Novel pyrazolo[1,5-a]pyridines as PI3K inhibitors: variation of the central linker group†
Jackie D.
Kendall
*ab,
Andrew J.
Marshall
a,
Anna C.
Giddens
a,
Kit Yee
Tsang‡
a,
Maruta
Boyd
a,
Raphaël
Frédérick§
a,
Claire L.
Lill
c,
Woo-Jeong
Lee
c,
Sharada
Kolekar
c,
Mindy
Chao
c,
Alisha
Malik
c,
Shuqiao
Yu
c,
Claire
Chaussade¶
bc,
Christina M.
Buchanan
bc,
Gordon W.
Rewcastle
ab,
Bruce C.
Baguley
ab,
Jack U.
Flanagan
ab,
William A.
Denny
ab and
Peter R.
Shepherd
bc
aAuckland Cancer Society Research Centre, School of Medical and Health Sciences, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: j.kendall@auckland.ac.nz
bMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
cDepartment of Molecular Medicine and Pathology, School of Medical and Health Sciences, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
First published on 30th October 2013
Abstract
As part of our investigation into the pyrazolo[1,5-a]pyridines as novel PI3K inhibitors, we report a range of analogues where the central linker portion of the molecule was varied while retaining the pyrazolo[1,5-a]pyridine and arylsulfonyl or arylcarbonyl groups. Isostere generating software BROOD was used to assist with producing ideas. The isoform selectivity of the compounds varied from pan-PI3K for compound 41 to p110α-selective for compound 58 or p110δ-selective for compound 57. The latter two compounds varied only in their sulphur oxidation state.
1. Introduction
Phosphoinositide 3-kinases (PI3Ks) are a group of lipid kinases which phosphorylate the 3′-hydroxyl group of phosphatidylinositol diphosphate (PIP2) to phosphatidylinositol triphosphate (PIP3). PIP3 then recruits protein kinase B (PKB, otherwise known as AKT) to the cell membrane where it is in turn phosphorylated and activated, leading to a cascade of cell signalling which controls a range of cellular processes like cell proliferation, growth and survival.1 PI3Ks are often over-expressed, and the lipid phosphatase PTEN (which dephosphorylates PIP3) is often deleted or inactivated in many cancer types, leading to increased levels of PIP3 and increased tumour survival.2The PI3Ks are split into three sub-families (class I, II and III), and class I is further split into class Ia and Ib based upon their mechanism of activation. The class Ia PI3Ks are heterodimeric, consisting of a catalytic subunit (p110α, p110β or p110δ) in complex with a regulatory subunit.1 PIK3CA, the gene encoding for p110α, is often over-expressed and mutated in many cancer types. Two of the most common of these mutations (E545K and H1047R) have been confirmed as activating mutations and hence increase levels of PIP3.3 Mutations in p110β and p110δ have not been reported.4,5 Inhibitors of PI3K, and in particular, selective inhibitors of p110α could prove to be an important new strategy in cancer treatment.6 A number of inhibitors of PI3K are currently in clinical trials, although none have yet reached market.7,8
We have shown previously that pyrazolo[1,5-a]pyridines such as 1 (ref. 9) and 2 (ref. 10) (Fig. 1) are potent and selective inhibitors of the p110α isoform of PI3K. However, since the sulfonohydrazide central portion of the molecule is a known structural alert,11 we were interested in replacing this with an alternative linker group. This has been investigated to a limited extent by Hayakawa et al.12 with their related imidazo[1,2-a]pyridine series of compounds, however PI3K isoform selectivity data was not reported for all compounds.
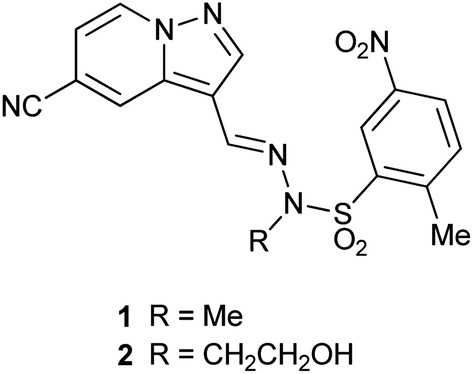 | ||
| Fig. 1 Structures of our previous pyrazolo[1,5-a]pyridine PI3K inhibitors. | ||
Molecular modelling of the pyrazolo[1,5-a]pyridines showed that the pyrazolo nitrogen formed a key hydrogen bond with Val851 of p110α;9 a residue interaction characteristic of many other PI3K inhibitor complexes.13 Furthermore the sulfonyl group formed a hydrogen bond with the p110α specific residue Gln859,9 and the nitro group was also critical for good potency and selectivity.10 We postulated that linking the pyrazolo[1,5-a]pyridine and nitroaryl sulfonyl moieties in an appropriate way would generate a p110α selective PI3K inhibitor with lower toxicity.
2. Results and discussion
2.1. Selection of sulfonohydrazide replacements
To increase our sulfonohydrazide replacement ideas, the bioisostere generating programme BROOD (v1.1.1; Openeye Scientific Software) was used select a set of alternative linking groups that fit the conformation of the sulfonohydrazide in a model of the pyrazolo[1,5-a]pyridines bound to p110α. Of the top 200 replacement fragments retrieved, only 51 unique isosteres were considered useful after visual inspection. Of these, only 42 hits contained a functional group we considered capable of hydrogen-bonding to the side chain amine of the p110α specific amino acid Gln859. The group was further narrowed down to 11 based on lipophilicity of the whole candidate compound, determined by comparison of the c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP values (calculated using ACD/PhysChem v. 7.10, Advanced Chemistry Development, Inc.) with the clogP value for 1 of 2.43. Compounds with clogP greater than 2.43 were discarded since such compounds would be more lipophilic and hence are likely to have lower aqueous solubility than 1. The group was finally reduced to 4 (Fig. 2) by assessment of the number of hydrogen-bond acceptors, in accordance with Lipinski rules.14
logP values (calculated using ACD/PhysChem v. 7.10, Advanced Chemistry Development, Inc.) with the clogP value for 1 of 2.43. Compounds with clogP greater than 2.43 were discarded since such compounds would be more lipophilic and hence are likely to have lower aqueous solubility than 1. The group was finally reduced to 4 (Fig. 2) by assessment of the number of hydrogen-bond acceptors, in accordance with Lipinski rules.14
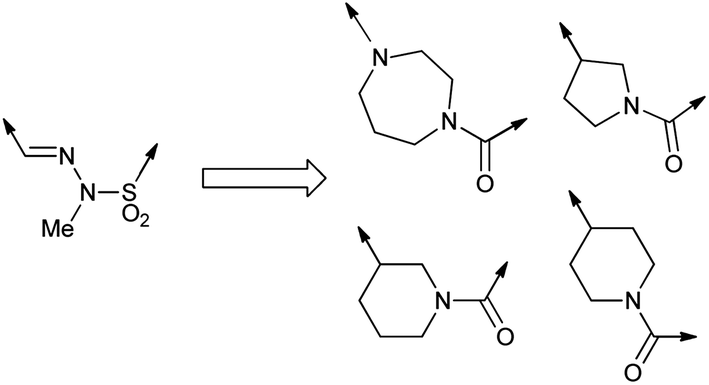 | ||
| Fig. 2 Filtered set of sulfonohydrazide isosteres generated by BROOD v1.1.1. | ||
We also chose some linker groups of our own devising: an allylic sulfone to mimic the shape of the sulfonohydrazide without containing the two nitrogens, a set of amine-containing linkers which could improve water solubility, and other heterocyclic linking groups.
2.2. Chemistry
Allylic sulfone 7 was made starting from pyrazolo[1,5-a]pyridine 3 (ref. 9) by iodination at the 3-position with N-iodosuccinimide (NIS) to form 4 (Scheme 1). Sulfinate 5 was reacted with acrolein to form aldehyde 6, which was subsequently reacted with the Grignard reagent derived from 4 followed by dehydration with MsCl and DBU to afford 7. The position of the double bond was confirmed by NOE interactions observed between the pyrazolo[1,5-a]pyridine C4–H (7.65 ppm) and alkene CH (6.52 ppm), and between the allylic CH2 (4.07 ppm) and aryl methyl protons (2.85 ppm).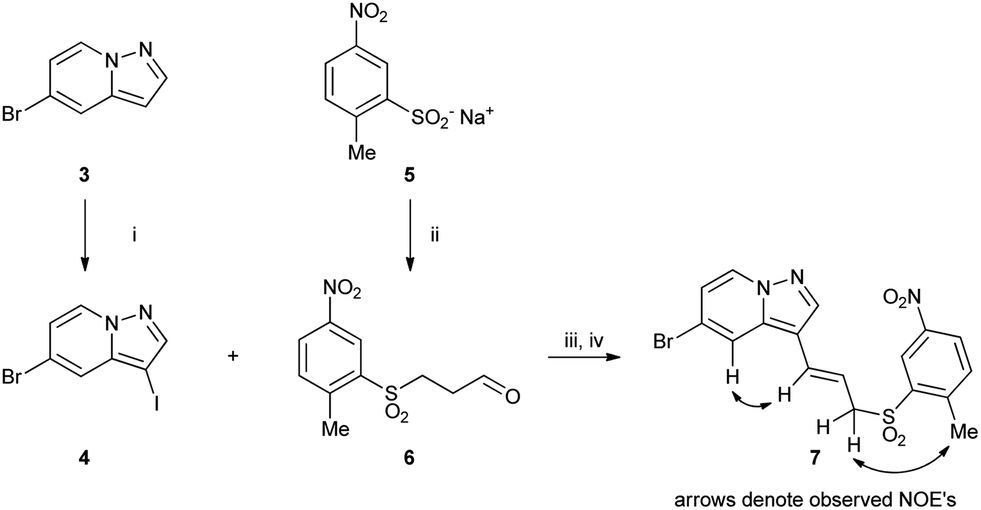 | ||
| Scheme 1 Reagents: (i) NIS, MeCN; (ii) acrolein, AcOH; (iii) iPrMgCl·LiCl, THF, −40 °C then 6; (iv) MsCl, DBU, MeCN. | ||
Cyclic amine derivatives 16–19 were made starting from amine 8 (ref. 9) (Scheme 2). Amine 8 was protected as either benzyl carbamate 9 or trifluoroacetamide 10, then iodinated at the 3-position to afford 11 and 12. Next, a palladium catalysed coupling with a cyclic vinyl boronate installed the amine ring. When 12 was used, the trifluoroacetamide protecting group was lost during this step. Hydrogenation over Pd/C then reduced the double bond from the amine ring and removed the benzyl carbamate to leave Boc protected compounds 13–15. Then diazotisation converted the free amine to a bromide and also removed the Boc group under the acidic conditions used, and finally sulfonylation or acylation afforded compounds 16–19.
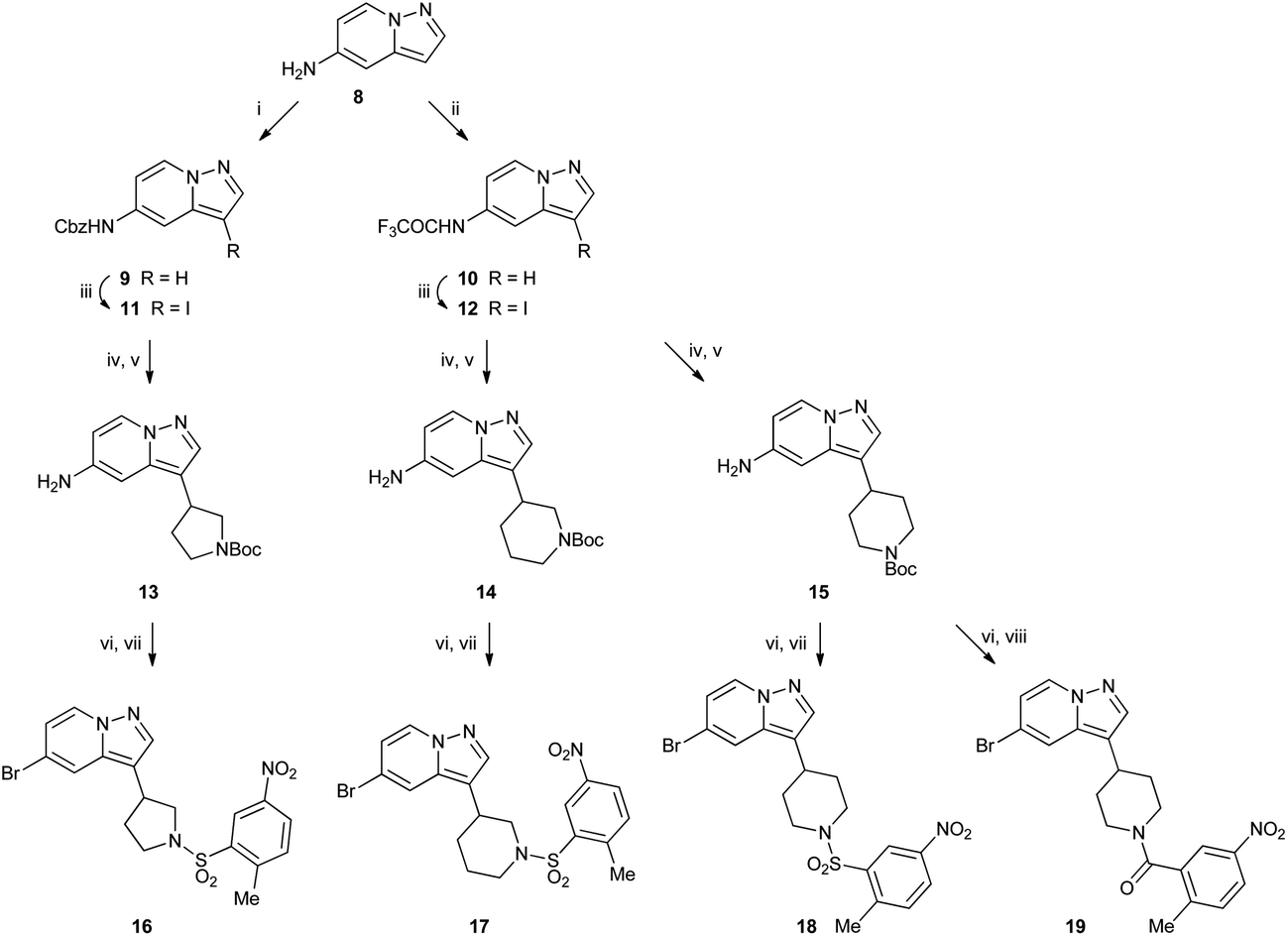 | ||
| Scheme 2 Reagents: (i) CbzCl, Na2CO3, acetone; (ii) TFAA, NEt3, CH2Cl2; (iii) NIS, MeCN; (iv) boronic ester, PdCl2(dppf), aq. Na2CO3, toluene, EtOH; (v) H2, Pd/C, DMF; (vi) NaNO2, H2O, 47% HBr then CuBr, 47% HBr; (vii) 2-methyl-5-nitrobenzenesulfonyl chloride, NEt3, CH2Cl2; (viii) 2-methyl-5-nitrobenzoyl chloride, NEt3, CH2Cl2. | ||
Aminomethylene-linked compounds 23a–h and 24a–g which contain a basic amine to increase aqueous solubility were made by reductive amination of aldehyde 20 (ref. 9) to afford amines 21a–h (Scheme 3). Removal of the Boc group was then followed by sulfonylation to give sulfonamides 23a–h or acylation to give carboxamides 24a–g.
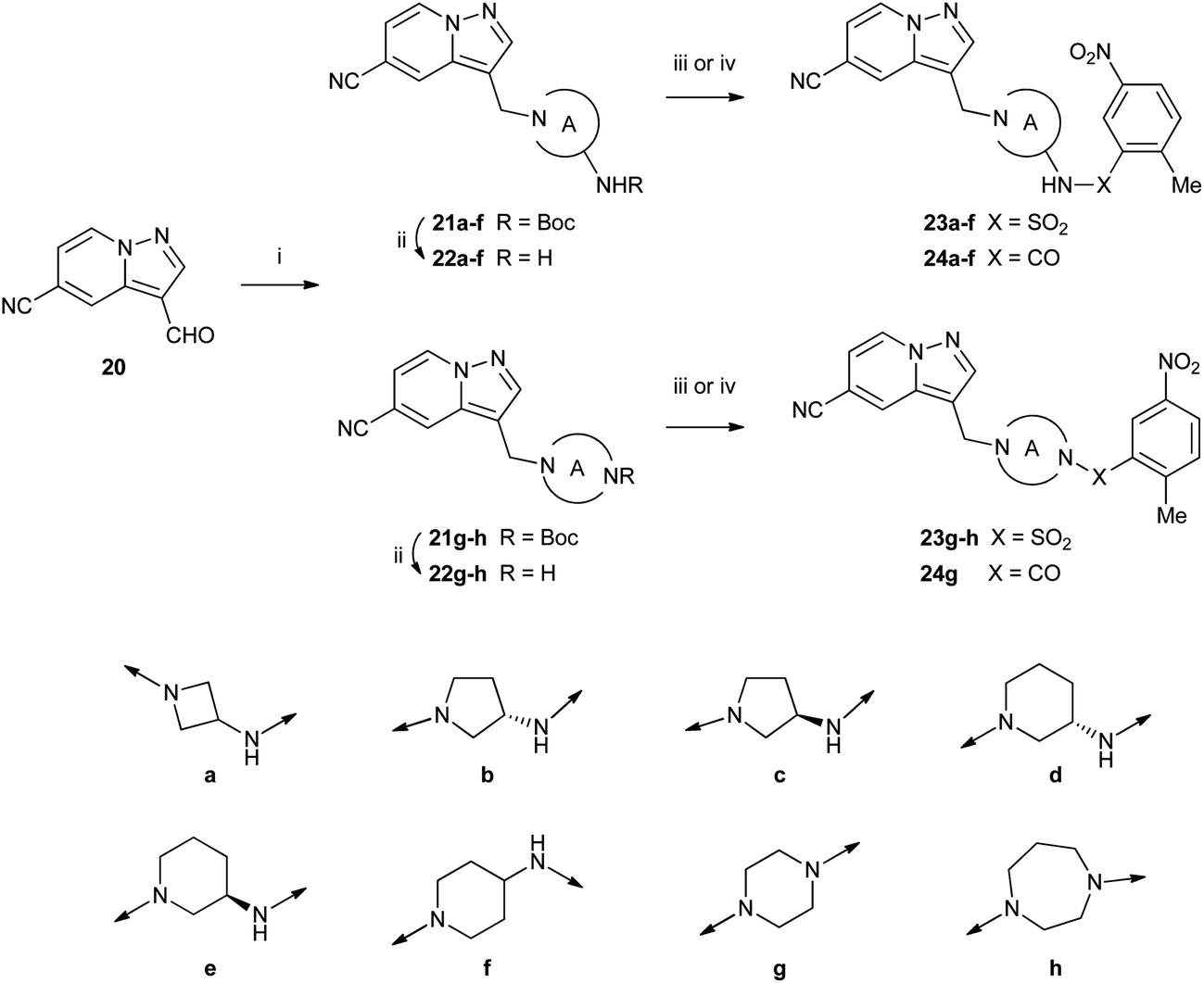 | ||
| Scheme 3 Reagents: (i) amine, AcOH, CH2Cl2 then Na(CN)BH3, EtOH; (ii) TFA, CH2Cl2; (iii) 2-methyl-5-nitrobenzenesulfonyl chloride, NEt3, DMF or MeCN or CH2Cl2; (iv) 2-methyl-5-nitrobenzoyl chloride, NEt3, DMF or MeCN or CH2Cl2. | ||
Thiazoles were made using a Hantzsch thiazole synthesis from an α-bromoketone and a thioamide. The syntheses of the required thioamides 29–34 are depicted in Scheme 4. Carboxylic acid 25 was converted to thioamide 29 by formation of the acid chloride followed by treatment with ammonia to give the primary amide, followed by thionation with Lawesson's reagent to give 29 in good yield. Benzylic chloride 26 was displaced with potassium cyanide to give the phenylacetonitrile intermediate, which was subsequently reacted with phosphorous pentasulfide in ethanol under mild conditions to give thioamide 30.15 Dithiocarbamate 31 was also prepared from 26 by firstly converting it to the more reactive bromide followed by displacement with a large excess of ammonium dithiocarbamate.16 Sulfonyl chloride 27 was reacted with cyanamide, followed by thiolysis using in situ generation of thiosulfuric acid to give thiourea 32. Thioamide 33 was accessed by displacement of bromoacetonitrile with the sulfinate salt of 27 to give the nitrile, which underwent thiolysis with hydrogen sulfide. Thiourea 34 was prepared from aniline 28 by reaction with benzoyl isothiocyanate followed by hydrolysis according to published routes.17,18
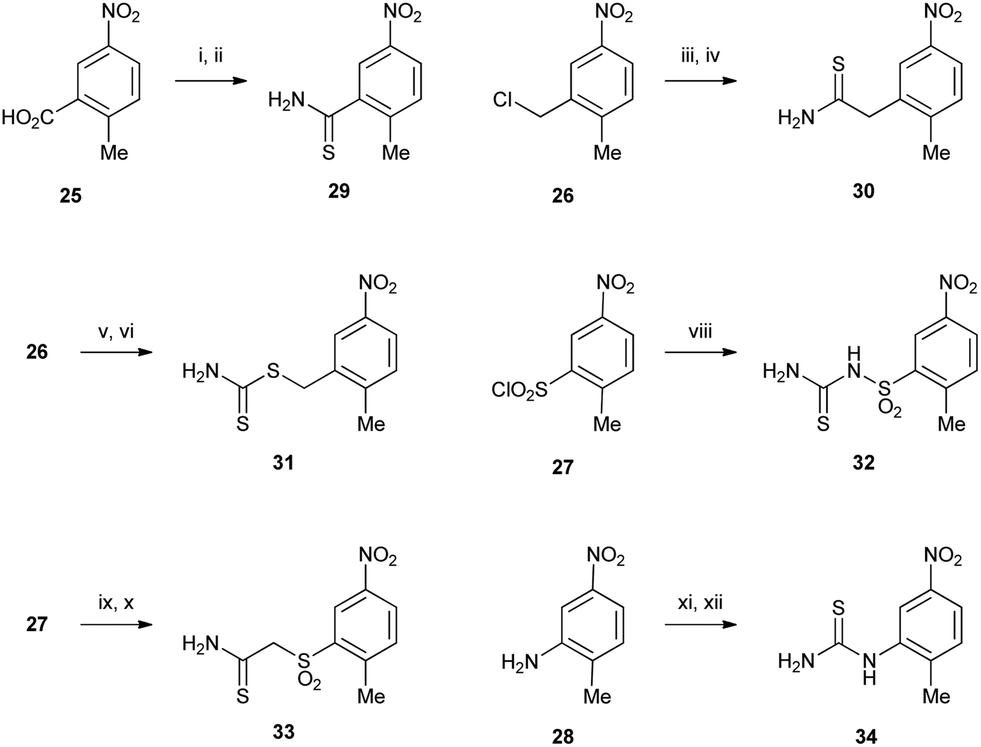 | ||
| Scheme 4 Reagents: (i) SOCl2 then aq. NH3, CH2Cl2; (ii) Lawesson's reagent, MeOH, reflux; (iii) KCN, DMSO; (iv) P2S5, EtOH, reflux; (v) LiBr, THF, reflux; (vi) H2NCS2− NH4+, EtOH, reflux; (viii) Na2NCN, H2O, 40 °C then Na2S2O3, 4.5 M aqueous H2SO4; (ix) Na2SO3, NaHCO3, H2O then bromoacetonitrile, DMF; (x) H2S, NEt3, THF; (xi) PhCONCS, acetone, reflux; (xii) KOH, MeOH, 40 °C. | ||
Methylketone 35 (ref. 9) was brominated under standard conditions to give versatile intermediate α-bromoketone 36, which underwent a modified Hantzsch reaction with thioamides 29–34 to give thiazoles 37–42 (Scheme 5). Thioether 39 was oxidized with MMPP (magnesium bis(monoperoxyphthalate)) to give sulfone 43. Sulfonamide 40 was methylated with methyl iodide under mild conditions to give two regioisomeric products 44 and 45. The sites of methylation were identified by NMR experiments. N-Methylsulfonamide 44 showed NOE interactions between the N-methyl protons (3.65 ppm) and those of the aryl methyl protons (2.67 ppm) and aryl C6–H (8.88 ppm), whereas N-methylthiazole 45 had NOE interactions between the N-methyl protons (3.46 ppm) and pyrazolo[1,5-a]pyridine C2–H (7.97 ppm) and C4–H (7.65 ppm).
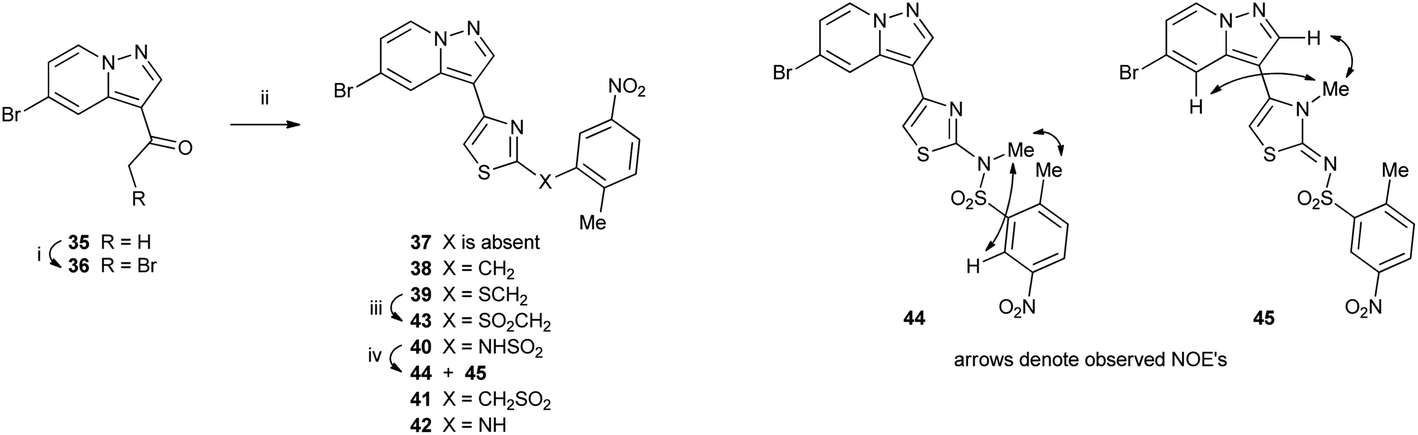 | ||
| Scheme 5 Reagents: (i) Br2, 33% HBr/AcOH, (ii) 29–34, EtOH; (iii) MMPP, CH2Cl2, EtOH, H2O, (iv) MeI, DMF. | ||
2-Thiothiazoles 47, 50, 52 and 55 were accessed via copper-mediated coupling19 of thione 46 with phenyl boronic acids to give moderate yields of the desired thioethers (Scheme 6). Thione 46 could be prepared in good yield by reaction of α-bromoketone 36 with fresh ammonium dithiocarbamate. Oxidation with either Oxone® or MMPP gave the sulfoxide and/or sulfone, depending on the substrate and choice of oxidant. Oxone® oxidation of 47 gave exclusively sulfoxide 48, and MMPP oxidation afforded sulfone 49. MMPP oxidation of the more sterically hindered 2-methylphenyl compound 50 gave only sulfoxide 51 under microwave heating, whereas 5-cyano-2-fluorophenyl compound 52 afforded a mixture of sulfoxide 53 and sulfone 54 after reaction with the same reagent at room temperature. In contrast, 55 which did not bear a 2-phenyl substituent gave exclusively sulfone 56, suggesting that the presence of an ortho-phenyl substituent had a detrimental effect on sulfur oxidation. Nucleophilic displacement of the activated 2- or 4-fluorobenzenes (53, 54 and 56) with alkyl amines at room temperature gave the corresponding amino derivatives 57–60.
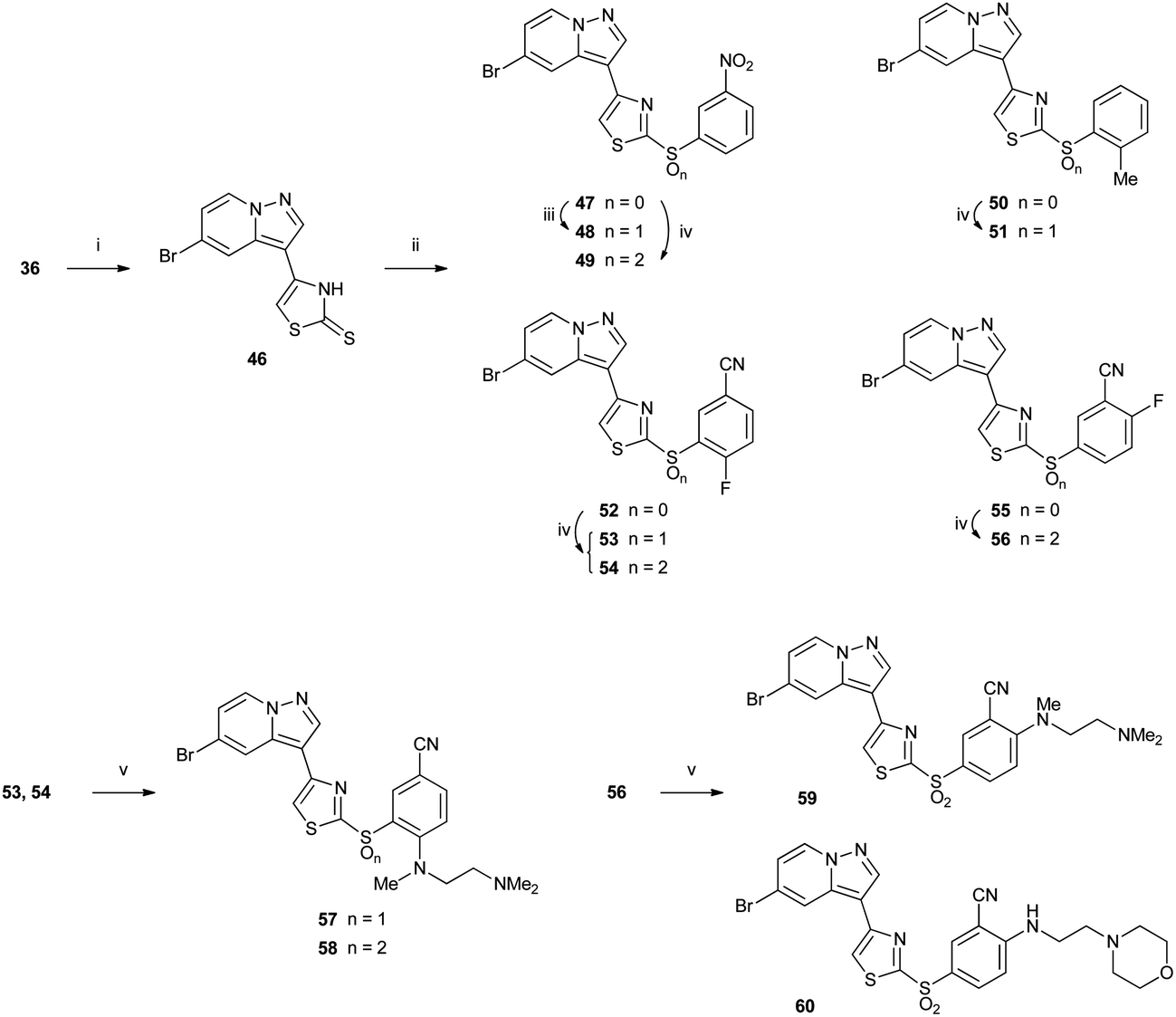 | ||
| Scheme 6 Reagents: (i) H2NCS2− NH4+, MeOH then AcOH, reflux; (ii) aryl boronic acid, Cu(OAc)2, 1,10-phenanthroline, 1,2-dichloroethane, reflux; (iii) Oxone®, solvent; (iv) MMPP, CH2Cl2, EtOH; (v) amine, THF. | ||
2.3. Biological activity
We have shown previously that both 5-bromo- and 5-cyanopyrazolo[1,5-a]pyridines are highly potent and selective inhibitors of the p110α isoform of PI3K,9,10 so these building blocks were chosen for further elaboration around the central hydrazone portion of the molecule. Compounds were assayed for their inhibition of the three class Ia isoforms of PI3K: p110α, p110β and p110δ (Table 1).| Cmpd | IC50 (μM) | Cmpd | IC50 (μM) | ||||
|---|---|---|---|---|---|---|---|
| p110α | p110β | p110δ | p110α | p110β | p110δ | ||
| a Data from ref. 9. | |||||||
| 1 | 0.0009 | 0.046 | 0.049 | 37 | >1 | >1 | >1 |
| 7 | 0.14 ± 0.05 | >1 | 3.35 ± 0.41 | 38 | >1 | >1 | >1 |
| 16 | 3.00 ± 0.80 | >1 | >1 | 39 | >1 | >1 | >1 |
| 17 | 0.59 ± 0.20 | >1 | >1 | 40 | 1.26 ± 0.41 | >1 | >1 |
| 18 | >1 | >1 | >1 | 41 | 0.24 ± 0.13 | 0.14 ± 0.01 | 0.09 ± 0.05 |
| 19 | >1 | >1 | >1 | 42 | >1 | >1 | >1 |
| 23a | >1 | >1 | >1 | 43 | >1 | >1 | >1 |
| 23b | >1 | >1 | >1 | 44 | >1 | >1 | 1.55 ± 0.27 |
| 23c | >1 | >1 | >1 | 45 | >1 | >1 | >1 |
| 23d | >1 | >1 | >1 | 47 | >1 | >1 | >1 |
| 23e | >1 | >1 | >1 | 48 | 0.74 ± 0.01 | 4.42 ± 1.83 | 0.32 ± 0.17 |
| 23f | >1 | >1 | >1 | 49 | 0.63 ± 0.18 | 5.45 ± 0.30 | 0.25 ± 0.05 |
| 23g | >1 | >1 | >1 | 51 | 1.28 ± 0.22 | >1 | >1 |
| 23h | >1 | >1 | >1 | 53 | 0.30 ± 0.01 | >1 | 0.20 ± 0.03 |
| 24a | >1 | >1 | >1 | 54 | 0.72 ± 0.02 | >1 | 0.44 ± 0.03 |
| 24b | >1 | >1 | >1 | 56 | 0.17 ± 0.02 | >1 | 0.15 ± 0.12 |
| 24c | >1 | >1 | >1 | 57 | 4.03 ± 1.65 | 4.34 ± 1.76 | 0.25 ± 0.03 |
| 24d | >1 | >1 | >1 | 58 | 0.37 ± 0.15 | 3.56 ± 0.08 | 2.00 ± 0.48 |
| 24e | >1 | >1 | >1 | 59 | 2.47 ± 0.69 | >1 | 0.64 ± 0.14 |
| 24f | >1 | >1 | >1 | 60 | 2.13 ± 0.29 | >1 | 0.24 ± 0.13 |
| 24g | >1 | >1 | >1 | ||||
Replacement of the hydrazone with allylic sulfone 7, where the hydrazone nitrogens atoms were replaced by carbons, was 150-fold less active against p110α than 1, while retaining some selectivity over p110β and p110δ. None of the cyclic amine derivatives 16–19, 23a–h and 24a–g had IC50 less than 1 μM for any of the class Ia PI3K isoforms, with the sole exception of 3-substituted piperidine 17 with a modest p110α IC50 of 0.59 μM. These compounds were not investigated further, and we instead focussed our efforts on aromatic heterocyclic linker groups.
Of the 2-methyl-5-nitrophenyl compounds 37–45, only 41 had p110α IC50 less than 1 μM, however this compound lost all selectivity for p110α over the other class Ia PI3K isoforms (p110α/β/δ IC50 0.24/0.14/0.09 μM). By comparison of 3-nitrophenyl compounds 47, 48 and 49, the addition of at least one oxygen on sulfur was necessary for p110α IC50 less than 1 μM although there was little difference between sulfoxide 48 and sulfone 49 (p110α IC5048: 0.74 μM and 49: 0.63 μM). Both 48 and 49 were less than 10-fold selective over p110β but were slightly more potent against p110δ than p110α.
The removal of the nitro group (51) decreased p110α activity in comparison to 48 by 2-fold. Benzonitrile derivatives 53, 54 and 56–60 all demonstrated PI3K IC50 of less than 1 μM against at least one of the class Ia isoforms, however the selectivity varied with the nature of the other phenyl substituent and sulfur oxidation state. Fluorophenyl compounds 53, 54 and 56 all had p110α IC50 under 1 μM and were all similarly active against p110δ. In contrast, amine-containing compounds 57, 59 and 60 all demonstrated modest selectivity, albeit for p110δ over p110α (by 16-fold, 4-fold and 9-fold, respectively). In contrast 58, the sulfone analogue of sulfoxide 57, was 5-fold selective for p110α over p110δ.
3. Conclusions
A range of pyrazolo[1,5-a]pyridines were prepared where the central portion of the molecule was varied with cyclic and acyclic, aromatic and aliphatic linking groups to investigate the PI3K potencies. We found that the thiazole group was preferred, however the isoform selectivity could be varied by changes of the sulfur oxidation state and phenyl ring substituents. These compounds were not sufficiently potent in comparison with our earlier sulfonohydrazides, and were not investigated further.4. Experimental
4.1. Molecular modelling
The bioisostere generating programme BROOD (v1.1.1; Openeye Scientific Software) was used with a 3-dimensional model of the sulfonohydrazide core based on the top ranked predicted binding mode of N′-((5-bromopyrazolo[1,5-a]pyridin-3-yl)methylene)-N,2-dimethyl-5-nitrobenzenesulfonohydrazide (compound 5m in ref. 9) in the ATP binding site of p110α. BROOD settings were kept at default values, and the electrostatic tanimoto similarity value was calculated. The f5 multi-conformer fragment library consisting of 3-dimensional models of 140000 fragments was searched. The electrostatic tanimoto similarity hit list was ranked according to the combined electrostatic shape and electrostatic tanimoto values then visually inspected. The 2D scheme of the full compounds was generated for further analysis.
4.2. Chemistry
Methods and characterisation for all novel compounds are detailed in the ESI.†4.3. Enzyme assays
The Class Ia PI3K assays were performed using a basic thin layer chromatography technique, as described previously.20 The p110δ/p85 PI3K was from Upstate and the p110α/p85 and p110β/p85 isoforms were prepared as described previously.20 Reactions were made containing 0.1 μg recombinant enzyme, 10 μg L-α-phosphatidylinositol, inhibitor (DMSO only or DMSO + inhibitor to a final concentration of 1%), 2× lipid kinase buffer (40 mM Tris–HCl pH 7.4, 200 mM NaCl, 1 mM EDTA), and activated upon the addition of an ATP mix (5 mM MgCl2, 100 μM ATP, 0.1 μL [γ33P]ATP). Reactions were incubated at room temperature for 1 h following which the reactions were stopped by the addition of 1 M HCl. The lipids were then extracted using a two step procedure. Firstly, 200 μL of chloroform:methanol (1:1) was added, the biphasic reactions mixed and centrifuged briefly, and the inorganic phase was removed and discarded. Following this 80 μL of methanol:hydrochloric acid (1:1) was added and the same procedure followed. Next, 70 μL of the organic phase was transferred to a clean 1.6 mL tube and the reactions were dried using a speed vac, with no heating, for 30 min. The reactions were spotted onto TLC plates (Merck Ltd) and developed for 1 h in 1-propanol:2 M acetic acid (13:7). The TLC plates were then dried at room temperature and quantified using a phosphorimager (StormImager, Amersham). Nine inhibitor concentrations were used to determine the IC50. Each experiment was performed twice and the average IC50 value used.
Acknowledgements
The authors would like to thank the Health Research Council of New Zealand, Maurice Wilkins Centre for Molecular Biodiscovery, and Pathway Therapeutics for their financial assistance.References
- S. Shuttleworth, F. Silva, C. Tomassi, A. Cecil, T. Hill, H. Rogers and P. Townsend, Prog. Med. Chem., 2009, 48, 81–131 CrossRef CAS.
- M. C. Hollander, G. M. Blumenthal and P. A. Dennis, Nat. Rev. Cancer, 2011, 11, 289–301 CrossRef CAS PubMed.
- C. Chaussade, K. Cho, C. Mawson, G. W. Rewcastle and P. R. Shepherd, Biochem. Biophys. Res. Commun., 2009, 381, 577–581 CrossRef CAS PubMed.
- J. M. Cleary and G. I. Shapiro, Curr. Oncol. Rep., 2010, 12, 87–94 CrossRef CAS PubMed.
- P. Liu, H. Cheng, T. M. Roberts and J. J. Zhao, Nat. Rev. Drug Discovery, 2009, 8, 627–644 CrossRef CAS PubMed.
- S. Jamieson, J. U. Flanagan, S. Kolekar, C. Buchanan, J. D. Kendall, W.-J. Lee, G. W. Rewcastle, W. A. Denny, R. Singh, J. Dickson, B. C. Baguley and P. R. Shepherd, Biochem. J., 2011, 438, 53–62 CrossRef CAS PubMed.
- P. Wu and Y. Hu, Med. Chem. Commun., 2012, 3, 1337–1355 RSC.
- B. Markman, J. J. Tao and M. Scaltriti, Curr. Pharm. Des., 2013, 19, 895–906 CrossRef CAS.
- J. D. Kendall, P. D. O'Connor, A. J. Marshall, R. Frédérick, E. S. Marshall, C. L. Lill, W.-J. Lee, S. Kolekar, M. Chao, A. Malik, S. Yu, C. Chaussade, C. Buchanan, G. W. Rewcastle, B. C. Baguley, J. U. Flanagan, S. M. F. Jamieson, W. A. Denny and P. R. Shepherd, Bioorg. Med. Chem., 2012, 20, 69–85 CrossRef CAS PubMed.
- J. D. Kendall, A. C. Giddens, K. Y. Tsang, R. Frédérick, E. S. Marshall, R. Singh, C. L. Lill, W.-J. Lee, S. Kolekar, M. Chao, A. Malik, S. Yu, C. Chaussade, C. Buchanan, G. W. Rewcastle, B. C. Baguley, J. U. Flanagan, S. M. F. Jamieson, W. A. Denny and P. R. Shepherd, Bioorg. Med. Chem., 2012, 20, 58–68 CrossRef CAS PubMed.
- S. D. Nelson, J. Med. Chem., 1982, 25, 753–765 CrossRef CAS.
- M. Hayakawa, H. Kaizawa, K.-i. Kawaguchi, N. Ishikawa, T. Koizumi, T. Ohishi, M. Yamano, M. Okada, M. Ohta, S.-i. Tsukamoto, F. I. Raynaud, M. D. Waterfield, P. Parker and P. Workman, Bioorg. Med. Chem., 2007, 15, 403–412 CrossRef CAS PubMed.
- A. Berndt, S. Miller, O. Williams, D. D. Le, B. T. Houseman, J. I. Pacold, F. Gorrec, W.-C. Hon, Y. Liu, C. Rommel, P. Gaillard, T. Rückle, M. K. Schwarz, K. M. Shokat, J. P. Shaw and R. L. Williams, Nat. Chem. Biol., 2010, 6, 117–124 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- B. Kaboudin and D. Elhamifar, Synthesis, 2006, 224–226 CrossRef CAS PubMed.
- M. Yokoyama and H. Hatanaka, Synthesis, 1985, 891–892 CrossRef CAS PubMed.
- C. R. Rasmussen, F. J. Villani, L. E. Weaner, B. E. Reynolds, A. R. Hood, L. R. Hecker, S. O. Nortey, A. Hanslin, M. J. Costanzo, E. T. Powell and A. J. Molinari, Synthesis, 1988, 456–459 CrossRef CAS.
- S. Mahboobi, S. Dove, A. Sellmer, M. Winkler, E. Eichhorn, H. Pongratz, T. Ciossek, T. Baer, T. Maier and T. Beckers, J. Med. Chem., 2009, 52, 2265–2279 CrossRef CAS PubMed.
- H. Prokopcová and C. O. Kappe, J. Org. Chem., 2007, 72, 4440–4448 CrossRef PubMed.
- C. Chaussade, G. W. Rewcastle, J. D. Kendall, W. A. Denny, K. Cho, L. M. Gronning, M. L. Chong, S. H. Anagnostou, S. P. Jackson, N. Daniele and P. R. Shepherd, Biochem. J., 2007, 404, 449–458 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3md00221g |
| ‡ Present address: Universal Display Corporation, Hong Kong. |
| § Present address: Université Catholique de Louvain (UCL), Louvain Drug Research Institute, Medicinal Chemistry Research Group, 73 Avenue E. Mounier, bte B1.73.10, B-1200 Brussels, Belgium. |
| ¶ Present address: Centre Hospitalier Universitaire de Nice, Nice, France. |
| This journal is © The Royal Society of Chemistry 2014 |