DOI:
10.1039/C3MD00259D
(Concise Article)
Med. Chem. Commun., 2014,
5, 64-67
Matched molecular pairs derived by retrosynthetic fragmentation
Received
10th September 2013
, Accepted 27th October 2013
First published on 22nd November 2013
Abstract
Matched molecular pairs (MMPs) are defined as pairs of compounds that only differ by a chemical change at a single site. MMPs have become popular in medicinal chemistry to support lead optimization, absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis, and other applications. Thus far, MMPs have been algorithmically defined and not on the basis of reaction information. This often limits the chemical interpretability and practical utility of MMPs. Therefore, we introduce synthetically accessible MMPs that are automatically generated by applying reaction rules following the retrosynthetic combinatorial analysis procedure (RECAP). A library of more than 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RECAP-MMPs was generated from public domain compounds active against 435 different targets exclusively utilizing high-confidence activity data. This library is made freely available for use in medicinal chemistry.
000 RECAP-MMPs was generated from public domain compounds active against 435 different targets exclusively utilizing high-confidence activity data. This library is made freely available for use in medicinal chemistry.
Introduction
MMPs have been introduced as pairs of compounds that only differ by a chemical change at a single site,1,2 a so-called chemical transformation.3 They are mostly generated by fragmentation-3 or maximum common substructure-based1,4 algorithms. In recent years, MMPs have become popular tools in medicinal chemistry for a variety of applications5,6 including structure–activity relationship (SAR)7,8 and activity profile9 analysis, lead optimization,10,11 ADMET analysis,11–13 or the exploration of bioisosterism.14 A major reason for the attractiveness of the MMP concept in medicinal chemistry is that chemical transformations such as R-group replacements or core structure modifications can directly be associated with defined property changes (e.g., activity, solubility, or stability) within the context of actual compounds,5,6 hence providing a basis for chemically intuitive analysis. By contrast, a shortcoming of current MMPs is that participating compounds are usually not related by chemical reactions. Hence, chemical transformations constituting MMPs are often not chemically interpretable and accessible, which limits their practical utility in medicinal chemistry, for example, when attempting to convert compounds into MMP partners with more favorable properties. Therefore, we introduce herein a new category of MMPs that are generated on the basis of retrosynthetic fragmentation employing the well-known RECAP reaction rules.15 Accordingly, these second-generation MMPs are termed RECAP-MMPs. The chemical transformation relating compounds forming RECAP-MMPs to each other results from a specific reaction. We show that RECAP-MMPs are a subset of original MMPs, with very few exceptions, and generate a large library of RECAP-MMPs for 435 different compound classes exclusively utilizing high-confidence activity data. This library is made available to the scientific community without restrictions.
Methods
All activity classes from ChEMBL16 (release 15) were collected that contained at least 5 compounds with available (assay-independent) Ki values. Equilibrium constants were exclusively used to ensure high confidence of activity data.17 A total of 435 target-specific datasets comprising 40650 unique active compounds were obtained. Compounds with multiple Ki values for the same target were only considered if all values fell within one order of magnitude. If this confidence criterion was met, the average value of independent measurements was used as the final potency annotation.
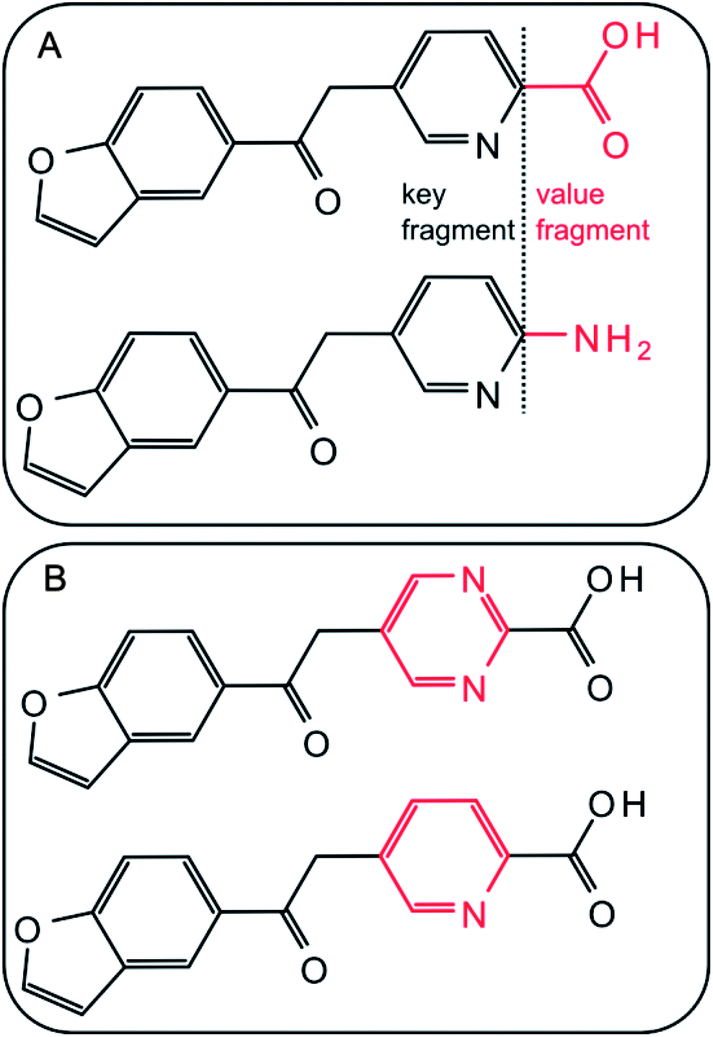
From each activity class, MMPs were systematically generated using an in-house implementation of the Hussain and Rea algorithm.3 Each compound was subjected to systematic single-, double-, and triple-cut fragmentation of all exocyclic single bonds between non-hydrogen atoms. During fragmentation, connectivity information was retained. Core structures and variable substituents resulting from fragmentation were stored in an index table as key and value fragments, respectively. Each pair of compounds having the same key and different value fragments formed an MMP. The size of a transformation was limited to a maximum of 13 non-hydrogen atoms and the size difference between exchanged fragments to 8 non-hydrogen atoms. In addition, keys were required to have at least twice the size of value fragments for each transformation. Application of these criteria yielded transformation size-restricted MMPs18 in which value fragments (substituents) were generally limited to relatively small substructures.18Fig. 1 shows exemplary MMPs. In the following, MMPs generated by systematic fragmentation are referred to as “standard MMPs”.
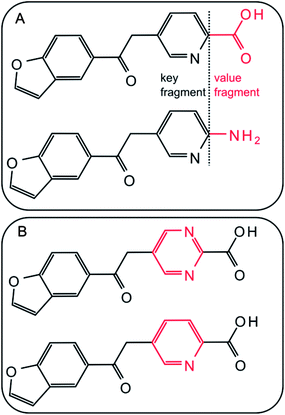 |
| | Fig. 1 MMPs. Two exemplary MMPs are shown. Exchanged fragments are highlighted in red. | |
For the generation of RECAP-MMPs, a RECAP rule-based fragmentation scheme was applied.15,19 Accordingly, bonds were only cut on the basis of retrosynthetic rules. In addition, a transformation was only accepted if the two exchanged fragments were generated by the same reaction. Transformation size restrictions were applied as specified above. Original RECAP rules were slightly modified for single bond fragmentation. The urea and thiourea rules were not utilized because they affect multiple bonds. In addition, quaternary amines were not distinguished from non-charged amines. All applied retrosynthetic rules are reported in Fig. 2. RECAP-MMPs were systematically generated using in-house Java code and the Open Eye Toolkit.20 For non-commercial applications source code is available upon request. Statistical analyses were carried out using R.21
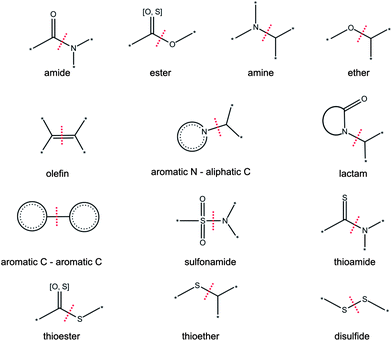 |
| | Fig. 2 RECAP rules. Thirteen retrosynthetic fragmentation rules are illustrated that were applied to generate RECAP-MMPs. The red line indicates the bond that is cut according to each reaction. In the case of amines, ethers, and thioethers the heteroatom should not be a part of any other functional group and not form exclusive bonds to multiple aromatic carbons. | |
Results and discussion
Standard versus RECAP-MMPs
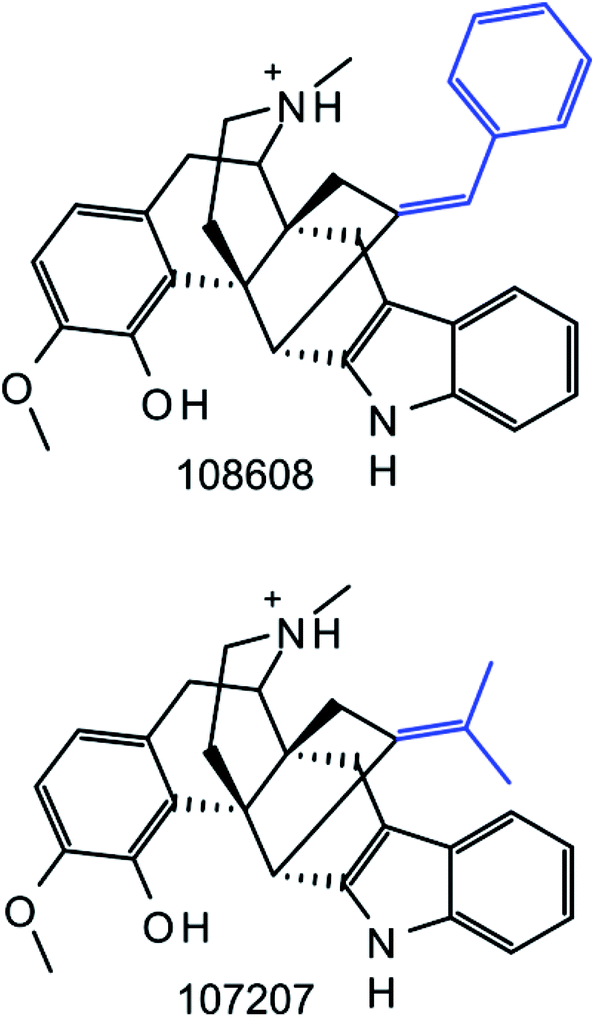
As reported in Table 1, we obtained 435 Ki-based datasets from ChEMBL with 40650 compounds. From these compounds, we systematically generated standard MMPs and RECAP-MMPs. A total of 223671 unique standard and 92743 unique RECAP-MMPs were obtained. Many MMPs originated from multiple datasets. For 86 datasets, no RECAP-MMP was obtained, due to small compound numbers (on average, these 86 datasets contained only 10.6 compounds). The application of a confined set of retrosynthetic rules yielded fewer MMPs than systematic fragmentation, as expected. Surprisingly, however, nearly half as many RECAP-MMPs were obtained. Moreover, we found that essentially all RECAP-MMPs were reproduced by systematic fragmentation. Only 11 instances of RECAP-MMPs were detected that were not obtained by systematic fragmentation. An example is shown in Fig. 3. In this pair of compounds, qualifying exocyclic single bonds were absent. Hence, systematic fragmentation did not yield an MMP. Because RECAP-MMPs were a subset of standard MMPs, with only very few exceptions, 42% of all standard MMPs were conserved when reaction-based fragmentation was applied, a larger proportion than anticipated.
Table 1 Datasets and MMP statisticsa
|
Statistics are reported for compound datasets, standard and RECAP-MMPs, and MMP cliffs.
|
| Datasets |
435 |
| Compounds |
40650 |
| Standard MMPs |
223671 |
| RECAP-MMPs |
92734 |
| Standard MMP cliffs |
13261 |
| RECAP-MMP cliffs |
4406 |
| Standard MMP cliff frequency |
5.9% |
| RECAP-MMP cliff frequency |
4.8% |
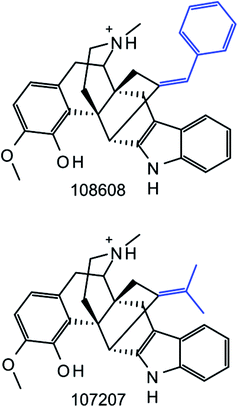 |
| | Fig. 3 Unique RECAP-MMP. Two compounds forming a RECAP-MMP are shown that was not generated by systematic fragmentation. RECAP-MMP value fragments are highlighted in blue. Compound ChEMBL IDs are given. | |
Chemical transformations
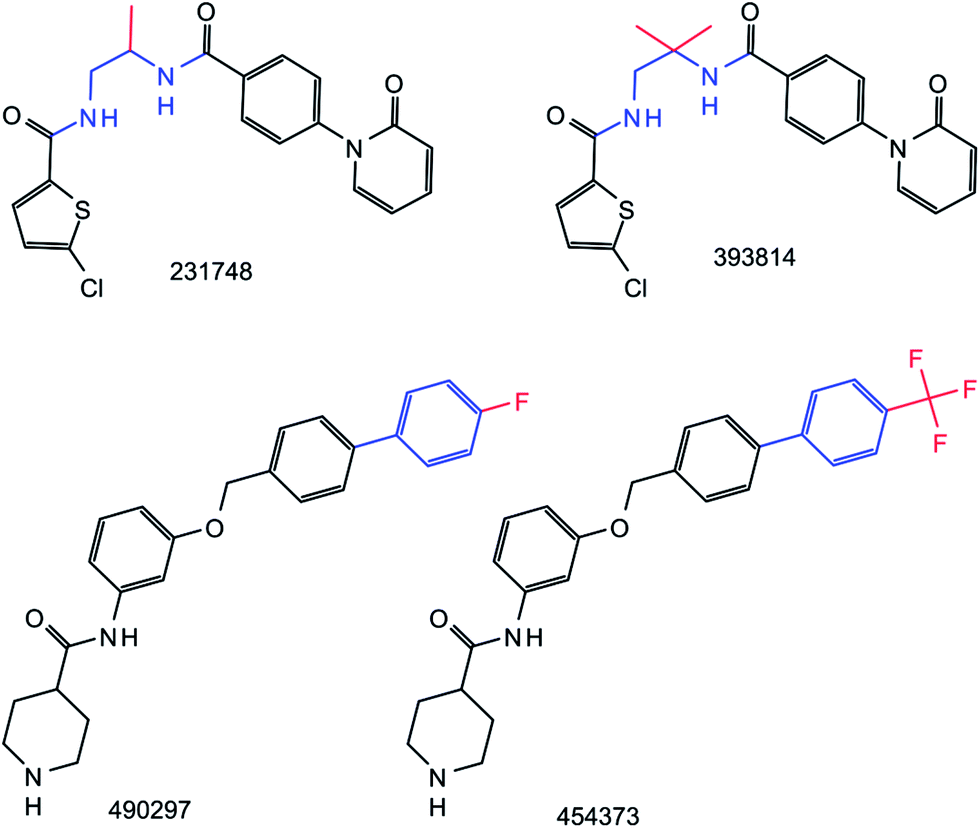
However, despite the high degree of MMP conservation, we generally observed that standard and RECAP-based transformations differed for a qualifying compound pair. Thus, although the same MMP was obtained on the basis of systematic or retrosynthetic fragmentation, the corresponding transformations were distinct. Examples are provided in Fig. 4. In general, RECAP-based transformations tended to be larger than standard transformations, on average by 3–5 non-hydrogen atoms per MMP depending on the dataset. From RECAP transformations, reagents could often be deduced for the given reaction. By contrast, exchanges of small fragments in standard MMPs were typically not interpretable in reaction terms. Thus, transformation information clearly distinguished RECAP-MMPs from standard MMPs.
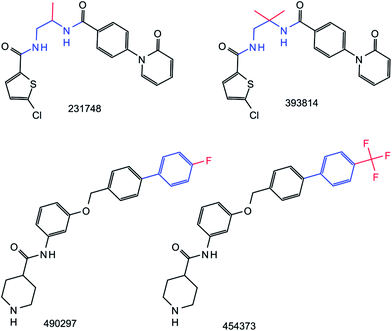 |
| | Fig. 4 Comparison of standard and RECAP-MMPs. Two pairs of compounds forming standard and RECAP-MMPs are shown. ChEMBL IDs are provided. Transformations in standard MMPs are highlighted in red and transformations in RECAP-MMPs in red and blue. The comparison illustrates that RECAP-based substructures representing a transformation were typically larger than substructures produced by systematic fragmentation. The RECAP-MMPs at the top and bottom were obtained through cuts of two amide bonds and an aromatic carbon–aromatic carbon bond, respectively. | |
Reaction distribution
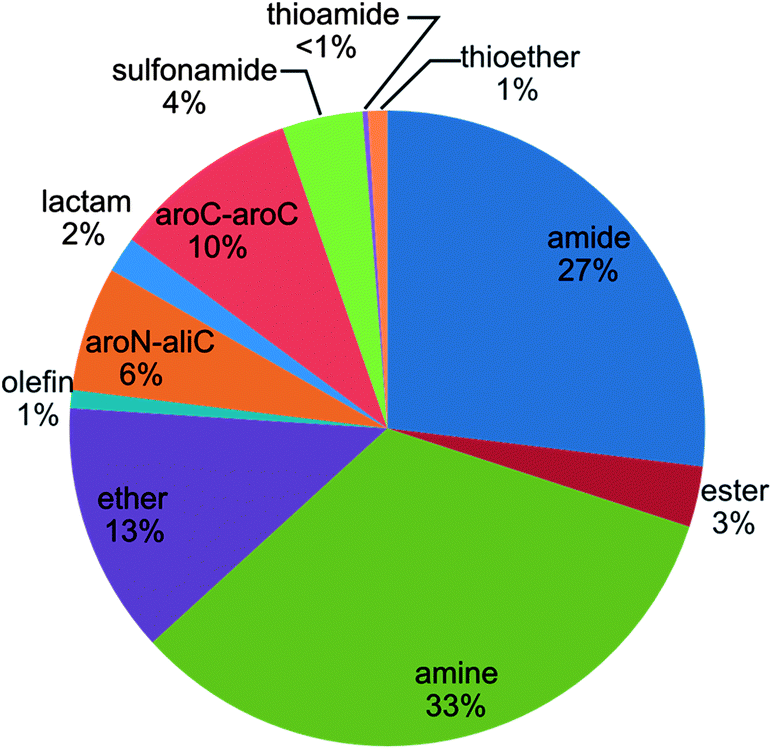
Fig. 5 reports the fractions of RECAP-MMPs that were defined by specific retrosynthetic rules according to Fig. 2. Interestingly, no instances of RECAP-MMPs were detected that resulted from fragmentation of thioester and disulfide bonds, and thioamide bond cleavage accounted for less than 1% of all RECAP-MMPs. By contrast, amine and amide chemistry dominated the distribution of RECAP-MMPs, with 33% and 27%, respectively, followed by ethers (13%) and aromatic carbon–aromatic carbon bonds (10%), hence reflecting the current compound portfolio in medicinal chemistry.22 In addition, between 6% and 1% of RECAP-MMPs resulted from fragmentation of aromatic nitrogen–aliphatic carbon bonds, esters, lactams and olefins.
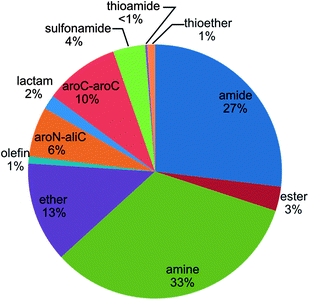 |
| | Fig. 5 Reaction frequency. The graph reports the proportions of RECAP-MMPs that were obtained on the basis of different retrosynthetic rules. | |
MMP cliffs
As an indicator of the SAR information content, we also determined the fraction of activity cliffs that were captured by standard and RECAP-MMPs, so-called MMP cliffs.18 Activity cliffs are generally defined as pairs of structurally similar or analogous compounds with a large difference in potency.23 Therefore, all MMPs were determined in which the two compounds displayed a potency difference (Ki values) of at least two orders of magnitude.18,23 As reported in Table 1, the frequency of occurrence of standard MMP and RECAP-MMP cliffs was 5.9% and 4.8%, respectively. Thus, systematic and retrosynthetic fragmentation captured activity cliffs with similar frequency.
RECAP-MMP library
The 92734 unique RECAP-MMPs identified in our study are made freely available as a machine-readable library organized on the basis of target sets (available at http://www.limes.uni-bonn.de/forschung/abteilungen/Bajorath/labwebsite/downloads). Given the target set organization, individual RECAP-MMPs might occur multiple times in different sets. This ensures that a complete set of RECAP-MMPs is available for each compound class. Furthermore, in the library, standard and retrosynthetic transformations are provided for each RECAP-MMP that was reproduced by systematic fragmentation to enable direct comparison of these transformations. Moreover, all RECAP-MMP cliffs are specified.
A randomly chosen sample of 50 RECAP-MMPs was traced back to compounds in original publications (via ChEMBL compound IDs) and it was examined whether the synthesis of these compounds was reported in the original publications. For more than 75% of these RECAP-MMPs, compounds were found to be synthesized by corresponding routes (in a number of original references, no compound synthesis was reported). Hence, in many cases, there was a direct link between RECAP-MMPs and synthetic routes of compounds from which these RECAP-MMPs originated.
Conclusions
Herein we have introduced second-generation MMPs defined on the basis of retrosynthetic rules and compared these RECAP-MMPs with standard MMPs. In RECAP-MMPs, chemical transformations are reaction-based and interpretable. Given the current popularity of the MMP concept, it is hoped that the library of RECAP-MMPs we provide will serve as a knowledge base to further improve the utility of matched molecular pairs in medicinal chemistry.
References
- R. P. Sheridan, J. Chem. Inf. Comput. Sci., 2002, 42, 103–108 CrossRef CAS PubMed.
-
P. W. Kenny and J. Sadowski, in Chemoinformatics in Drug Discovery, ed. T. I. Oprea, Wiley-VCH, Weinheim, Germany, 2004, pp. 271–285 Search PubMed.
- J. Hussain and C. Rea, J. Chem. Inf. Model., 2010, 50, 339–348 CrossRef CAS PubMed.
- D. J. Warner, E. J. Griffen and S. A. St-Gallay, J. Chem. Inf. Model., 2010, 50, 1350–1357 CrossRef CAS PubMed.
- E. Griffen, A. G. Leach, G. R. Robb and D. J. Warner, J. Med. Chem., 2001, 54, 7739–7750 CrossRef PubMed.
- A. M. Wassermann, D. Dimova, P. Iyer and J. Bajorath, Drug Dev. Res., 2012, 73, 518–527 CrossRef CAS.
- R. P. Sheridan, P. Hunt and J. C. Culberson, J. Chem. Inf. Model., 2006, 46, 180–192 CrossRef CAS PubMed.
- J. E. J. Mills, A. D. Brown, T. Ryckmans, D. C. Miller, S. E. Skerratt, C. M. Barker and M. E. Bunnage, Med. Chem. Commun., 2011, 3, 174–178 RSC.
- Y. Hu and J. Bajorath, ACS Med. Chem. Lett., 2011, 2, 523–527 CrossRef CAS.
- P. J. Hajduk and D. R. Sauer, J. Med. Chem., 2008, 51, 553–564 CrossRef CAS PubMed.
- G. Papadatos, M. Alkarouri, V. J. Gillet, P. Willett, V. Kadirkamanathan, C. N. Luscombe, G. Bravi, N. J. Richmond, S. D. Pickett, J. Hussain, J. M. Pritchard, A. W. Cooper and S. J. Macdonald, J. Chem. Inf. Model., 2010, 50, 1872–1876 CrossRef CAS PubMed.
- A. G. Leach, H. D. Jones, D. A. Cosgrove, P. W. Kenny, L. Ruston, P. MacFaul, J. M. Wood, N. Colclough and B. Law, J. Med. Chem., 2006, 46, 6672–6682 CrossRef PubMed.
- M. L. Lewis and L. Cuchurall-Sanchez, J. Comput.-Aided Mol. Des., 2009, 23, 97–103 CrossRef CAS PubMed.
- A. M. Wassermann and J. Bajorath, Future Med. Chem., 2011, 3, 425–436 CrossRef CAS PubMed.
- X. Q. Lewell, D. B. Judd, S. P. Watson and M. M. Hann, J. Chem. Inf. Comput. Sci., 1998, 38, 511–522 CrossRef CAS.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed.
- Y. Hu and J. Bajorath, J. Chem. Inf. Model., 2012, 52, 2550–2558 CrossRef CAS PubMed.
- X. Hu, Y. Hu, M. Vogt, D. Stumpfe and J. Bajorath, J. Chem. Inf. Model., 2012, 52, 1138–1145 CrossRef CAS PubMed.
- E. Lounkine and J. Bajorath, J. Chem. Inf. Model., 2009, 49, 162–168 CrossRef CAS PubMed.
- OpenEye Scientific Software Inc., Santa Fe, NM.
- R Foundation for Statistical Computing, Vienna, Austria.
- W. P. Walters, J. Green, J. R. Weiss and M. A. Murcko, J. Med. Chem., 2011, 54, 6405–6416 CrossRef CAS PubMed.
- D. Stumpfe and J. Bajorath, J. Med. Chem., 2012, 55, 2932–2942 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.