DOI:
10.1039/C3MD00119A
(Concise Article)
Med. Chem. Commun., 2014,
5, 32-40
Synthesis and biological evaluation of tetrahydropyridinepyrazoles (‘PFPs’) as inhibitors of STAT3 phosphorylation†
Received
19th April 2013
, Accepted 26th October 2013
First published on 30th October 2013
Abstract
The transcription factor STAT3 is constitutively overexpressed in many human tumors and hence represents a putative target for anticancer drug design. In this work, we describe the synthesis and biological evaluation of a novel chemotype, pyridine-fused pyrazoles (‘PFPs’) as inhibitors of STAT3 phosphorylation. The effect of the compounds synthesized was evaluated in cell proliferation assays of MCF-7 and HepG2 cancer cell lines and two of the compounds tested (12g and 12k) were found to show significant activity. Both compounds were also found to inhibit the proliferation of Hep3B, HUH-7 and PLC/PRF5 HCC cells in a dose- and time-dependent manner. Furthermore, we established in a DNA binding assay that one of the compounds (12g) was able to significantly inhibit the DNA binding ability of STAT3. Cytotoxicity of 12g against PC3 cells, which do not constitutively phosphorylate STAT3, was found to be minimal, hence lending further support for our mode-of-action hypothesis of this compound. We established for this structure a complete inhibition of CXCL12-induced cell invasion and associated wound healing in HCCLM3 cells, corroborating the proposed modulation of the STAT3 axis by 12g. Finally, molecular modeling was employed to evaluate the hypothesis of PFPs to bind to the SH2 domain of STAT3. Given the efficacy of PFPs in the biological systems studied here we propose their further evaluation in the context of STAT3-mediated cancer therapy.
Signal Transducer and Activator of Transcription proteins (STATs) are involved in both developmental and oncogenic signaling pathways and comprise seven members, namely STAT1 to STAT4 and STAT6, as well as STAT5a and STAT5b which are closely related. STAT3, which belongs to a mammalian STAT family, is frequently activated in many human cancers, including those of liver, lung, head and neck, prostate and breast, as well as myeloma and leukemia.1,2 It is involved in the expression of a variety of genes in response to cell stimuli, and thus plays a key role in many cellular parameters like cell growth and apoptosis, as well as multiple steps of metastasis including cell survival, invasion, angiogenesis, and the evasion of tumor-cells to the endothelium.3,4 Functionally STAT3 can be activated by certain growth factors, cytokines and hormones and this activation leads to intracellular phosphorylation followed by dimerisation. The STAT3 dimer translocates into the nucleus, binds to DNA and promotes gene transcription, leading in turn (when functioning as an oncogene) to cell growth, cell survival, angiogenesis and resistance to apoptosis.5–7 STAT3 expression is predominant in hepatocellular carcinoma (HCC), a fatal liver cancer affecting 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people annually and thus one of the leading causes of cancer-related lethality.8,9
000 people annually and thus one of the leading causes of cancer-related lethality.8,9
Given its functions outlined above, inhibiting the phosphorylation of STAT3 was proposed as a target for cancer therapy, either directly or indirectly, and this concept was supported by work involving viral vectors which significantly inhibited STAT3 signaling.10 Also, given the increasing interest in the area, several small-molecule inhibitors of this protein have recently been published.11–13 In order to block STAT3 indirectly the inhibition of the STAT signaling pathway by blocking upstream tyrosine kinases has been proposed. For example, small-molecule inhibitors of Proto-Oncogene Tyrosine-Protein Kinase (SRC), Janus Kinase (JAK), Breakpoint Cluster Region Protein-V-Abelson Murine Leukemia Viral Oncogene Homolog 1 (BCR-ABL), Fms-like tyrosine kinase 3 (FLT3), and Epidermal growth factor receptor (EGFR), have all been shown to block STAT3 or STAT5 signaling and induce tumor-cell apoptosis.14–16 Also some traditional medicines (such as cucurbitacin I) are known to be selective inhibitors of JAK and hence to modulate STAT3 signaling.17
The main drawback of inhibitors of upstream tyrosine kinases is that they have many targets (both due to kinase inhibitor promiscuity as well as cross-talk in signaling pathways), which is different with compounds that target STAT3 directly. Although the selective direct inhibition by means of small molecules is certainly not trivial some progress has already been made in this direction. Most of those involve targeting the Src Homology2 (SH2) domain of STAT3 which is essential for both tyrosine-phosphorylation and dimerisation, with the hope that inhibition of dimerisation also translates to efficacy in the clinic.18 Several dimerisation disruptors of STAT3 have been reported and are in various phases of clinical development; still the area is a field of active research and no compounds have been approved as of yet.
Given the biological relevance of targeting STAT3, we in this work report a new class of inhibitors for the phosphorylation of STAT3 by pyridine-fused pyrazoles (‘PFPs’, see Fig. 1 and Table 1 for structural and synthetic details) and we show that the most active compounds are able to inhibit the proliferation, invasion, and migration of cancer cells (MCF-7, HepG2, HUH-7, PLC/PRF/5, and Hep3B cell lines) as well as inhibiting the DNA-binding ability of STAT3 in HCC cells.
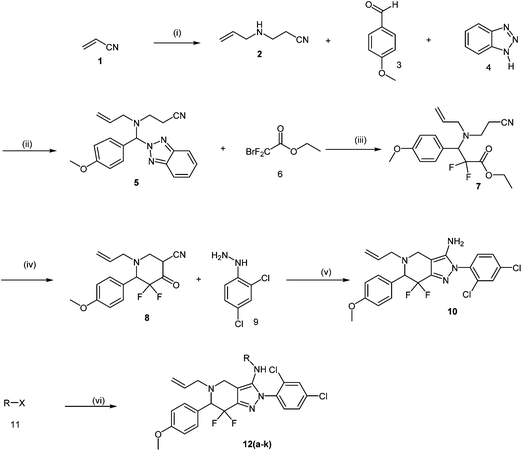 |
| | Fig. 1 Synthesis scheme of target compounds (see Material and Methods section and ESI† section for details). (i) Allyl amine, EtOH, RT; (ii) MeOH, RT; (iii) zinc dust, trimethylsilyl chloride, THF, RT; (iv) diisopropyl amine, N-butyl lithium, THF; (v) EtOH, reflux; (vi) pyridine, RT. | |
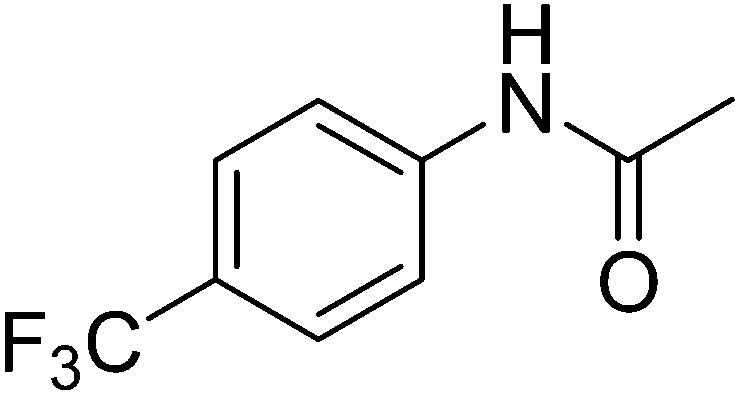
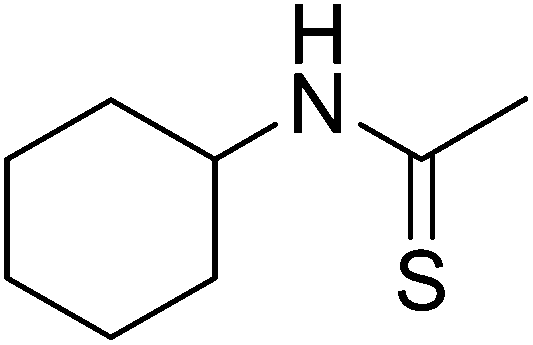
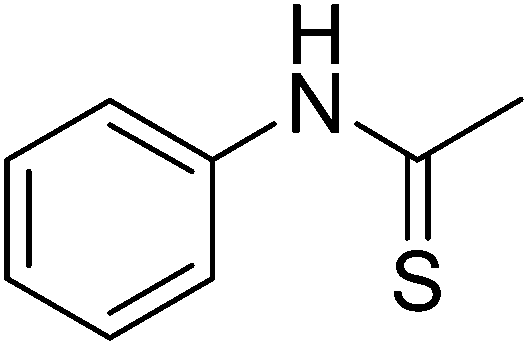
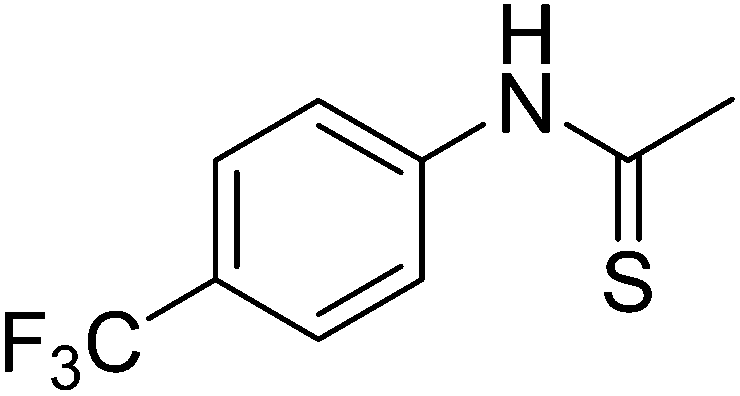
Table 1 R-groups of the compounds synthesized and evaluated for bioactivity against STAT3 in this work
| Compounds |
R |
MW |
|
12a
|
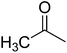
|
507.37 |
|
12b
|
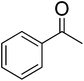
|
545.25 |
|
12c
|
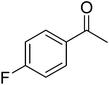
|
587.43 |
|
12d
|
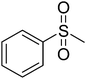
|
605.48 |
|
12e
|
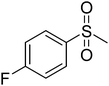
|
623.47 |
|
12f
|
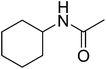
|
590.5 |
|
12g
|
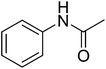
|
584.45 |
|
12h
|
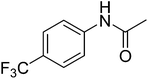
|
652.45 |
|
12i
|
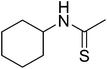
|
606.56 |
|
12j
|
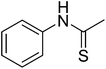
|
600.51 |
|
12k
|
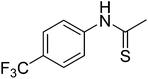
|
668.51 |
Results and discussion
Effect of PFPs on the proliferation of cancer cells
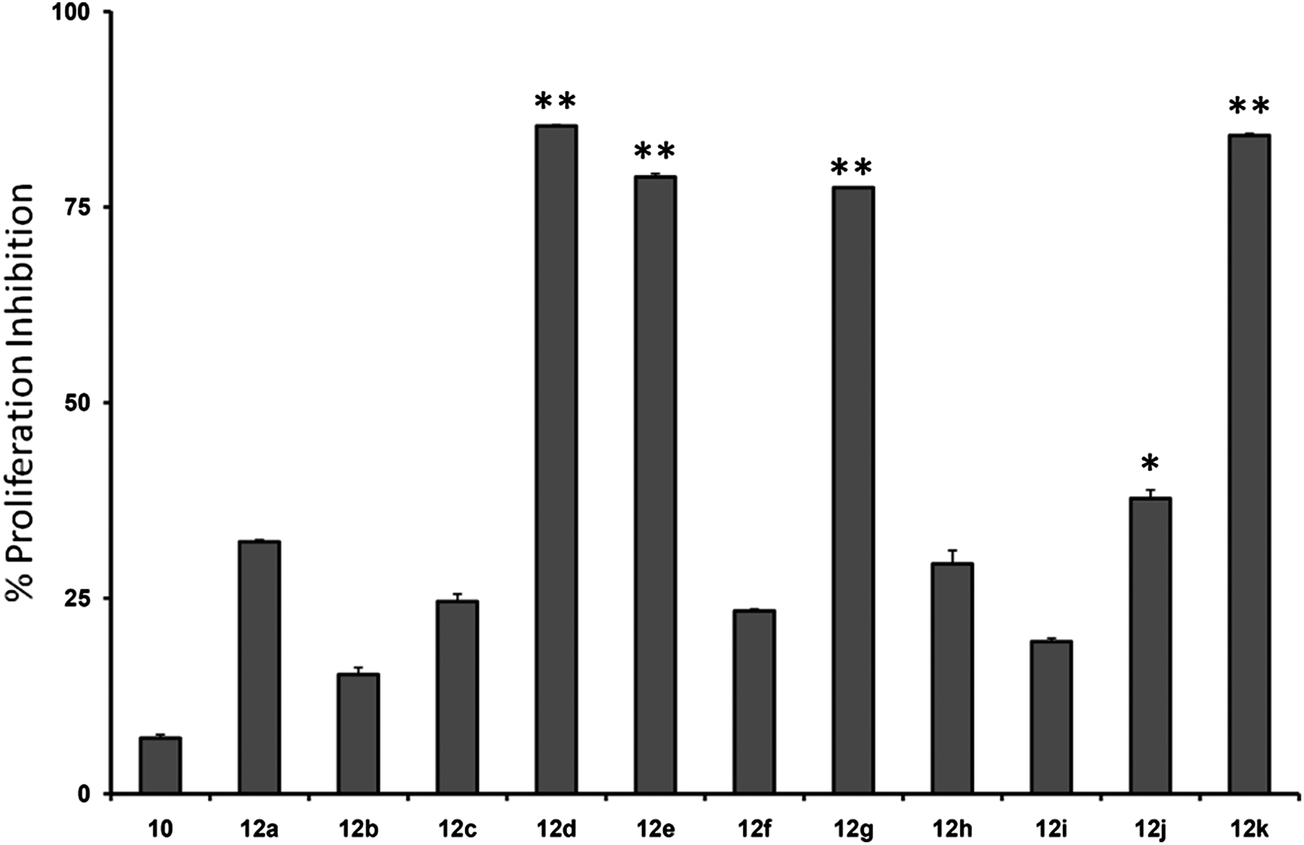
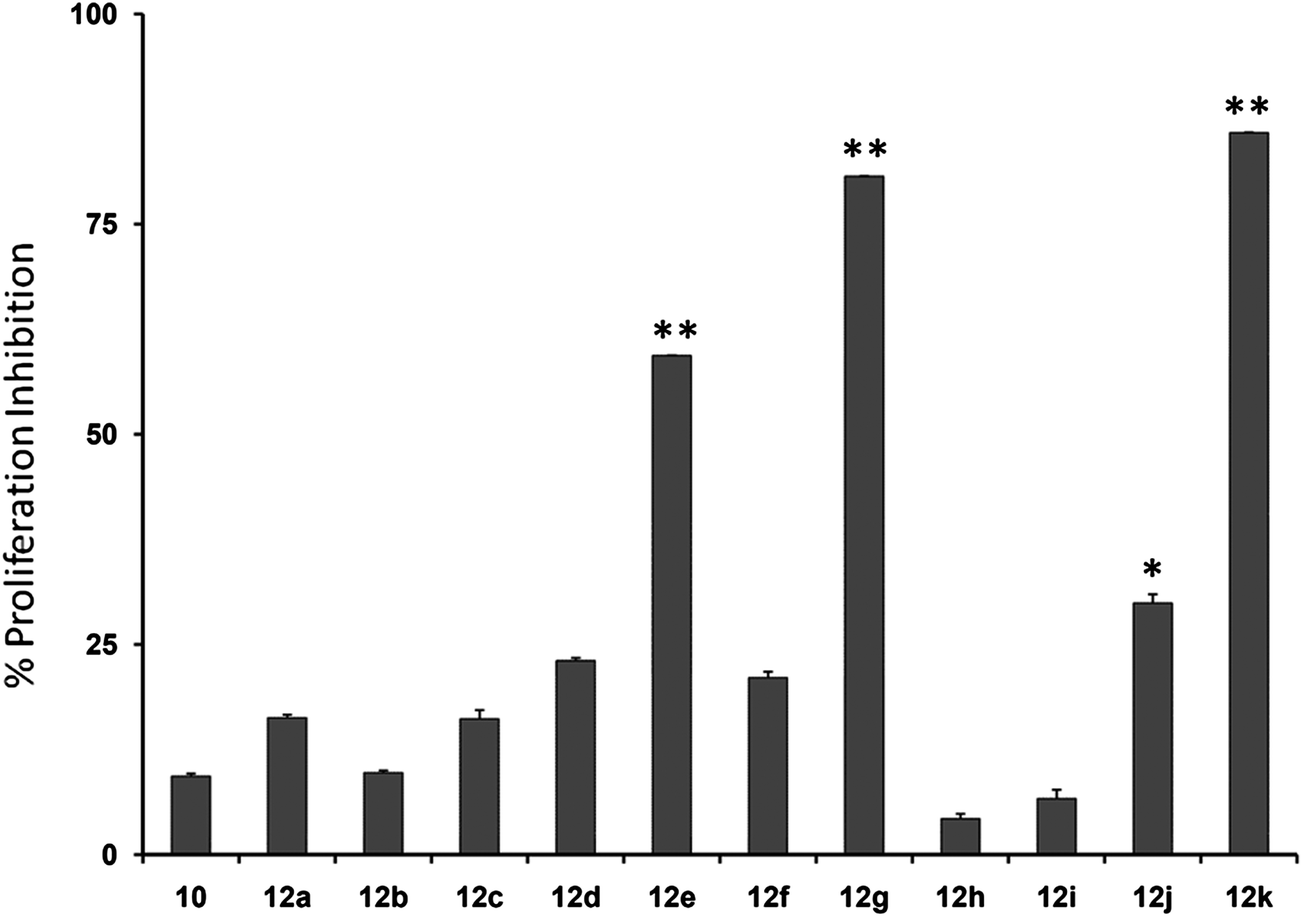
Initially, we investigated the anti-proliferative effect of PFPs on MCF-7 cancer cells and it was found that 12d, 12e, 12g, and 12k were able to inhibit the proliferation of MCF-7 cells by 85.3%, 78.8%, 77.4%, and 84.1%, at 100 μM, respectively (Fig. 2). Furthermore, 12g and 12k also inhibited the proliferation of HCC cancer cells (HepG2) by 80% and 85.1%, respectively (Fig. 3). These data show that 12g and 12k, where R = arylated urea and thiourea, are most potent among all PFPs tested. The sulphonamide substituted PFPs 12d and 12e could in principle work as prodrugs/bioprecursors, however they failed to inhibit the proliferation of HepG2 cells in our study.
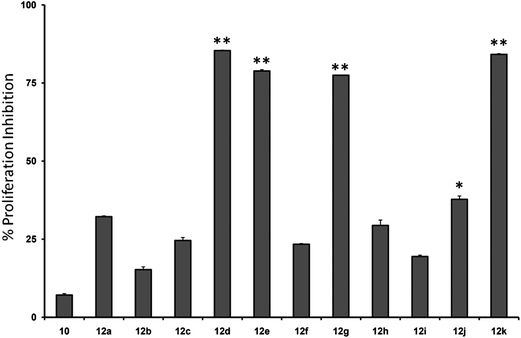 |
| | Fig. 2 Cytotoxic effects effect of the compounds described here on MCF-7 cells (plotted as % proliferation inhibition at a concentration of 10 μM), where four of the compounds measured obtained >75% growth inhibition. Standard deviations between the triplicates are indicated. ** indicates p value <0.01; * indicates p value <0.05. | |
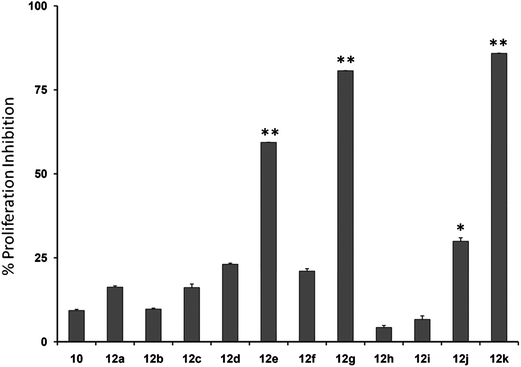 |
| | Fig. 3 Cytotoxic effects of PFPs on HepG2 cells (plotted as % proliferation inhibition at a concentration of 10 μM), where two of the compounds measured obtained >25% growth inhibition. ** indicates p value <0.01; * indicates p value <0.05. | |
MTT dye uptake assay of HCC cells
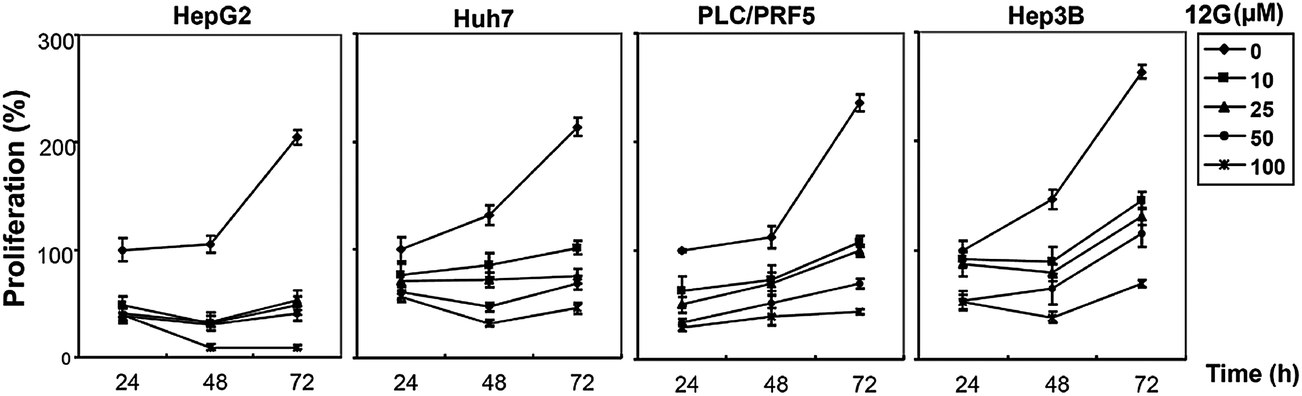
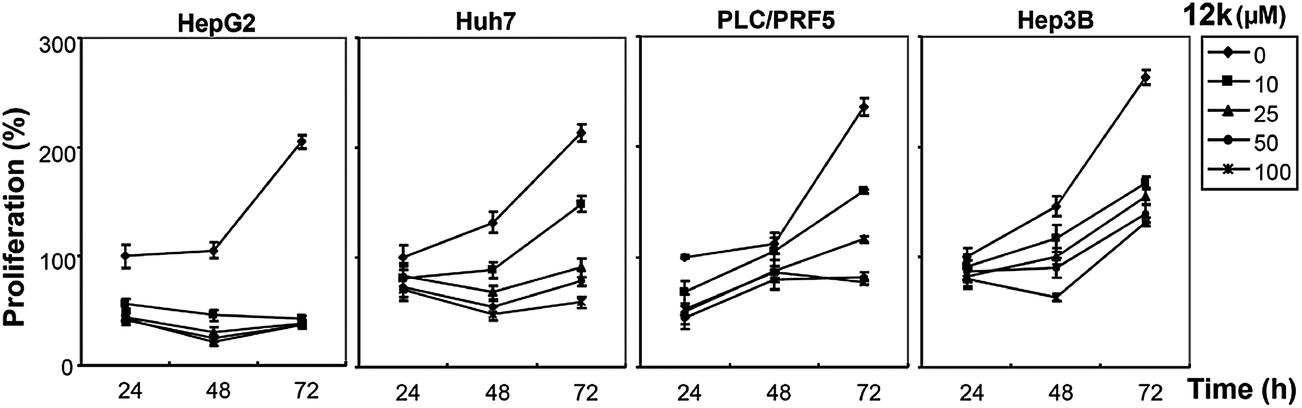
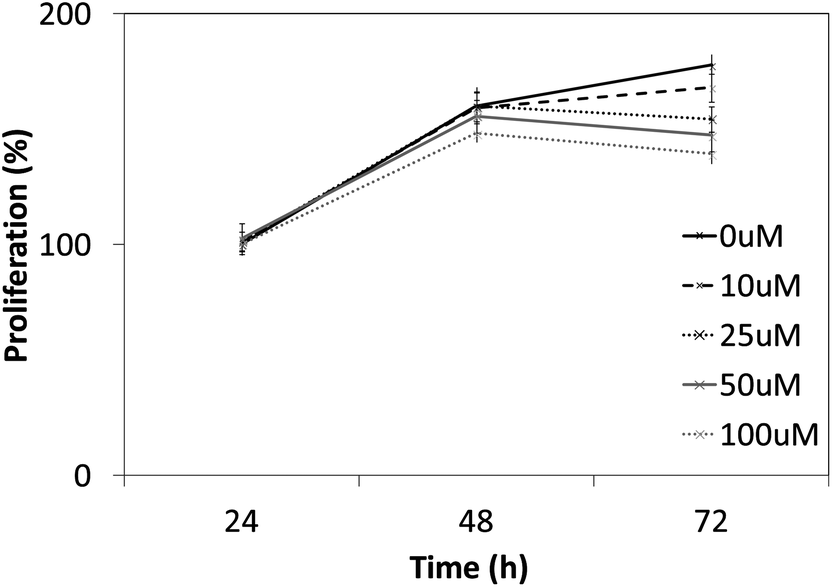
We next investigated whether the most potent compounds against MCF-7 and HepG2 cells can also inhibit the proliferation of other HCC cells via MTT dye uptake assay as described in detail in the Material and Methods section. Here it was found that both 12g and 12k were able to inhibit the proliferation of HepG2, HUH-7, PLC/PRF/5, and Hep3B cells in a dose- and time-dependent manner, as shown in Fig. 4 and 5.
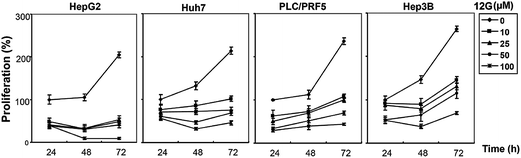 |
| | Fig. 4 The effect of 12g on the proliferation of HCC cells is both time- and dose-dependent. HepG2, HUH-7, PLC/PRF/5, and Hep3B cells (5 × 103 ml−1) were plated in triplicate, treated with indicated concentrations of 12g and then subjected to MTT assay after 24, 48 and 72 hours to analyze proliferation of cells. Standard deviations between the triplicates are indicated. * indicates p value <0.05. | |
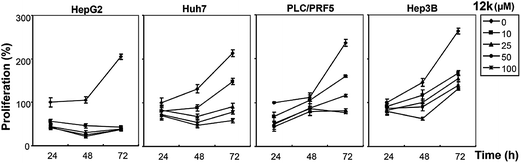 |
| | Fig. 5 The effect of 12k on the proliferation of HCC cells is both time- and dose-dependent. HepG2, HUH-7, PLC/PRF/5, and Hep3B cells (5 × 103 ml) were plated in triplicate, treated with indicated concentrations of 12k, and then subjected to MTT assay after 24, 48 and 72 hours to analyze proliferation of cells. Standard deviations between the triplicates are indicated. * indicates p value <0.05. | |
STAT3 DNA binding assay
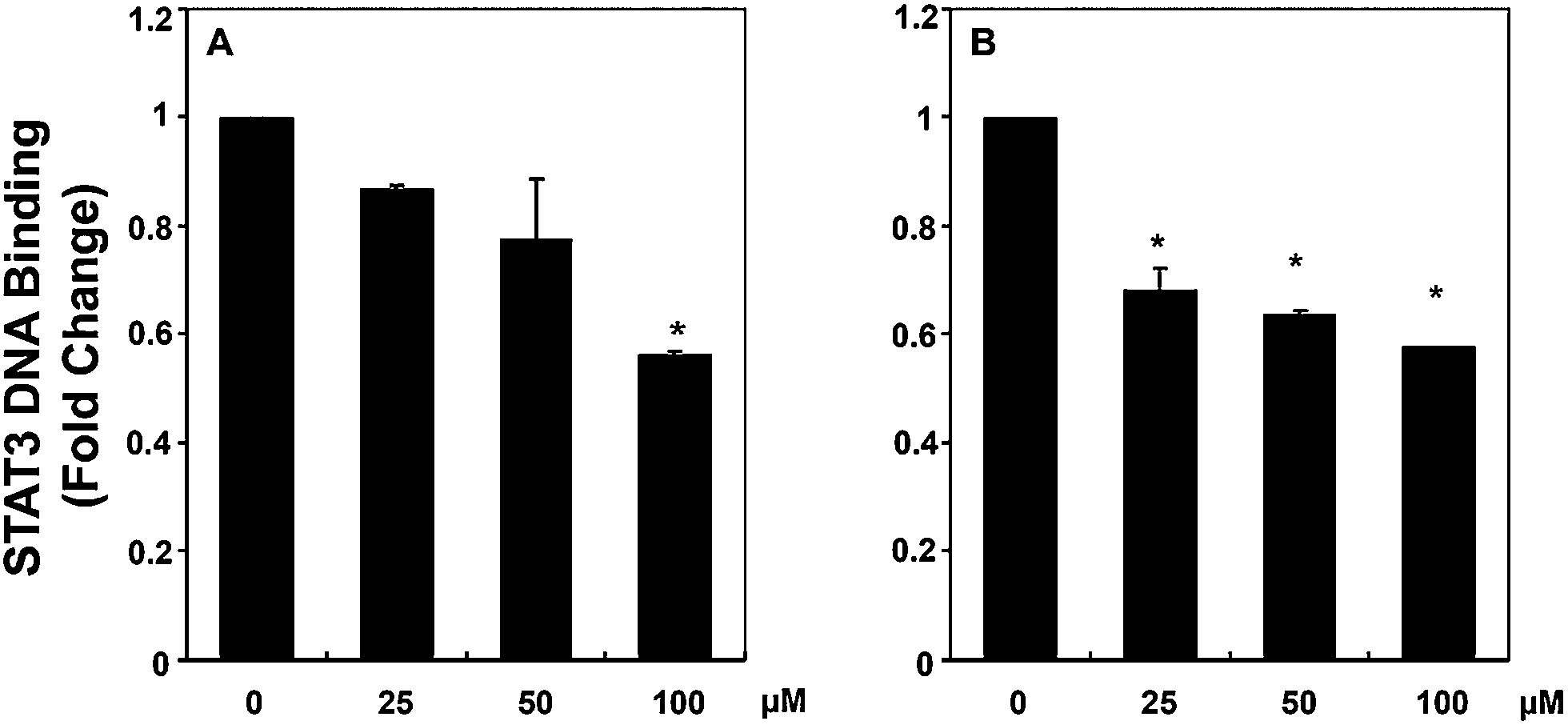
In order to validate the mode of action of the compounds found to be active in cell proliferation assays and also in affecting STAT3 phosphorylation, we further functionally analyzed the influence of 12g and 12k on the ability of STAT3 to bind DNA in HCC cells. Analysis of nuclear extracts prepared from HepG2 cells using ELISA-based TransAM™ STAT3 assay kits showed that 12g inhibited the DNA binding abilities of STAT3 in a dose-dependent manner (see Fig. 6a). The effect of 12k on STAT3–DNA binding activity is similar in magnitude but not linearly related to dose when compared to 12g (Fig. 6b), which may also be related to stability differences between the thiourea in 12k and the urea in 12g. These results suggest that 12g can inhibit the ability of STAT3 to bind DNA in HCC cells significantly, giving insight into the mode of action of this compound.
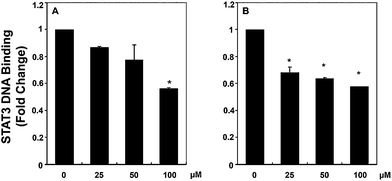 |
| | Fig. 6 Effect of compound treatment at a concentration of 10 μM on the ability of STAT3 to bind DNA where both compounds were significantly active. HepG2 cells were treated with indicated concentrations of 12k (A) and 12g (B) for 6 h. The nuclear extracts were prepared, and 5 μg of the nuclear extract protein was used for ELISA based DNA binding assay as described in “Material and methods”. The results shown are representative of three independent experiments, *p < 0.05. | |
Effect of compound 12g on STAT3 phosphorylation
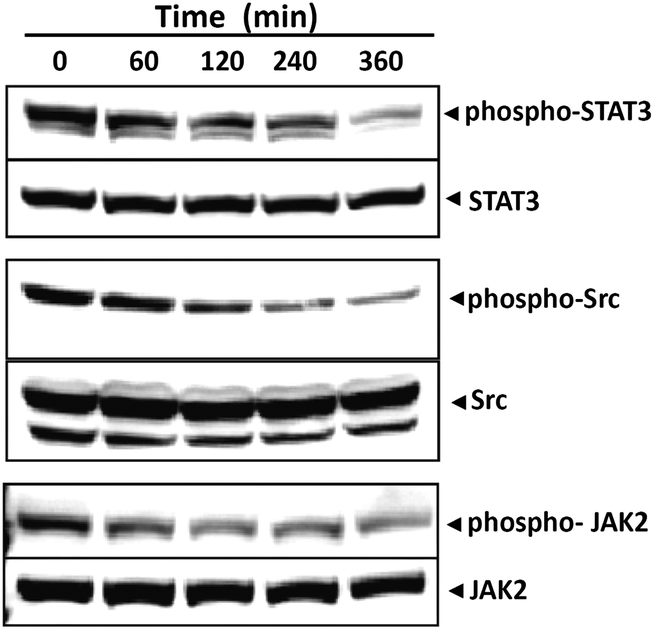
Given that the presence of phospho-STAT3 at Tyrosine 705 is known to be a key residue in STAT3 dimerization we further investigated the ability of 12g to modulate constitutive STAT3 activation in HCC cells via inhibition of phosphorylation at this residue. For this purpose, HepG2 cells were incubated with 12g for different time intervals, whole cell extracts were prepared and the phosphorylation of STAT3 was examined by western blot analysis using antibodies of STAT3 Tyr705 phosphorylation. We found that 12g inhibits the constitutive activation of STAT3 phosphorylation in HepG2 cells in a time-dependent manner (Fig. 7). Maximum inhibition occurs at 4–6 h, whereas its effect on the expression of STAT3 protein is negligible.
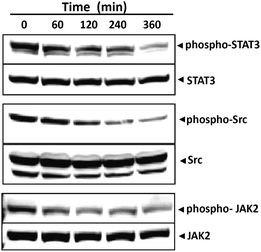 |
| | Fig. 7 Compound 12g inhibits STAT3 activation in HepG2 cells in a time-dependent manner. Maximum inhibition occurs at 4–6 h, whereas its effect on the expression of STAT3 protein is negligible. Compound 12g also suppresses the constitutive phosphorylation of c-Src kinase and JAK2 in a time-dependent manner. The levels of both non-phosphorylated kinases remained unchanged under the same conditions. (All data has been obtained at a compound concentration of 10 μM.) | |
Compound 12g suppresses constitutive activation of c-Src
STAT3 has been reported to be activated by soluble tyrosine kinases of the Src kinase families.5,6 Hence, we determined the effect of 12g on the constitutive activation of Src kinase in HepG2 cells and found that 12g suppresses the constitutive phosphorylation of c-Src kinase in a time-dependent manner (Fig. 7). The levels of non-phosphorylated Src kinases remained unchanged under the same conditions.
Compound 12g suppresses the constitutive activation of JAK2
Since STAT3 is also activated by soluble tyrosine kinases of the Janus family (JAKs)5,6 we next determined whether 12g affects constitutive activation of JAK2 in HepG2 cells. Also, the JAK2/STAT3 pathway is well-established in HCC carcinoma which provided additional support for the importance of this particular experiment. Here we found that 12g suppresses the constitutive phosphorylation of JAK2 in a time-dependent manner (see Fig. 7). The levels of non-phosphorylated JAK2 remained unchanged under the same conditions.
Effect of compound 12g on prostate cancer (PC3) cells
In order to further lend support to our hypothesis of compound 12g targeting STAT3 directly, we measured its effect on the proliferation of PC3 cancer cells which do not phosphorylate STAT3 constitutively using the MTT method. In agreement with our hypothesis, we found that 12g had a minimal effect on the proliferation of this cell line (Fig. 8), indicating that the bioactivity of this compound is indeed mediated via STAT3.
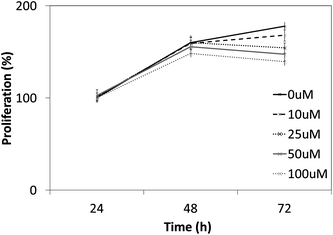 |
| | Fig. 8 Cytotoxic effects of 12g on PC3 cells which do not express STAT3 constitutively. As it can be seen, much less cytotoxicity as compared to other cancer cell lines is observed (compared to Fig. 4 and 5), lending further support that 12g targets the STAT3 signaling axis. | |
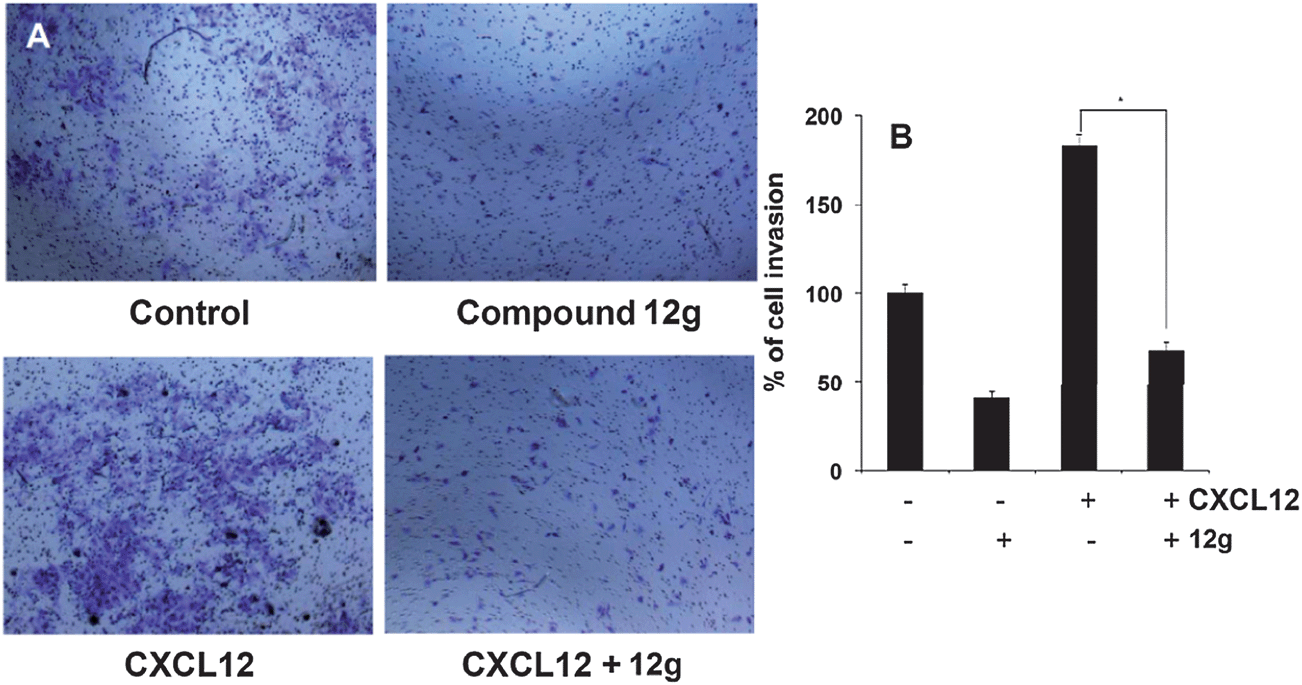
Effects of compound 12g on HCCLM3 cell migration and invasion in the presence or absence of CXCL12
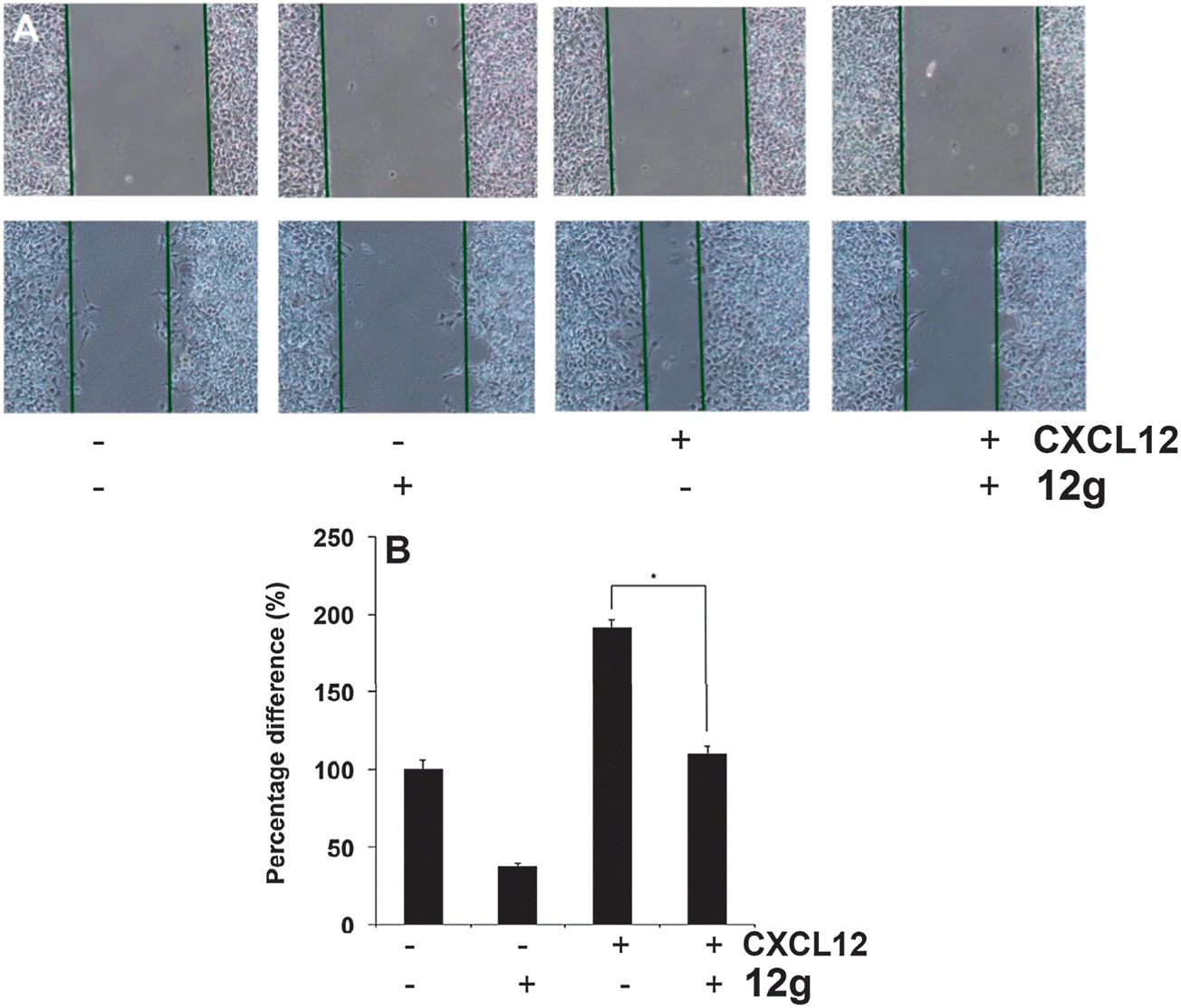
It has been reported that STAT3 target gene that promotes motility, migration, and invasion of tumor cells,19,20 and for further target validation hence we next investigated the effect of 12g on CXCL12-induced HCCLM3 cell migration. Using an in vitro wound healing assay, we found that HCCLM3 cells migrated faster under the influence of CXCL12, and this effect was not observed upon treatment with 12g (Fig. 9). We further elucidated the effect of compound 12g on CXCL12-induced cell invasion, a key hallmark of cancer, and it was found that 12g indeed suppressed CXCL12-induced invasion of HCCLM3 cells (Fig. 10). Additionally, we also found that the CXCL12 untreated HCCLM3 cells migrated and invaded more slowly and these effect were abolished upon treatment with 12g (Fig. 9 and 10), with all of those observations being consistent of this compound targeting the STAT3 axis.
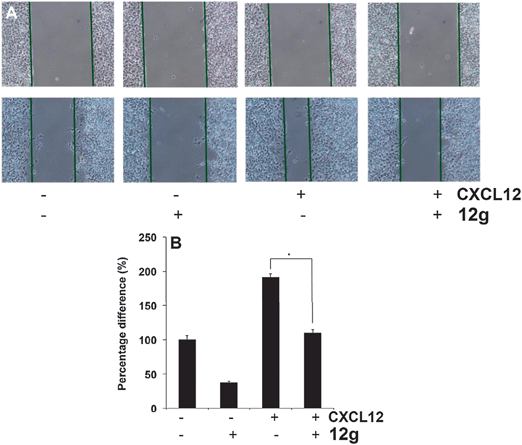 |
| | Fig. 9 Inhibition of HCC cell migration by 12g, where compound treatment significantly inhibits CXCL12 induced cell migration. Confluent monolayers of HCCLM3 cells were scarred, and repair was monitored microscopically after 6 h of pre-treatment with compound 12g, before being exposed to 100 ng ml−1 CXCL12 for 24 h. Width of wound was measured at time zero and 48 h of incubation with and without the compound. The representative photographs showed the same area at time zero and after 24 h of incubation. Graphs, mean (n = 3); bars, SE. *, P < 0.05. All data has been obtained at a compound concentration of 10 μM. | |
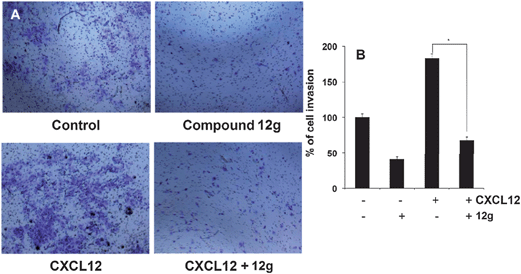 |
| | Fig. 10 A HCC cell invasion assay. It can be seen that 12g significantly reduces CXCL12 induced cell invasion (lower right hand corner of panel A), compared to untreated cells. B Columns: percentage cell invasion; bars S.E., *p < 0.05. Representative results of two independent experiments are shown and observations are found to be consistent with 12g targeting the STAT3 signaling axis. All data has been obtained at a compound concentration of 10 μM. | |
Molecular modeling of PFPs binding to the SH2 domain of STAT3
Molecular modeling was performed to investigate the structural interaction of the most active STAT3 inhibitors (12g and 12k) with the target in more detail (see Fig. S1 and S2,† respectively). It appears that two different orientations of the ligands are possible in the binding site, placing the substitutes phenyl moieties into different subpockets in each case, and that the activity of 12g may be attributed to hydrogen bond/charge interactions between the carbonyl group and the nitrogen from the tetrahydropyridine ring and Lys591 of STAT3 SH2 domain.
Conclusion
In this work we described the synthesis and biological evaluation of a novel class of inhibitors of STAT3 signaling – pyridine-fused pyrazoles (‘PFPs’) – that showed antiproliferative effects in MCF-7, HepG2, Hep3B and PLC/PRF5 cell lines. One of the most potent compounds, 12g, inhibited DNA binding of STAT3, whereas expression itself remains unaltered. In addition this compound showed complete inhibition of CXCL12-induced cell invasion and associated wound healing. 12g failed to inhibit the proliferation of PC3 prostate cancer cells, a cell line which does not have constitutively phosphorylated STAT3, which corroborates our hypothesis of 12g inhibition of the STAT3 signaling pathway. HCCLM3 cell migration and invasion however are inhibited by this compound, in line with the previously reported involvement of STAT3 in cell motility. Molecular modeling studies suggest different binding modes of 12g and 12k, giving a possible explanation for the different effects observed experimentally. Given the importance of STAT3 modulation in various cancers the results presented here hence warrant further study of those compounds in more complex systems.
Materials and methods
Target site and scaffold choice
In order to narrow down chemical space for the synthesis of STAT3 inhibitors we used both the crystal structure of the recently published STAT3β homodimer (PDBID: 1BG1)21 as well as a number of heterocycles known to target this protein as a starting point, such as the pyrazole moiety in Celecoxib, the benzimidazole in Dovitinib, the indazole in ABT-869 and the purine ring.22–26 Our attempt was here to emulate similar ligand–protein interactions with the SH2 domain of STAT3 as suggested in a previous study for cryptotanshinone27 and to hence target the polar/basic region around Lys591, Arg609, Glu612 and Ser613 (including p-Tyr705, which is important for the dimerisation event following phosphorylation), the hydrophobic region around Ile597, Trp623, Ile634 and Val637, as well as the charged amino acids (surrounded by hydrophobic residues) Glu594, Glu592 and Arg595. The manual design process led us conclude that the pyridine-fused pyrazole (‘PFP’) scaffold was suitable both from an activity perspective, as well as not bearing any obvious liabilities from the medicinal chemistry perspective.
Synthesis
The synthesis of PFPs involved six steps via Reformatsky reaction, Dickman cyclysation, and cyclocondensation of 2,4-phenyl hydrazine (9) with cyano ketone (8) (Fig. 1). Finally, compound 10 was derivatised using various acid chlorides, sulphonyl chloride, isocyanates, and thiocyanates in the presence of pyridine to give the corresponding title compounds such as tetrahydroindazole carboxymides 12(a–c), sulfonamides 12(d–e), urea derivatives 12(f–h) and thiourea derivatives 12(i–k). Characterization was performed via IR, 1H NMR, 13C NMR, and Elemental Analysis. For individual compound syntheses see ESI† section.
Analytical chemistry.
All analytical thin layer chromatography was performed with E. Merck silica gel 60F254 aluminum sheets and was visualized with UV light. The following mobile phases were employed for TLC: chloroform, methanol and hexane, ethyl acetate in different ratios. The instrumental techniques employed for the characterization of the newly synthesized compounds includes IR NMR and mass spectroscopy. IR spectra were recorded on an FT-IR spectrometer. 1H NMR spectra were recorded on CDCl3, DMSO-d6 solution using a 400 and 300 MHz Fourier Transform NMR with tetramethylsilane (TMS) as internal standard. Mass and purity were recorded on a LC-MSD-Trap-XCT.
Biology
Biological reagents.
MTT, Tris, glycine, NaCl, SDS, and BSA, were purchased from Sigma-Aldrich (St. Louis, MO). Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), 0.4% trypan blue vital stain, and antibiotic–antimycotic mixture were obtained from Invitrogen. Rabbit polyclonal antibodies to STAT3 and mouse monoclonal antibodies against phospho-STAT3 (Tyr 705) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to phospho-specific Src (Tyr416), Src, phospho-specific JAK2 (Tyr1007/1008) and JAK2 were purchased from Cell Signalling Technology (Beverly, MA). CXCL12 was purchased from ProSpec-Tany TechnoGene Ltd. (Rehovot, Israel). Goat anti-rabbit-horse radish peroxidase (HRP) conjugate and goat anti-mouse HRP were purchased from Sigma-Aldrich (St Louis, MO).
Cell lines.
Human hepatocellular carcinoma (HCC) cell lines (HepG2, Hep3B and PLC/PRF5), and prostate cancer PC3 cells were obtained from American Type Culture Collection (Manassass, VA). HUH-7 and HCCLM3 cells were kindly provided by Prof. Kam Man Hui, National Cancer Centre, Singapore. All the five HCC cell lines were cultured in DMEM containing 1× antibiotic–antimycotic solution with 10% FBS. PC3 cells were cultured in Roswell Park Memorial Institute-1640 (RPMI-1640) medium containing 1× antibiotic–antimycotic solution with 10% FBS.
DNA binding assay.
DNA binding was performed using a STAT3 DNA binding ELISA kit (Active Motif, Carlsbad, CA, USA). Briefly, nuclear extracts (5 μg) from compound treated cells were incubated in a 96-well plate coated with oligonucleotide containing the STAT3 specific DNA probe. Bound STAT3 was then detected using a specific primary antibody. An HRP-conjugated secondary antibody was then applied to detect the bound primary antibody and provided the basis for colorimetric quantification. The enzymatic product was measured at 450 nm with a microplate reader (Tecan Systems, San Jose, CA, USA).
MTT assay.
The antiproliferative effect of compounds against tumor cells was determined by the MTT dye uptake method as described previously.18 Briefly, the cells (5 × 103 ml−1) were incubated in triplicate in a 96-well plate in the presence or absence of indicated concentration of compounds in a final volume of 0.2 ml for different time intervals at 37 °C. Thereafter, 20 μl MTT solution (5 mg ml−1 in PBS) was added to each well. After a 2 h incubation at 37 °C, 0.1 ml lysis buffer (20% SDS, 50% dimethylformamide) was added; incubation was done for 1 h at 37 °C and the optical density (OD) at 570 nm was measured by Tecan plate reader.
Western blotting.
For detection of various proteins, compound 12g-treated whole-cell extracts were lysed in lysis buffer (20 mM Tris (pH 7.4), 250 mM NaCl, 2 mM EDTA (pH 8.0), 0.1% Triton X-100, 0.01 mg ml−1 aprotinin, 0.005 mg ml−1 leupeptin, 0.4 mM PMSF, and 4 mM NaVO4). Lysates were spun at 14000 rpm for 10 min to remove insoluble material and resolved on a 7.5% SDS gel. After electrophoresis, the proteins were electrotransferred to a nitrocellulose membrane, blocked with 5% non-fat milk, and probed with various antibodies (1:1000) overnight at 4 °C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence (ECL; GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Invasion assay.
The in vitro invasion assay was performed using the Bio-Coat Matrigel invasion assay system (BD Biosciences, San Jose, CA), according to the manufacturer's instructions. HCCLM3 cells (2 × 105 cells) were suspended in serum-free DMEM medium and seeded into the Matrigel transwell chambers consisting of polycarbonate membranes with 8 μm pores. After pre-incubation with or without the compound 12g, for 6 h, the transwell chambers were then placed into appropriate wells of a 24-well plate, in which either the basal medium only or basal medium containing CXCL12 had been added. After pre-incubation with or without 12g for 6 h, the transwell chambers were then placed into appropriate wells of a 24-well plate, in which only the basal medium had been added. After incubation for 24 h, the upper surfaces of the transwell chambers were wiped with cotton swabs and the invading cells were fixed and stained with crystal violet solution. The invading cell numbers were counted in five randomly selected microscope fields (×100).
Wound healing assay.
HCCLM3 cells were treated as described above. Before plating the cells, two parallel lines were drawn at the underside of the wells, to serve as fiducial marks demarcating the wound areas to be analyzed. Prior to inflicting the wound, the cells should be fully confluent. The growth medium was aspirated off and replaced by calcium-free PBS to prevent killing of the cells at the edge of the wound by exposure to high calcium concentrations before two parallel scratch wounds were made perpendicular to the marker lines with a sterile 1000 μl automated pipette tip. Thereafter, the calcium-free medium was then changed to medium with or without the compound 12g. After incubation for 6 h, the growth medium was changed to basal medium with or without CXCL12. After incubation for 6 h, the growth medium was then changed to basal medium. 24 h later the wounds were observed using bright field microscopy and multiple images were taken at areas flanking the intersections of the wound and the marker lines at the start and end of the experiment. Gap distance of the wound was measured at three different sites using Photoshop, and the data were normalized to the average of the control. Graphs were plotted against the percentage of migration distance the cells moved before and after treatment, normalized to control.
Acknowledgements
This research was supported by the Singapore Ministry of Health's National Medical Research Council to GS under its IRG funding scheme, University Grants Commission to Basappa (41-257-2012-SR) and Bhadregowda (41-316-2012-SR), Vision Group Science and Technology to Basappa, Government of Karnataka, INDIA, and Department of Science and Technology (no. SR/FT/LS-142/2012) to Basappa. AB thanks Unilever for funding. We thank Dr P. Sathyashankar, and Dr Subhendu Kumar Mohanty of Syngene International limited, India for valuable suggestions.
References
- J. E. Darnell, Jr, Science, 1997, 277, 1630–1635 CrossRef.
- E. Devarajan and S. Huang, Curr. Mol. Med., 2009, 9, 626–633 CrossRef CAS.
- J. E. Darnell, Jr, Nat. Rev. Cancer, 2002, 2, 740–749 CrossRef PubMed.
- J. Turkson, Expert Opin. Ther. Targets, 2004, 8, 409–422 CrossRef CAS PubMed.
- J. E. Darnell, Jr, I. M. Kerr and G. R. Stark, Science, 1994, 264, 1415–1421 Search PubMed.
- G. Niu, T. Bowman, M. Huang, S. Shivers, D. Reintgen, A. Daud, A. Chang, A. Kraker, R. Jove and H. Yu, Oncogene, 2002, 21, 7001–7010 CrossRef CAS PubMed.
- J. Turkson and R. Jove, Oncogene, 2000, 19, 6613–6626 CrossRef CAS PubMed.
- J. Whang-Peng, A.-L. Cheng, C. Hsu and C.-M. Chen, J. Exp. Clin. Med., 2010, 2, 93–103 CrossRef CAS.
- P. Rajendran, T. H. Ong, L. Chen, F. Li, M. K. Shanmugam, S. Vali, T. Abbasi, S. Kapoor, A. Sharma, A. P. Kumar, K. M. Hui and G. Sethi, Clin. Cancer Res., 2011, 17, 1425–1439 CrossRef CAS PubMed.
- O. A. Timofeeva, N. I. Tarasova, X. Zhang, S. Chasovskikh, A. K. Cheema, H. Wang, M. L. Brown and A. Dritschilo, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 267–272 CrossRef PubMed.
- A. K. Lakkaraju and F. G. van der Goot, Mol. Cell, 2013, 51, 386–396 CrossRef CAS PubMed.
- K. Takakuma, N. Ogo, Y. Uehara, S. Takahashi, N. Miyoshi and A. Asai, PLoS One, 2013, 8, e71646 CAS.
- A. Samsonov, N. Zenser, F. Zhang, H. Zhang, J. Fetter and D. Malkov, PLoS One, 2013, 8, e68391 CAS.
- M. Huang, F. Dorsey Jay, P. K. Epling-Burnette, R. Nimmanapalli, T. H. Landowski, L. B. Mora, G. Niu, D. Sinibaldi, F. Bai, A. Kraker, H. Yu, L. Moscinski, S. Wei, J. Djeu, W. S. Dalton, K. Bhalla, T. P. Loughran, J. Wu and R. Jove, Oncogene, 2002, 21, 8804–8816 CrossRef CAS PubMed.
- M. Levis, J. Allebach, K. F. Tse, R. Zheng, B. R. Baldwin, B. D. Smith, S. Jones-Bolin, B. Ruggeri, C. Dionne and D. Small, Blood, 2002, 99, 3885–3891 CrossRef CAS PubMed.
- D. H. Albert, P. Tapang, T. J. Magoc, L. J. Pease, D. R. Reuter, R. Q. Wei, J. Li, J. Guo, P. F. Bousquet, N. S. GhoreishiHaack, B. Wang, G. T. Bukofzer, Y. C. Wang, J. A. Stavropoulos, K. Hartandi, A. L. Niquette, N. Soni, E. F. Johnson, J. O. McCall, J. J. Bouska, Y. Luo, C. K. Donawho, Y. Dai, P. A. Marcotte, K. B. Glaser, M. R. Michaelides and S. K. Davidsen, Mol. Cancer Ther., 2006, 5, 995–1006 CrossRef CAS PubMed.
- M. A. Blaskovich, J. Sun, A. Cantor, J. Turkson, R. Jove and S. M. Sebti, Cancer Res., 2003, 63, 1270–1279 CAS.
- S. J. Schreiner, A. P. Schiavone and T. E. Smithgall, J. Biol. Chem., 2002, 277, 45680–45687 CrossRef CAS PubMed.
- A. Subramaniam, M. K. Shanmugam, E. Perumal, F. Li, A. Nachiyappan, X. Dai, S. N. Swamy, K. S. Ahn, A. P. Kumar, B. K. Tan, K. M. Hui and G. Sethi, Biochim. Biophys. Acta, 2013, 1835, 46–60 CAS.
- P. Rajendran, T. H. Ong, L. Chen, F. Li, M. K. Shanmugam, S. Vali, T. Abbasi, S. Kapoor, A. Sharma, A. P. Kumar, K. M. Hui and G. Sethi, Clin. Cancer Res., 2011, 17, 1425–1439 CrossRef CAS PubMed.
- S. Becker, B. Groner and C. W. Muller, Nature, 1998, 394, 145–151 CrossRef CAS PubMed.
- S. Reed, H. Li, C. Li and J. Lin, Biochem. Biophys. Res. Commun., 2011, 407, 450–455 CrossRef CAS PubMed.
- V. M. Shahani, P. Yue, S. Haftchenary, W. Zhao, X. Zhang, P. T. Gunning, J. Turkson, L. L. Julie, D. Ball and C. Nona, ACS Med. Chem. Lett., 2011, 2, 79–84 CrossRef CAS PubMed.
- Z. M. Nofal, E. A. Soliman, S. S. Abd El-Karim, M. I. El Zahar, A. M. Srour, S. Sethumadhavan and T. J. Maher, Acta Pol. Pharm., 2011, 68, 519–534 CAS.
- K. A. Z. Siddiquee, P. T. Gunning, J. Turkson, M. Glenn, W. P. Katt, S. Zhang, C. Schroeck, S. M. Sebti, R. Jove and A. D. Hamilton, ACS Chem. Biol., 2007, 2, 787–798 CrossRef CAS PubMed.
- K. F. Chen, W. T. Tai, J. W. Huang, C. Y. Hsu, W. L. Chen, A. L. Cheng, P. J. Chen and C. W. Shiau, Eur. J. Med. Chem., 2011, 46, 2845–2851 CrossRef CAS PubMed.
- D. S. Shin, H. N. Kim, K. D. Shin, Y. J. Yoon, S. J. Kim, D. C. Han and B. M. Kwon, Cancer Res., 2009, 69, 193–202 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3md00119a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.