Differential activation of soluble guanylate cyclase by a series of aryl disulfanyl dinitrate esters
Adrian C.
Nicolescu
*a and
Gregory R. J.
Thatcher
b
aFaculty of Health Sciences, Queen’s University, Kingston, Ontario, K76 3N6, Canada. E-mail: nicolesc@queensu.ca
bDepartment of Medicinal Chemistry and Pharmacognosy, University of Illinois College of Pharmacy, University of Illinois at Chicago, 833 S. Wood Street, Chicago, Illinois 60612, USA
First published on 22nd October 2013
Abstract
Classic organic nitrates, such as nitroglycerin (GTN), are important vasodilators classed as NO donors, since biological activity is assumed to result entirely from bioactivation to NO and activation of soluble guanylate cyclase (sGC). Para-substituted aryl disulfanyl nitrate esters (SS-nitrates), designed based on a sulfhydryl-dependent mechanism of NO release, were investigated for their sGC activation. To obtain further insights into their interaction with hemoproteins, SS-nitrates were reacted with oxyhemoglobin (O2-Hb). SS-nitrates activated sGC with higher efficacy than GTN in the presence of cysteine. Some SS-nitrates, unlike GTN, activated sGC in the absence of cysteine. Linear correlations between the σp Hammett parameters and sGC activation efficacies or rates of O2-Hb oxidation by SS-nitrates in the presence of cysteine were found. Our results validate the design of SS-nitrates to circumvent the need for cysteine-mediated bioactivation and hint at NO-dependent and NO-independent mechanisms of sGC activation.
Introduction
Organic nitrates, such as nitroglycerin (GTN), have been used in cardiovascular therapy as potent vasodilators for over 130 years. Classical nitrate therapeutics have inconveniences, including peripheral hypotension at high doses and the development of tolerance, which are argued to limit their potential benefits in other pathological conditions. Organic nitrates are postulated to generate NO in vivo.1–3 NO, an important cell messenger physiologically produced by nitric oxide synthase, exerts its multiple vascular, neuronal and immunological functions by directly activating heme-containing soluble guanylate cyclase (sGC), which catalyzes the conversion of guanosine triphosphate to the secondary cell messenger cyclic guanosine-3′,5′-monophosphate (cGMP).4–6 Formation of NO from GTN has been detected in intact arteries,7 cultured smooth muscle cells,8 arterial homogenates,3 or from the reaction of GTN with thiols in blood plasma.9 However, the yield and rate of NO release from GTN under aerobic conditions are very low6 and cannot account for GTN's nanomolar vasodilatory potency.6,10 Thus, classical organic nitrates have been proposed to act as NO mimetics rather than NO donors, since they produce biological activity associated with NO without generating significant amounts of NO.6,10,11 Direct activation of sGC by nitrates and bioactivity resulting from the release of NO2−, the primary product of nitrate biotransformation,12,13 are two possible explanations.Recently, the potential for the expanded therapeutic use of novel organic nitrates in pathologies of the respiratory, gastrointestinal, and the central nervous systems has been demonstrated.10,14,15 Several clinical trials on a family of nitrates termed cyclooxygenase inhibitory NO donors (CINODs, or NO-NSAIDS), as well as animal studies on SS-nitrates illustrate the potential for use in cardiovascular therapy and beyond.16–19 The use in neurodegenerative disorders, such as Alzheimer's and Parkinson's disease, is also of significant interest due to organic nitrates ability of acting as NO-donors and/or NO-mimetics,19,20 and the cytoprotective actions of NO in counteracting the deleterious effects of oxidative stress to cells.21
Sulfhydryl-dependent and heme-dependent pathways have been proposed in the bioactivation of organic nitrates.3,10 Applying the principle of the sulfhydryl-dependent mechanism, we reported the synthesis of 2,3-dinitrooxy-propane-1-disulfanylbenzene derivatives para-substituted with electron withdrawing or donating substituents (SS-nitrates) (Fig. 1), and showed that SS-nitrates are capable of releasing significant NO amounts in non-enzymatic reactions with thiols in simple buffered solutions.22 In the present work we report for the first time that in the presence of cysteine, these SS-nitrates activated sGC from rat aorta homogenates with efficacy values (Emax) that were superior to GTN and approaching that of the genuine NO-donor spermine NONOate (SPE/NO). A reasonable linear correlation was found between the Hammet parameter σp of the para-substituents and the efficacies of sGC activation by SS-nitrates in the presence of cysteine. Remarkably and in contrast to GTN, some SS-nitrates activated sGC in the absence of cysteine. The reaction of SS-nitrates with oxyhemoglobin (O2-Hb), often quoted as a model for the interaction of nitrates with hemoproteins, has also been investigated to gain further possible mechanistic insights. Our findings further show that novel nitrates can be engineered to both release controlled amounts of NO and activate sGC by different mechanisms than classical organic nitrates such as GTN.
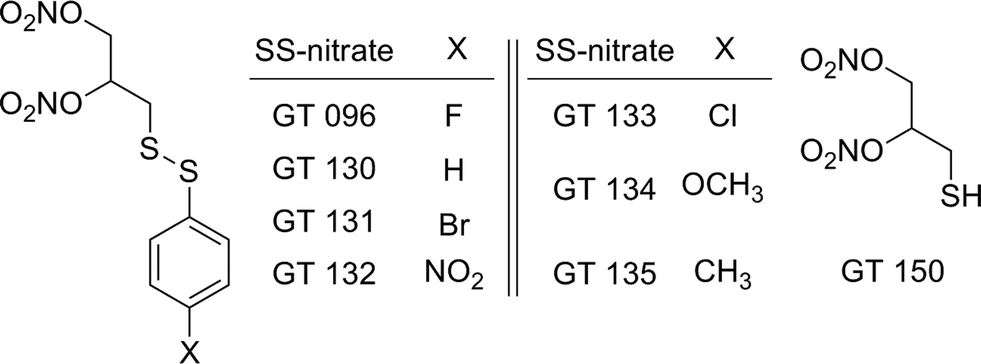 | ||
| Fig. 1 Chemical structures of the SS-nitrates and β-mercaptodinitrate GT 150. | ||
Results
sGC activation by SS-nitrates
In the presence of cysteine, all SS-nitrates activated sGC from rat aorta homogenates in a concentration-dependent manner (Fig. 2A and B), with up to a three-fold increased in the efficacy (maximum activity, Emax) compared to GTN (Table 1). The potency (EC50) of sGC activation by SS-nitrates was comparable to that of GTN (2 × 10−4 M). No activity above the basal levels was observed for GTN in the absence of cysteine in the concentration range studied. GT 135 and GT 131 activated sGC beyond the enzyme basal activity in the absence of cysteine (Fig. 2C), with EC50 and efficacy of 1.7 × 10−4 M and 114 ± 17%, and 4 × 10−5 M and 60 ± 4%, respectively. GT 150 fully activated sGC both in the presence and absence of cysteine, with an efficacy comparable to that of the genuine NO donor SPE/NO and up to almost four-fold that of GTN (Fig. 2D). GT 150 was more potent than GTN in activating sGC both in the presence (EC50 = 7.1 × 10−5 M) and absence (EC50 = 4.3 × 10−5 M) of cysteine, but less potent than the relatively slow NO donor SPE/NO (EC50 = 1.2 × 10−5). Emax values for sGC activation by SS-nitrates in the presence of cysteine plotted against the σp Hammett parameter for the para-substituents yielded a significant (p = 0.0033) linear correlation for the semilogarithmic plot (Fig. 3A). The correlation coefficient (R2 = 0.846) was not surprising considering the variability associated with the complexity of the reaction mixture.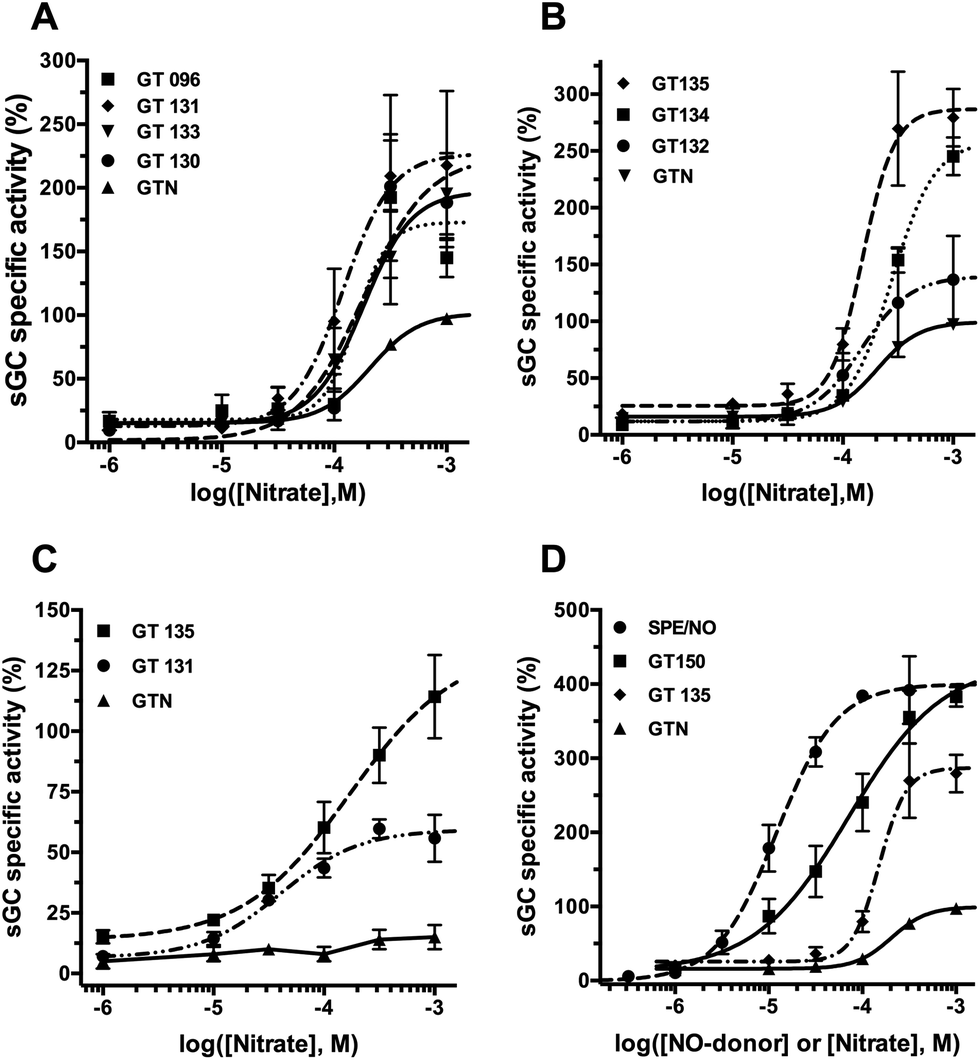 | ||
| Fig. 2 Concentration-response curves for sGC activation by SS-nitrates and GTN in the presence (A) and (B), or absence (C) of 1 mM cysteine. (D) Comparison between sGC activation by SPE/NO, GT 150, GT 135 and GTN. Enzyme activity was assayed after 10 minutes incubation at 37 °C. Data points represent means ± SEM of 3 to 5 different enzyme preparations. | ||
| Nitrate | σ p | E max a (%) | V 0 b (au/s) × 105 |
|---|---|---|---|
| a Data represent mean ± SD of 3 to 5 independent experiments. b Data represent mean ± SD of triplicates. c Significant difference from GTN, P < 0.05, one-way ANOVA with Tukey's post hoc test. | |||
| GT 132 | 0.72 | 116 ± 48c | ND |
| GT 131 | 0.23 | 218 ± 58c | 1.7 ± 0.02c |
| GT 133 | 0.22 | 195 ± 32c | 1.7 ± 0.06c |
| GT 096 | 0.06 | 192 ± 50c | 1.2 ± 0.08c |
| GT 130 | 0.00 | 201 ± 72c | 1.8 ± 0.10c |
| GT 135 | −0.17 | 279 ± 25c | 3.4 ± 0.01c |
| GT 134 | −0.28 | 245 ± 17c | 6.3 ± 0.12c |
| GTN | NA | 97 ± 3 | 3.7 ± 0.05 |
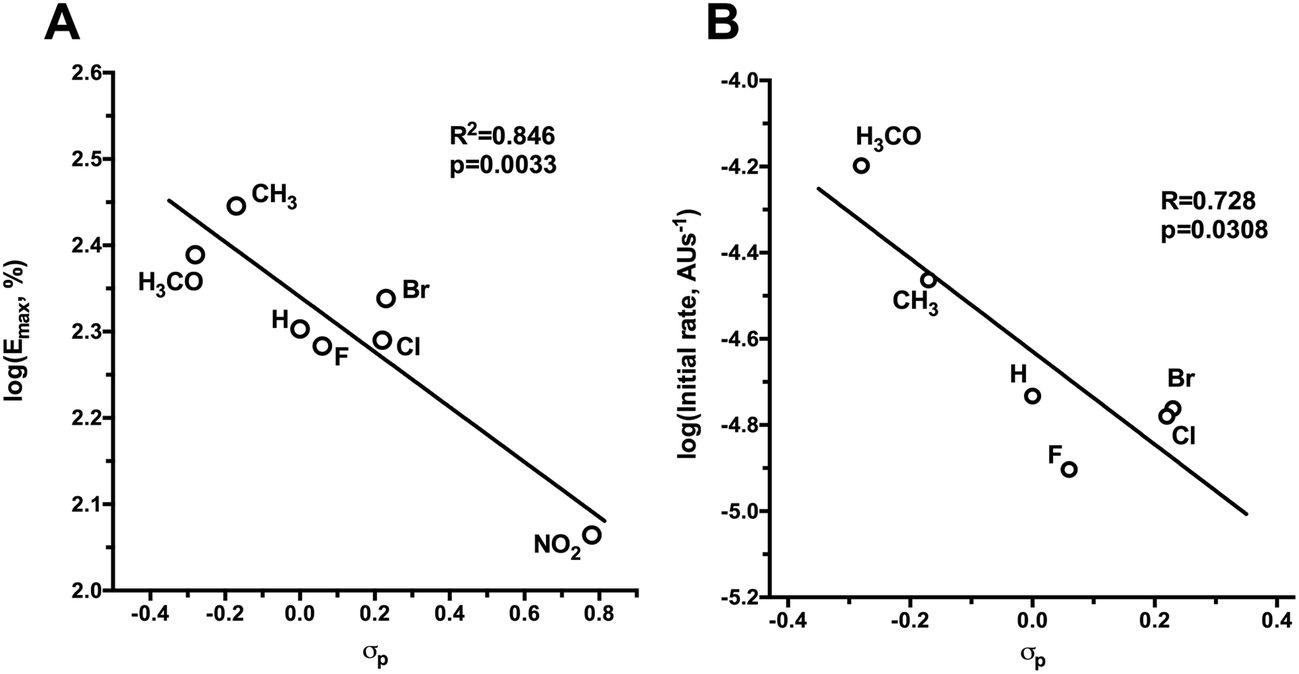 | ||
| Fig. 3 Semilogarithmic plots of sGC activation efficacy (A) and initial rates of O2-Hb oxidation (B) by SS-nitrates in the presence of cysteine versus the σp Hammett parameter of the para-substituents. | ||
O2-Hb oxidation by SS-nitrates
All nitrates oxidized O2-Hb in the presence cysteine. In the absence of cysteine, the initial rates of O2-Hb oxidation by SS-nitrates were not significantly different from that of GTN (data not shown). An almost two-fold increase over the initial rate of GTN was observed for GT 134, whereas the other SS-nitrates showed smaller rates of O2-Hb oxidation compared to GTN (Table 1). The O2-Hb oxidation rate for GT 132 could not be determined because of the intense yellow color that rapidly developed during the reaction due to the formation of p-nitrothiophenol. A significant linear correlation (R2 = 0.728, p = 0.0308) was obtained between the initial rates of O2-Hb oxidation and the Hammett parameter σp of the para-substituents for the semilogarithmic plot (Fig. 3B).Discussion
Classical organic nitrates, such as GTN, activate sGC and increase cGMP formation in both intact tissue and tissue homogenates.3,23,24 In contrast to intact tissue, organic nitrates activate sGC in tissue homogenates only in the presence of specific thiols such as cysteine, thiosalicylic acid and N-acetyl cysteine, and not other thiols such as dithiothreitol or glutathione.6,24 Nevertheless, in simple, non-enzymatic systems GTN reacts slowly with cysteine to release small amounts of NO, detectable only with the highly sensitive chemi-luminescence analyzer, which is difficult to correlate with the rapid onset of its biological effect at very low pharmacological concentrations.6,25Mechanism-based pathways leading to an sGC activator (e.g., NO) have been proposed for the biotransformation of organic nitrates: (i) sulfhydryl-dependent, and (ii) heme-dependent pathways.3,26 The biotransformation of nitrates to NO is a 3e− redox process, possibly involving organic nitrites and inorganic nitrite (NO2−) as intermediates,10,24,27 and not inorganic nitrate.10,22 Organic nitrites are vasodilators that can spontaneously generate NO in neutral aqueous solution and rapidly react with thiols to yield nitrosothiols.10,28 Interestingly, NO2− has recently emerged as a biological pool of NO,29 since its biotransformation to NO is accomplished by reductase along the physiological O2 gradient, potentially relevant in hypoxic states.30 The possibility that nitrates also act as NO2−-donors that deliver NO2− intracellularly to specific locations has been suggested.10
The design of SS-nitrates relies on the sulfhydryl-dependent biotransformation hypothesis and on the concept that the presence of both thiol and nitrate groups in the same molecule would greatly increase the rate of their interaction and release of NO.22 We showed previously that four SS-nitrates and the β-mercaptodinitrate GT 150, in neutral, air-equilibrated aqueous solution, release high micromolar NO concentrations, whereas GTN releases low nanomolar NO concentrations,6,22 indicating that SS-nitrates represent a distinct, novel class of NO donor organic nitrates. The potencies of genuine NO donors for sGC activation correlated very well with their rates of NO release.6 However, the potencies for sGC activation by SS-nitrates were comparable, possibly reflecting comparable rates of NO released by SS-nitrates under the reaction conditions. The observed correlation between the σp Hammet parameter of the para-substituents and the efficacies of sGC activation by SS-nitrates (Fig. 3) can be explained in terms of GT 150 formed during a thiol-disulfide reaction.22,31,32 The para-substituents will affect the reaction rate and nature of the products (Fig. 4). Electron-withdrawing groups with high σp (e.g., NO2) would stabilize a thiophenolate intermediate and react faster with cysteine to release the para-nitrothiophenolate (a better leaving group) and an asymmetric alkyl disulfanyl dinitrate (the “non-productive” pathway in Fig. 4). In contrast, electron-donating groups with low σp (e.g., OCH3 and CH3) would not favor a thiophenolate intermediate and react slower with cysteine to release GT 150 (the NO “productive” pathway in Fig. 4). Notably, the reaction of aliphatic thiols with asymmetrical disulfides such as R–SS–p-C6H4NO2 occurs rapidly (e.g., 0.17–9.51 × 104 M−1 s−1) with the release of the para-nitrothiophenolate.33
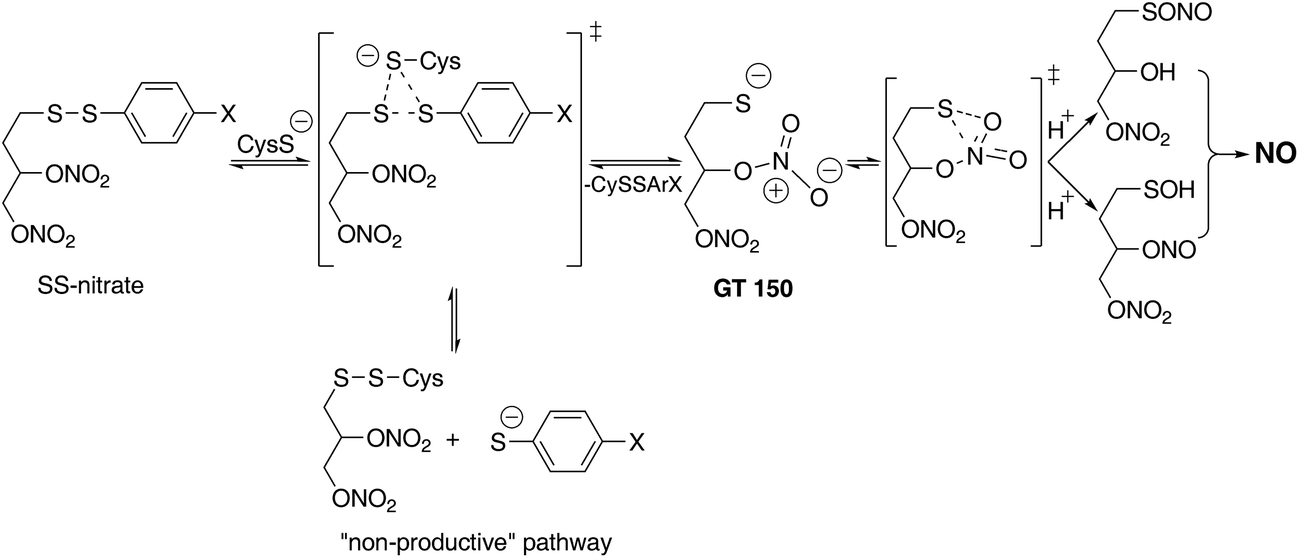 | ||
| Fig. 4 Proposed mechanism for NO release by SS-nitrates. | ||
The proposed mechanism of NO release from SS-nitrates (Fig. 4) involves an initial nucleophilic attack of cysteine at the aryl sulfur atom and the formation of GT 150.22 The nucleophilic thiolate in GT 150 would then attack the proximal nitrate group and generate, via a favorable six-member ring transition state, a sulfenate and a nitrite ester.22 A sulfenyl nitrite (RSONO) could also form, since the migration of oxygen from nitrogen to sulfur has been reported, e.g., in the reaction of glutathione with nitrosoarenes to yield sulfinamides (RSONHR').34 The nitrite intermediate can spontaneously release NO in aqueous solutions,28 or rapidly transfer the NO group to thiols and generate nitrosothiols, which in the presence of trace transition metal ions quantitatively release NO.35,36
GTN does not activate sGC in the absence of cysteine, and even acts as an antagonist of the sGC activation by NO,6 an effect possibly due to GTN's capability to oxidize the ferrous-heme of sGC.37 The remarkable sGC activation by some SS-nitrates in the absence of added cysteine would imply an interaction with sGC cysteine residues. Indeed, sGC has 20 and 14 free cysteine residues in the α1- and β1-subunit, respectively.38–40 Thus, an NO-independent activation of sGC by SS-nitrates, occurring either by a direct interaction with the regulatory ferrous-heme group of the enzyme or by binding to an allosteric site, cannot be completely dismissed. Accordingly, SS-nitrates would behave as partial agonists that induce conformational changes of the enzyme, resulting in submaximal activation.
Despite inherent inconveniences, the O2-Hb assay could offer insights into the interaction of organic nitrates with hemoproteins. The O2-Hb oxidation assay has been widely used to quantify NO release by NO donors and nitrate esters,2,41 and a linear dependence of O2-Hb oxidation rates on the concentration of various organic nitrates was reported.42 Artz and Thatcher showed, however, that such linear correlations occur only for compounds that rapidly release NO such as the very reactive NONOates (e.g., DEA/NO), but not for nitrosothiols, alkyl nitrites or more stable NONOates (e.g., SPE/NO or DETA/NO).43 Although significantly different among each other, the rates of O2-Hb oxidation by SS-nitrates in the presence of cysteine varied over a small range, largely agreeing with the reported rates of NO release by SS-nitrates in the presence of cysteine.22 The observed linear correlation between the O2-Hb oxidation rates by SS-nitrates in the presence of cysteine and the σp Hammett parameter of the para-substituents in the SS-nitrates can be understood in terms of GT 150 formation and subsequent release of NO.
The differential sGC activation by SS-nitrates is important since the clinical effectiveness of therapeutics does not depend on EC50, but rather on efficacy and ability to reach the site of action.44 Our results further posit the possibility of different substituents to partition between two competing mechanisms for the reaction of SS-nitrates with thiols. One immediate implication is that modest modifications in the structure of SS-nitrates can result in significantly different activations of sGC. Thus, the putative interactions of novel nitrate esters with different pathological pathways become of real therapeutic interest for diseases in which NO signaling is relevant (e.g., Alzheimer's and Parkinson's diseases). The superior efficacies in sGC activation by SS-nitrates compared to GTN may prove beneficial in neurodegeneration, since it has been demonstrated that increased levels of cGMP are neuroprotective.45 Indeed, the related novel nitrates GT 715 and GT 061 were more potent and efficacious in activating hippocampal sGC with minimal systemic vascular effects compared to GTN,19 and were neuroprotective in various animal models of neurological disorders.16,19,20 Thus, novel organic nitrates that dissociate the neuromodulatory from cardiovascular properties of classic nitrates can be developed. Our results also reveal the possibility that SS-nitrates are novel chemical entities with an action mechanism in their own right, which may also differentiate them from GTN's undesired side effects, including tolerance. Considering the biological significance of NO and the therapeutic benefits of organic nitrates, the properties exhibited by SS-nitrates can represent a platform for designing new therapeutics that extend their applications beyond those of classical organic nitrates.
Experimental procedures
Materials
The SS-nitrates were obtained from GB Therapeutics (Kingston, ON) and were ≥99% pure by 1H-NMR spectroscopy and combustion analysis. The synthesis of SS-nitrates has been fully described.22 GTN (Tridil®, Du Pont Pharmaceuticals, Scarborough, ON) was used as an injectable solution (5 mg ml−1). Stock solutions of SS-nitrates were prepared in DMSO. Dilutions of stock solutions were prepared with osmosis purified distilled H2O and the organic solvent in the final reaction mixture was less than 1%. All other chemicals were obtained from Sigma-Aldrich Chemicals (Milwaukee, WI).Activation of soluble guanylate cyclase
Tissue homogenates (10%, w/v) from aorta of adult male Sprague-Dawley rats (200–250 g) were centrifuged at 105![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 1 h at 4 °C, and the supernatant containing sGC used for the enzyme activity assay as previously described.6,46 The GTN concentration-activity curve was obtained for each individual tissue preparation, and the sGC specific activity values of SS-nitrates were normalized to the corresponding maximal sGC activation values of GTN.
000 × g for 1 h at 4 °C, and the supernatant containing sGC used for the enzyme activity assay as previously described.6,46 The GTN concentration-activity curve was obtained for each individual tissue preparation, and the sGC specific activity values of SS-nitrates were normalized to the corresponding maximal sGC activation values of GTN.
Oxyhemoglobin oxidation
O2-Hb preparation methodology was adapted from previously described methods.42,47 Briefly, bovine hemoglobin (25 mg ml−1) was completely dissolved in oxygenated 0.1 M phosphate buffer pH 7.4. An excess of sodium dithionite over the iron content of hemoglobin was added and the solution was slowly purged for 5 min with pure oxygen. The O2-Hb solution was desalted and purified by passing it through a Sephadex G-25 column. The concentration of the O2-Hb solution was calculated considering ε415 = 131 mM−1 cm−1.47 The time-course of the absorbance difference ΔA(A404–A408) for O2-Hb oxidation was monitored in the presence of 1 mM GTN or SS-nitrates with or without 2 mM cysteine. All kinetic measurements were performed in triplicate on a SpectraMax PLUS384 (Molecular Devices) microplate reader using a quartz 96-well plate. At least 30 experimental data points (between 5 and 6 min) from the initial part of the differential absorbance versus time curves were fitted to straight lines to determine initial rates of O2-Hb oxidation by GTN or SS-nitrates.43Statistical analysis
Data are reported as means ± standard errors (SEM) or standard deviations (SD). The relationship between σp Hammett parameter and sGC efficacies or the O2-Hb oxidation rates was assessed using linear regression and Pearson correlation analysis. EC50s were determined from nonlinear fitting of sGC activation data to sigmoidal concentration–response curves with variable slopes using GraphPad Prism 4.03 software. One-way analysis of variance (ANOVA) followed by Tukey's post-hoc test was used for comparing multiple groups. Two-tailed p values < 0.05 were considered statistically significant.Abbreviations
| cGMP | Cyclic guanosine-3′,5′-monophosphate |
| DEA/NO | Diethylamine NONOate |
| DETA/NO | Diethylenetriamine NONOate |
| GTN | Nitroglycerin (glyceryl trinitrate) |
| NO | Nitric oxide |
| NONOate | Diazeniumdiolate salt |
| sGC | Soluble guanylate cyclase |
| SPE/NO | Spermine NONOate. |
Acknowledgements
Ms. Diane Anderson is acknowledged for technical assistance with the sGC experiments, and GB Therapeutics Ltd for past support.References
- C. A. Gruetter, P. J. Kadowitz and L. J. Ignarro, Can. J. Physiol. Pharmacol., 1981, 59, 150–156 CrossRef CAS.
- E. Noack and M. Feelisch, Basic Res. Cardiol., 1991, 86, 37–50 Search PubMed.
- B. M. Bennett, B. J. McDonald, R. Nigam and W. C. Simon, Trends Pharmacol. Sci., 1994, 15, 245–249 CrossRef CAS.
- S. Katsuki, W. Arnold, C. Mittal and F. Murad, J. Cyclic Nucleotide Res., 1977, 3, 23–35 CAS.
- J. W. Denninger and M. A. Marletta, Biochim. Biophys. Acta, 1999, 1411, 334–350 CrossRef CAS.
- J. D. Artz, V. Toader, S. I. Zavorin, B. M. Bennett and G. R. Thatcher, Biochemistry, 2001, 40, 9256–9264 CrossRef CAS PubMed.
- G. S. Marks, B. E. McLaughlin, K. Nakatsu and J. F. Brien, Can. J. Physiol. Pharmacol., 1992, 70, 308–311 CrossRef CAS.
- S. J. Chung and H. L. Fung, J. Pharmacol. Exp. Ther., 1990, 253, 614–619 CAS.
- S. Chong and H. L. Fung, Biochem. Pharmacol., 1991, 42, 1433–1439 CrossRef CAS.
- G. R. Thatcher, A. C. Nicolescu, B. M. Bennett and V. Toader, Free Radicals Biol. Med., 2004, 37, 1122–1143 CrossRef CAS PubMed.
- G. R. Thatcher, B. M. Bennett and J. N. Reynolds, Curr. Alzheimer Res., 2005, 2, 171–182 CrossRef CAS.
- Z. Chen, J. Zhang and J. S. Stamler, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8306–8311 CrossRef CAS PubMed.
- Z. Chen and J. S. Stamler, Trends Cardiovasc. Med., 2006, 16, 259–265 CrossRef CAS PubMed.
- Y. C. Hou, A. Janczuk and P. G. Wang, Curr. Pharm. Des., 1999, 5, 417–441 CAS.
- I. L. Megson, Drugs Future, 2000, 25, 701–715 CrossRef CAS.
- S. Smith, H. C. Dringenberg, B. M. Bennett, G. R. Thatcher and J. N. Reynolds, Neuroreport, 2000, 11, 3883–3886 CrossRef CAS PubMed.
- J. L. Burgaud, E. Ongini and P. Del Soldato, Ann. N.Y. Acad. Sci., 2002, 962, 360–371 CrossRef CAS.
- M. P. Moya, A. J. Gow, R. M. Califf, R. N. Goldberg and J. S. Stamler, Lancet, 2002, 360, 141–143 CrossRef CAS.
- J. N. Reynolds, B. M. Bennett, R. J. Boegman, K. Jhamandas, J. D. Ratz, S. I. Zavorin, D. Scutaru, A. Dumitrascu and G. R. Thatcher, Bioorg. Med. Chem. Lett., 2002, 12, 2863–2866 CrossRef CAS.
- G. R. Thatcher, B. M. Bennett, H. C. Dringenberg and J. N. Reynolds, J. Alzheimers Dis., 2004, 6, S75–S84 CAS.
- V. Calabrese, C. Mancuso, M. Calvani, E. Rizzarelli, D. A. Butterfield and A. M. Stella, Nat. Rev. Neurosci., 2007, 8, 766–775 CrossRef CAS PubMed.
- S. I. Zavorin, J. D. Artz, A. Dumitrascu, A. C. Nicolescu, D. Scutaru, S. V. Smith and G. R. Thatcher, Org. Lett., 2001, 3, 1113–1116 CrossRef CAS PubMed.
- L. J. Ignarro and C. A. Gruetter, Biochim. Biophys. Acta, 1980, 631, 221–231 CrossRef CAS.
- H. Schroder, E. Noack and R. Muller, J. Mol. Cell. Cardiol., 1985, 17, 931–934 CrossRef CAS.
- J. F. Brien, B. E. McLaughlin, K. Nakatsu and G. S. Marks, J. Pharmacol. Methods, 1991, 25, 19–27 CrossRef CAS.
- G. R. J. Thatcher and H. Weldon, Chem. Soc. Rev., 1998, 27, 331–337 RSC.
- L. J. Ignarro, Hypertension, 1990, 16, 477–483 CrossRef CAS.
- A. C. Nicolescu, J. N. Reynolds, L. R. C. Barclay and G. R. J. Thatcher, Chem. Res. Toxicol., 2004, 17, 00 CrossRef CAS PubMed.
- F. M. Gonzalez, S. Shiva, P. S. Vincent, L. A. Ringwood, L. Y. Hsu, Y. Y. Hon, A. H. Aletras, R. O. Cannon, 3rd, M. T. Gladwin and A. E. Arai, Circulation, 2008, 117, 2986–2994 CrossRef CAS PubMed.
- M. T. Gladwin, N. J. Raat, S. Shiva, C. Dezfulian, N. Hogg, D. B. Kim-Shapiro and R. P. Patel, Am. J. Physiol.: Heart Circ. Physiol., 2006, 291, H2026–H2035 CrossRef CAS PubMed.
- R. P. Szajewski and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 2011–2026 CrossRef CAS.
- S. Oae, Organic sulfur chemistry: structure and mechanism, CRC Press, Boca Raton, FL, 1991 Search PubMed.
- D. J. Hupe and D. Wu, J. Org. Chem., 1980, 45, 3100–3103 CrossRef CAS.
- D. Gallemann, A. Greif, P. Eyer, J. Dasenbrock, E. Wimmer, J. Sonnenbichler, I. Sonnenbichler, W. Schafer and I. Buhrow, Chem. Res. Toxicol., 1998, 11, 1423–1433 CrossRef CAS PubMed.
- D. L. Williams, Methods Enzymol., 1996, 268, 299–308 CAS.
- R. J. Singh, N. Hogg, J. Joseph and B. Kalyanaraman, J. Biol. Chem., 1996, 271, 18596–18603 CrossRef CAS PubMed.
- J. D. Artz, B. Schmidt, J. L. McCracken and M. A. Marletta, J. Biol. Chem., 2002, 277, 18253–18256 CrossRef CAS PubMed.
- A. Friebe, B. Wedel, C. Harteneck, J. Foerster, G. Schultz and D. Koesling, Biochemistry, 1997, 36, 1194–1198 CrossRef CAS PubMed.
- D. Koesling, Methods, 1999, 19, 485–493 CrossRef CAS PubMed.
- N. B. Fernhoff, E. R. Derbyshire and M. A. Marletta, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21602–21607 CrossRef CAS PubMed.
- K. Schror, S. Forster and I. Woditsch, Naunyn-Schmiedeberg's Arch. Pharmacol., 1991, 344, 240–246 CrossRef CAS.
- M. Feelisch and E. A. Noack, Eur. J. Pharmacol., 1987, 139, 19–30 CrossRef CAS.
- J. D. Artz and G. R. Thatcher, Chem. Res. Toxicol., 1998, 11, 1393–1397 CrossRef CAS PubMed.
- B. G. Katzung, Basic & clinical pharmacology, McGraw Hill, 8th edn, 2000 Search PubMed.
- C. C. Chiueh, Ann. N.Y. Acad. Sci., 1999, 890, 301–311 CrossRef CAS.
- C. K. Mittal, H. Kimura and F. Murad, J. Biol. Chem., 1977, 252, 4384–4390 CAS.
- M. E. Murphy and E. Noack, Methods Enzymol., 1994, 233, 240–250 CAS.
| This journal is © The Royal Society of Chemistry 2014 |