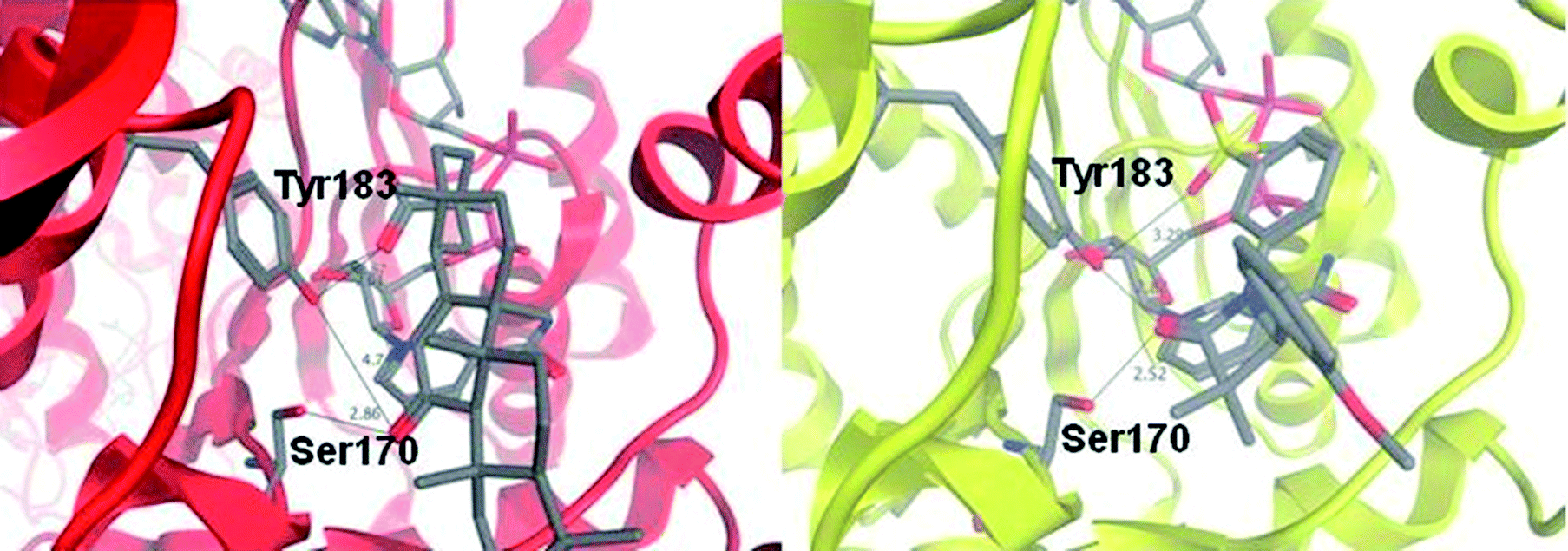 | ||
| Fig. 5 Binding modes for carbenoxolone in the active site of human 11β-HSD1 (red ribbon) and for azetidin-2-one 30 in the active site of murine 11β-HSD1 (yellow ribbon), as revealed by X-ray crystal structures. Carbenoxolone interacts with Ser170 via its ketone group (2.9 Å) and with Tyr183 via the tertiary acid (2.6 Å); there is also a contact between the acid and a ribose hydroxyl group of NADP+ (2.76 Å). By contrast, the amide group of compound 30 acts as an isostere of the ketone from CBX, but interacts with both Ser170 (2.5 Å); and Tyr183 (2.8 Å). The sulfone acts as an isostere for the tertiary acid, but does not make an optimum contact with either Tyr183 (3.3 Å); or the ribose group. This is despite the similar pharmacophore pattern that we observe in these two ligands, with a 6 Å separation between the two acceptor groups in 30, similar to CBX (Fig. 2). | ||
We obtained the murine structure because human structures were not accessible at the time of this work, although they did subsequently become available for a range of ligands.19,20,23 Given our restriction to a murine construct, we compared the available human and murine structures to explore side-chain variations within the active site. Fig. 6 shows an overlay of the 2bel human binding site (red ribbon) and our murine structure (yellow ribbon). It is clear that variations are relatively minor in the active site, especially in regions that ligand 30 contacts. This suggests that the limited size of the scaffold facilitates binding to both enzymes; the two active sites differ very little within the region that this ligand can access. There were clearly regions of the active site which would increase human affinity at the expense of mouse, and this has been confirmed in other disclosed series.19
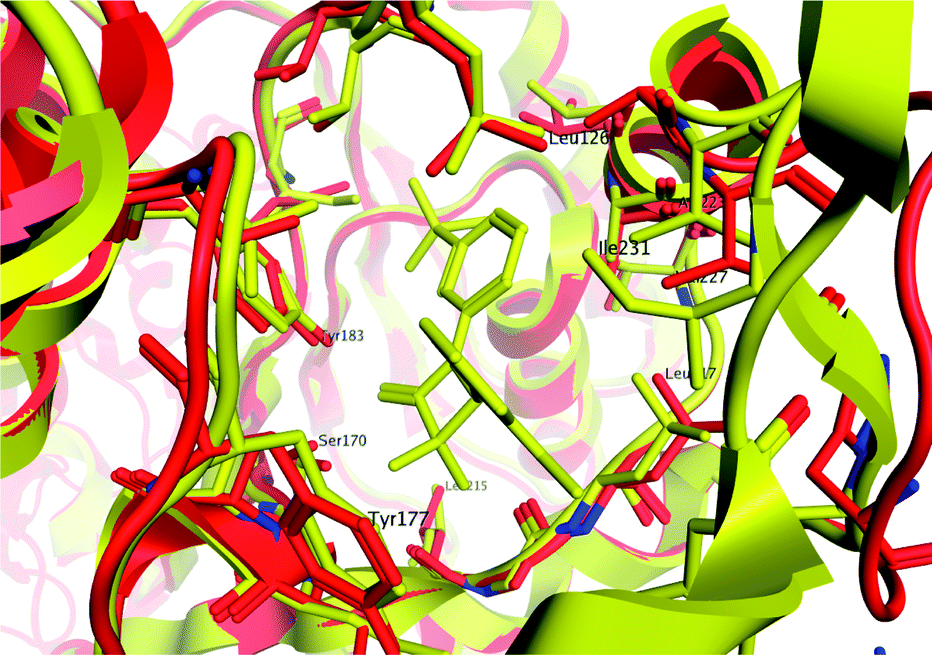 | ||
| Fig. 6 Overlay of active site for human 11β-HSD1 (red ribbon) and murine 11β-HSD1 (yellow ribbon) demonstrating close homology between side-chains in the region contacted by ligand 30 (shown in yellow). Contacts available to this ligand do not favour binding to the human enzyme over the murine form. | ||
Furthermore, the crystal structure confirmed that the gem-di-methyl group fits into a hydrophobic depression in the enzyme surface that would not accommodate larger groups (flanked by Leu171 and Tyr177) and it is the (4S)-enantiomer that exists in the crystal structure in line with higher potency than the (4R)-enantiomer.
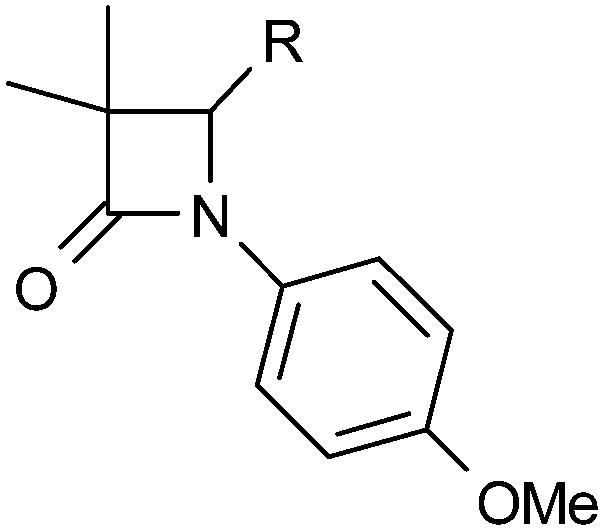
The substituents at the 1 and 4 positions which gave the highest LLE were combined and it was found that the 1-meta-sulfone-4-quinoxaline combination in 45 gave the most advantageous potency/lipophilicity balance with an LLE of 4.7 (Table 5). The enantiomer pair 46 and 47 confirmed the dependence of potency on stereochemistry at the 4-position, although in this case the absolute stereochemistry was not unambiguously determined. Selectivity against 11β-HSD2 (>30 μM) was maintained throughout this work and 45 and 48 were confirmed as potent against murine 11β-HSD1 (0.35 μM and 0.68 μM respectively) and inactive against rat 11β-HSD1 (>10 μM). All compounds containing a methoxy-phenyl had high in vitro metabolism (Clint > 200 μL per min per 106 cells), except 31 (Clint = 82 μL per min per 106 cells), 12 (Clint = 86 μL per min per 106 cells), and 13 (Clint = 130 μL per min per 106 cells). However, all combination compounds 44–48 showed lower in vitro metabolism (Table 5) which translated into low to moderate clearance in vivo (Table 6). Two compounds (44 and 48) were progressed to oral dosing in rat and this confirmed that acceptable oral exposure could be obtained for this series. Mouse hepatocyte data (Table 5) showed a similar trend to rat and in vivo pharmacokinetic data (Table 6) confirmed that 45 had acceptable oral exposure in mouse suitable for use in pre-clinical models.
|
|
||||||
|---|---|---|---|---|---|---|
| R1 | R4 | h 11β-HSD1 IC50a (μM) | logD7.4 |
LLE | Rat/mouse hep Clint (μL per min per 106 cells) | |
| a Mean value of at least 3 experiments; NT = not tested. | ||||||
| 44 | 3-MeSO2-Ph | 4-F-Ph | 0.91 | 2.8 | 3.2 | 16/46 |
| 45 | 3-MeSO2-Ph | 5-Quinoxalyl | 0.074 | 2.4 | 4.7 | 32/<3 |
| 46 | 4-F-Ph | (R)-4-F-Ph | >30 | 4.2 | — | 30/18 |
| 47 | 4-F-Ph | (S)-4-F-Ph | 0.28 | 4 | 2.6 | 25/NT |
| 48 | 4-F-Ph | 5-Quinoxalyl | 0.091 | 3.7 | 3.3 | 117/125 |
| Species | Clp (mL min−1 kg−1) | V dss (L kg−1) | IV half-life (h) | PO Cmax (μM) | Bioavailability (%) | |
|---|---|---|---|---|---|---|
|
a Compound was dosed to rats IV at 0.4 mg kg−1 and PO at 2 mg kg−1, to mouse IV at 2 mg kg−1 and PO at 2.5 mg kg−1, in 5% DMSO:95% hydroxypropyl beta cyclodextrin; NT = not tested.
|
||||||
| 44 | Rat | 2.9 | 1.2 | 5.4 | 0.95 | 27 |
| 45 | Rat | 52 | 3.2 | 0.77 | NT | NT |
| 47 | Rat | 19 | 9.7 | 7.2 | NT | NT |
| 48 | Rat | 57 | 2.6 | 0.76 | 0.11 | 39 |
| 45 | Mouse | 42 | 1.0 | 0.3 | 0.7 | 22 |
Conclusions
In summary, we identified a 3,3-di-methyl-azetidin-2-one series of 11β-HSD1 inhibitors which are potent against both the human and mouse enzymes and selective against 11β-HSD2. Optimisation of LLE was conducted, using the CBX/11β-HSD1 crystal structure to guide incorporation of a sulfone to pick up a polar interaction with the enzyme that was confirmed by a ligand/protein crystal structure. The preference for the (4S) stereoisomer being more potent was predicted by docking and confirmed in practice. Incorporation of less metabolically labile functional groups allowed oral exposure in rat and mouse to be realised. The potential of the 3,3-di-methyl-azetidin-2-ones series to deliver pre-clinical tools has been demonstrated.Acknowledgements
This research was sponsored by AstraZeneca. The authors thank Steve Bloor, Rob Garcia and Sally Johnson for the testing of compounds in biological assays and colleagues in physical chemistry, DMPK and safety assessment for the provision of secondary ADMET data.Notes and references
- S. M. Grundy, H. B. J. Brewer, J. I. Cleeman, S. C. J. Smith and C. Lenfant, Circulation, 2004, 109, 433–438 CrossRef PubMed.
- P. Björntorp and R. Rosmond, Nutrition, 2000, 16, 924–936 CrossRef.
- M. Wamil and J. R. Seckl, Drug Discovery Today, 2007, 12, 504–520 CrossRef CAS PubMed.
- G. Arnaldi, A. Angeli, A. B. Atkinson, X. Bertagna, F. Cavagnini, G. P. Chrousos, G. A. Fava, J. W. Findling, R. C. Gaillard, A. B. Grossman, B. Kola, A. Lacroix, T. Mancini, F. Mantero, J. Newell-price, L. K. Nieman, N. Sonino, M. L. Vance, A. Giustina and M. Boscaro, J. Clin. Endocrinol. Metab., 2003, 88, 5593–5602 CrossRef CAS PubMed.
- G. W. Strain, B. Zumoff, J. J. Strain, J. Levin and D. K. Fukushima, Metab., Clin. Exp., 1980, 29, 980–985 CrossRef CAS PubMed.
- R. Thieringer and A. Hermanowski-Vosatka, Expert Rev. Cardiovasc. Ther., 2005, 3, 911–924 CrossRef CAS PubMed.
- P. Björntorp, G. Holm and R. Rosmond, Diabetic Med., 1999, 16, 373–383 CrossRef.
- B. R. Walker, Eur. J. Endocrinol., 2007, 157, 545–559 CrossRef CAS PubMed.
- J. W. Tomlinson, E. A. Walker, I. J. Bujalska, N. Draper, G. G. Lavery, M. S. Cooper, M. Hewison and P. M. Stewart, Endocr. Rev., 2004, 25, 831–866 CrossRef CAS PubMed.
- C. R. W. Edwards, R. Benediktsson, R. S. Lindsay and J. R. Seckl, Steroids, 1996, 61, 263–269 CrossRef CAS PubMed.
- E. A. Walker and P. M. Stewart, Trends Endocrinol. Metab., 2003, 14, 334–339 CrossRef CAS PubMed.
- R. C. Wilson, S. Dave-Sharma, J. Wei, V. R. Obeyesekere, K. Li, P. Ferrari, Z. S. Krozowski, C. H. L. Shackleton, L. Bradlow, T. Wiens and M. I. New, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10200–10205 CrossRef CAS.
- Y. Kotelevtsev, R. W. Brown, S. Fleming, C. Kenyon, C. R. W. Edwards, J. R. Seckl and J. J. Mullins, J. Clin. Invest., 1999, 103, 683–689 CrossRef CAS PubMed.
- S. Diederich, C. Grossmann, B. Hanke, M. Quinkler, M. Herrmann, V. Bahr and W. Oelkers, Eur. J. Endocrinol., 2000, 142, 200–207 CrossRef CAS PubMed.
- R. C. Andrews, O. Rooyackers and B. R. Walker, J. Clin. Endocrinol. Metab., 2003, 88, 285–291 CrossRef CAS PubMed.
- S. P. Webster and T. D. Pallin, Expert Opin. Ther. Pat., 2007, 17, 1407–1422 CrossRef CAS.
- C. D. Boyle and T. J. Kowalski, Expert Opin. Ther. Pat., 2009, 19, 801–825 CrossRef CAS PubMed.
- S. A. Morgan and J. W. Tomlinson, Expert Opin. Invest. Drugs, 2010, 19, 1067–1076 CrossRef CAS PubMed.
- J. S. Scott, S. S. Bowker, J. deSchoolmeester, S. Gerhardt, D. Hargreaves, E. Kilgour, A. Lloyd, R. M. Mayers, W. McCoull, N. J. Newcombe, D. Ogg, M. J. Packer, A. Rees, J. Revill, P. Schofield, N. Selmi, J. G. Swales and P. R. O. Whittamore, J. Med. Chem., 2012, 55, 5951–5964 CrossRef CAS PubMed.
- J. S. Scott, J. deSchoolmeester, E. Kilgour, R. M. Mayers, M. J. Packer, D. Hargreaves, S. Gerhardt, D. J. Ogg, A. Rees, N. Selmi, A. Stocker, J. G. Swales and P. R. O. Whittamore, J. Med. Chem., 2012, 55, 10136–10147 CrossRef CAS PubMed.
- J. S. Scott, A. L. Gill, L. Godfrey, S. D. Groombridge, A. Rees, J. Revill, P. Schofield, P. Sörme, A. Stocker, J. G. Swales and P. R. O. Whittamore, Bioorg. Med. Chem. Lett., 2012, 22, 6756–6761 CrossRef CAS PubMed.
- J. S. Scott, P. Barton, S. N. L. Bennett, J. deSchoolmeester, L. Godfrey, E. Kilgour, R. M. Mayers, M. J. Packer, A. Rees, P. Schofield, N. Selmi, J. G. Swales and P. R. O. Whittamore, Med. Chem. Commun., 2012, 3, 1264–1269 RSC.
- F. W. Goldberg, A. G. Leach, J. S. Scott, W. L. Snelson, S. D. Groombridge, C. S. Donald, S. N. L. Bennett, C. Bodin, P. M. Gutierrez and A. C. Gyte, J. Med. Chem., 2012, 55, 10652–10661 CrossRef CAS PubMed.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed.
- X. Wu, K. Kavanagh, S. Svensson, B. Elleby, M. Hult, F. Von Delft, B. Marsden, H. Jornvall, L. Abrahmsen, U. Oppermann, Structure of 11-beta hydroxysteroid dehydrogenase in complex with NADP and carbenoxolone (PDB deposition 2bel).
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- Docking protocol: the 2bel X-ray structure was used as template for a docking study of compound 43. We used the Maestro protein preparation wizard from Schrodinger to prepare the binding site, after removing the CBX ligand but retaining the NADP+ co-factor; the wizard was allowed to flip HIS, GLN and ASN residues and to optimize the position of hydroxyl protons; protonations and tautomers were set to their preferred states for pH 7.0. The co-factor was modeled as NADP+, consistent with its state in the active form of the enzyme. Glide26 was used for the docking. A constraint was placed on the ligand docking pose, such that a hydrogen bond contact must be present with Tyr183, Ser170 or both. This was consistent with the observed binding mode of CBX and was a design criterion for the series. The best scoring pose is shown in Fig. 3, based on the GlideScore function.
- B. Alcaide, Y. Martin-Cantalejo, J. Perez-Castells, J. Rodriguez-Lopez, M. A. Sierra, A. Monge and V. Perez-Garcia, J. Org. Chem., 1992, 57, 5921–5931 CrossRef CAS.
- D. R. Kronenthal, C. Y. Han and M. K. Taylor, J. Org. Chem., 1982, 47, 2765–2768 CrossRef CAS.
- J. W. Clader, D. A. Burnett, M. A. Caplen, M. S. Domalski, S. Dugar, W. Vaccaro, R. Sher, M. E. Browne, H. Zhao, R. E. Burrier, B. Salisbury and H. R. Davis, J. Med. Chem., 1996, 39, 3684–3693 CrossRef CAS PubMed.
- J. Yin and S. L. Buchwald, Org. Lett., 2000, 2, 1101–1104 CrossRef CAS PubMed.
- J. Yin and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 6043–6048 CrossRef CAS PubMed.
- V. S. C. Yeh, R. Kurukulasuriya, S. Fung, K. Monzon, W. Chiou, J. Wang, D. Stolarik, H. Imade, R. Shapiro, V. Knourek-Segel, E. Bush, D. Wilcox, P. T. Nguyen, M. Brune, P. Jacobson and J. T. Link, Bioorg. Med. Chem. Lett., 2006, 16, 5555–5560 CrossRef CAS PubMed.
- Crystal structure of (4R)-40: molecular formula = C18H18BrNO2, formula weight = 360.23, crystal system = orthorhombic, space group = P212121, a = 5.9750(2) Å, b = 11.6900(3) Å, c = 23.7850(5) Å, V = 1661.33(8) Å3, T = 200 K, Z = 4, Dc = 1.440 mg m−3, (Mo-Kα) = 2.481 mm−1, 3815 reflections measured, 3788 independent reflections, 2938 observed reflections [I > 2.0σ(I)], R[F2 > 2σ(F2)] = 0.037, goodness of fit = 1.054. The absolute configuration around carbon C4 was determined to (R). Flack's x = 0.007(9).
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for the preparation of compounds; experimental protocols and X-ray coordinates for the crystal structure of 30 (CCDC: 882283) bound to 11β-HSD1; 11β-HSD1 & 2 assay protocols. See DOI: 10.1039/c3md00234a |
| This journal is © The Royal Society of Chemistry 2014 |