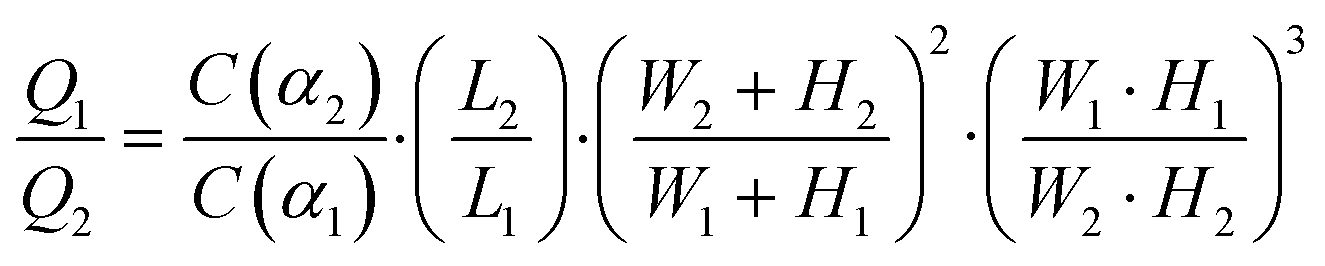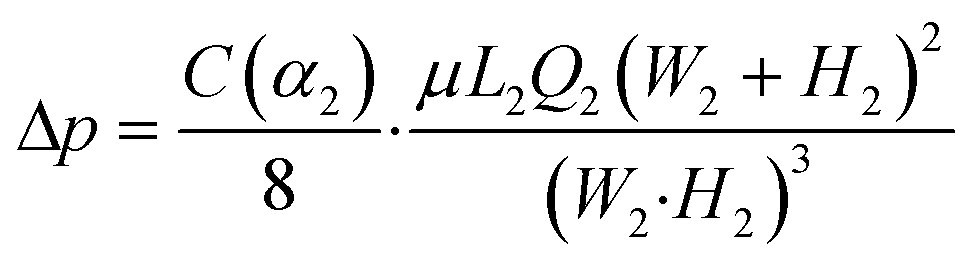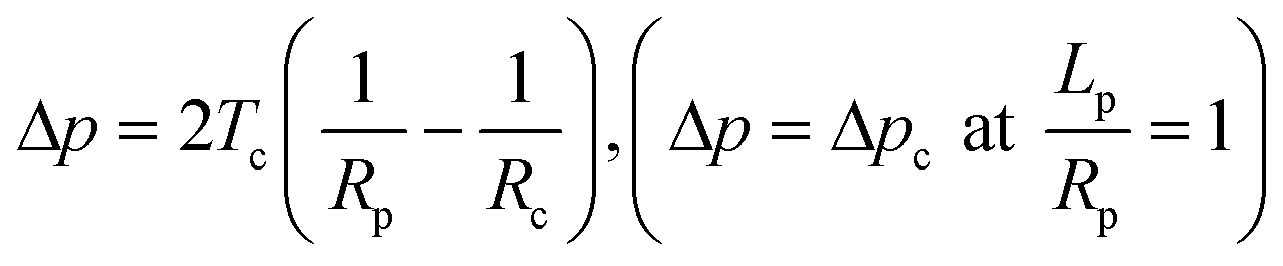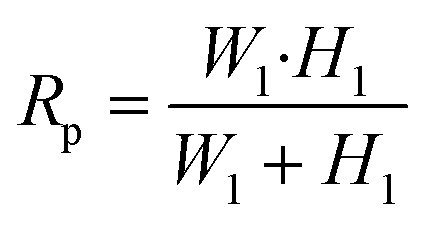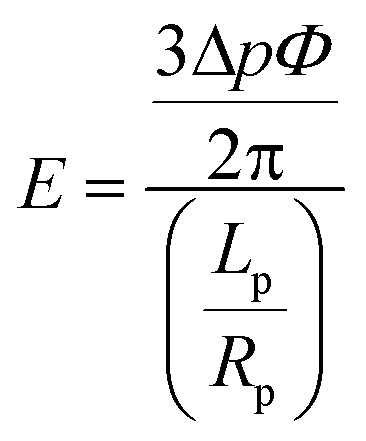DOI:
10.1039/C4LC01218F
(Paper)
Lab Chip, 2015,
15, 264-273
A microfluidic pipette array for mechanophenotyping of cancer cells and mechanical gating of mechanosensitive channels†
Received
14th October 2014
, Accepted 16th October 2014
First published on 16th October 2014
Abstract
Micropipette aspiration measures the mechanical properties of single cells. A traditional micropipette aspiration system requires a bulky infrastructure and has a low throughput and limited potential for automation. We have developed a simple microfluidic device which is able to trap and apply pressure to single cells in designated aspiration arrays. By changing the volume flow rate using a syringe pump, we can accurately exert a pressure difference across the trapped cells for pipette aspiration. By examining cell deformation and protrusion length into the pipette under an optical microscope, several important cell mechanical properties, such as the cortical tension and the Young's modulus, can be measured quantitatively using automated image analysis. Using the microfluidic pipette array, the stiffness of breast cancer cells and healthy breast epithelial cells was measured and compared. Finally, we applied our device to examine the gating threshold of the mechanosensitive channel MscL expressed in mammalian cells. Together, the development of a microfluidic pipette array could enable rapid mechanophenotyping of individual cells and for mechanotransduction studies.
Introduction
Several tools have been developed to study cell or molecular mechanics,1 including atomic force microscopy,2 magnetic twisting cytometry,3 acoustic tweezing cytometry,4 optical tweezers,5 micropipette aspiration,6 shear-flow,7 and cell stretching.8 Within these specialized techniques, optical tweezers and micropipette aspiration are two major approaches to study biomechanics at a single-cell level, which rely on the observation of cell deformation upon force perturbation to extract mechanical properties of single cells. Optical tweezer systems have been successfully developed and extensively applied to manipulate cells to study single-cell mechanics since the 1980s.9 In recent years, the advancement of micro/nanofabrication of integrated optical fibers, waveguides, and photonic crystals within microfluidic channels has enabled the exciting development and applications of optofluidic devices in single-cell trapping/manipulation/sorting or biological/chemical detection.10,11 The fabrication of integrated optofluidic devices usually involves a standard semiconductor manufacturing process, which can be very costly and time-consuming. The operation of optofluidic devices may also involve sophisticated optical setup and experimentation.
Micropipette aspiration relies on suction pressure exerted on a single cell to study its mechanical properties. A typical micropipette aspiration system consists of a pressure generator (typically a pair of water columns or a manometer), a pressure transducer, a glass micropipette, an x–y–z micro-manipulator and an optical microscope. During operation, the micro-manipulator is positioned in close proximity to a cell in the cell suspension chamber. Negative suction pressure generated by downward displacement of the water reservoir is exerted to the single cell aspirated into the micropipette tip and measured by a pressure transducer. Several mechanical properties can be measured based on the cell size, the magnitude of deformation, the size of the micropipette and the applied pressure. A traditional micropipette aspiration system requires skilled manual operation. Environmental factors cause fluctuation in the cell suspension, making manipulation of the micropipette challenging to approach cells for aspiration. Furthermore, the cell has to be well-positioned at the focal plane of an optical microscope. The difficulty to systematically determine the “end-point” by manual observation of the cell boundary results in random errors. A recent study has reported an effort to automate micropipette systems for single-cell mechanical characterization.12 Yet, sophisticated computer vision position control, motorized translation stage and pressure system with real-time visual feedback have to be implemented for operation. Water evaporation in the reservoir and mechanical fluctuation of external tubing/connections can still hinder the accuracy of measurement. Traditional micropipette aspiration systems can only study a single cell one at a time. Typically, it takes about 10 minutes to complete one measurement. This highly limits its throughput and capability for time-sensitive live-cell studies.
To address the problems associated with conventional micropipette aspiration, several microfluidic devices have been developed to perform micropipette aspiration with better measurement accuracy, improved throughput rate, and in an automated manner. Dudani et al. implemented a pinched-flow mechanism to hydrodynamically stretch single cells with an exceptionally high throughput of 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells s−1 (ref. 13). Cross-flows from branched side channels impinge on cells flowing along the main microfluidic channel. It acts as a virtual hydrodynamic micropipette to exert stress on cells and cause their deformations. Since there is no physical contact between the cells and the channel surface, the effect of cell adhesion and the chance of clogging are minimized. Another work from the same group measured cell deformability of leukocytes, malignant cells in pleural effusions, and pluripotent stem cells using an extensional flow from both directions.14 Shelby et al. constructed microfluidic channels with different constricted channel widths to evaluate Plasmodium falciparum-infected red blood cells at different stages based on deformation and clogging.15 Rosenbluth et al. developed a network of bifurcating microfluidic channels and used the transit time as a quantitative metric to measure cell stiffness of neutrophils with hemotalogic diseases.16 Using a similar principle, others have examined breast cancer cells in constricted microfluidic channels. The transit time and velocity were recorded as a measure of deformability to compare the stiffness of cancer cells with different metastatic potentials.17–19 Mak et al. developed a microfluidic device with serial subnucleus-scaled constrictions to study the cancer invasion process upon a sequence of deformation and relaxation events.20,21 The transit time required for the cancer cells to migrate through these constrictions and the cancer cells' strain rates and viscoelasticities were studied. One drawback with this device is that it does not require any external actuation system and hence the pressure difference is not quite precisely controlled. Guo et al. implemented a multi-layer microfluidic micropipette aspiration device for measuring single-cell deformability.22 Based on multi-layer soft lithography,23 the microfluidic flow channels were integrated with control valves for cell infusion, measurement, and removal. Single cells were flowed into a series of different sized constrictions, and cortical tensions of single cells were measured based on Haines' jump principle. This device has been used to measure cortical tensions of normal and abnormal red blood cells damaged by oxidation and parasitized by Plasmodium falciparum.24,25 These microfluidic devices measure single-cell mechanical properties by flowing cells through constrictions and observing their magnitudes of deformation. This one-time measurement approach has an advantage of higher throughput. However, more complex cell dynamic behavior and mechanical heterogeneity of different components, such as the nucleus, the cytosol, and the cytoskeletal network within a cell cannot be captured by this approach. Time lapse studies of inter- or intracellular transport phenomena are difficult to be implemented in the same single cell. The accuracy of measurement is affected by the resolution of an external pressure generator. Yet, environmental fluctuation, such as water evaporation in the inlet reservoir and leakage in the connections, can lead to measurement inaccuracy.
000 cells s−1 (ref. 13). Cross-flows from branched side channels impinge on cells flowing along the main microfluidic channel. It acts as a virtual hydrodynamic micropipette to exert stress on cells and cause their deformations. Since there is no physical contact between the cells and the channel surface, the effect of cell adhesion and the chance of clogging are minimized. Another work from the same group measured cell deformability of leukocytes, malignant cells in pleural effusions, and pluripotent stem cells using an extensional flow from both directions.14 Shelby et al. constructed microfluidic channels with different constricted channel widths to evaluate Plasmodium falciparum-infected red blood cells at different stages based on deformation and clogging.15 Rosenbluth et al. developed a network of bifurcating microfluidic channels and used the transit time as a quantitative metric to measure cell stiffness of neutrophils with hemotalogic diseases.16 Using a similar principle, others have examined breast cancer cells in constricted microfluidic channels. The transit time and velocity were recorded as a measure of deformability to compare the stiffness of cancer cells with different metastatic potentials.17–19 Mak et al. developed a microfluidic device with serial subnucleus-scaled constrictions to study the cancer invasion process upon a sequence of deformation and relaxation events.20,21 The transit time required for the cancer cells to migrate through these constrictions and the cancer cells' strain rates and viscoelasticities were studied. One drawback with this device is that it does not require any external actuation system and hence the pressure difference is not quite precisely controlled. Guo et al. implemented a multi-layer microfluidic micropipette aspiration device for measuring single-cell deformability.22 Based on multi-layer soft lithography,23 the microfluidic flow channels were integrated with control valves for cell infusion, measurement, and removal. Single cells were flowed into a series of different sized constrictions, and cortical tensions of single cells were measured based on Haines' jump principle. This device has been used to measure cortical tensions of normal and abnormal red blood cells damaged by oxidation and parasitized by Plasmodium falciparum.24,25 These microfluidic devices measure single-cell mechanical properties by flowing cells through constrictions and observing their magnitudes of deformation. This one-time measurement approach has an advantage of higher throughput. However, more complex cell dynamic behavior and mechanical heterogeneity of different components, such as the nucleus, the cytosol, and the cytoskeletal network within a cell cannot be captured by this approach. Time lapse studies of inter- or intracellular transport phenomena are difficult to be implemented in the same single cell. The accuracy of measurement is affected by the resolution of an external pressure generator. Yet, environmental fluctuation, such as water evaporation in the inlet reservoir and leakage in the connections, can lead to measurement inaccuracy.
In this work, we present a novel microfluidic pipette array (μFPA) device, which enables the study of cell mechanical properties and cellular transport phenomena in a parallel manner. Our μFPA device infrastructure is very simple and it does not require a pneumatic control system and integrated control valves for cell loading, measurement, and cell removal. Our device is able to autonomously trap single cells to designated chamber arrays and performs aspiration measurement using a syringe pump and an optical microscope. In this paper, we first explain the design and operation of our μFPA device through theoretical modeling, numerical simulations, and experimental demonstrations. Then, as a demonstration of its utility, we applied our μFPA device to measure mechanical properties of single cells. Deformability measurement of human breast non-tumorigenic and metastatic cancer cells using our μFPA device showed that breast cancer cells were less stiff than their healthy counterparts. Lastly, we applied our μFPA device to study the gating properties of the mechanosensitive channel of large conductance (MscL) expressed in mammalian cells.
Materials and methods
Design and operation
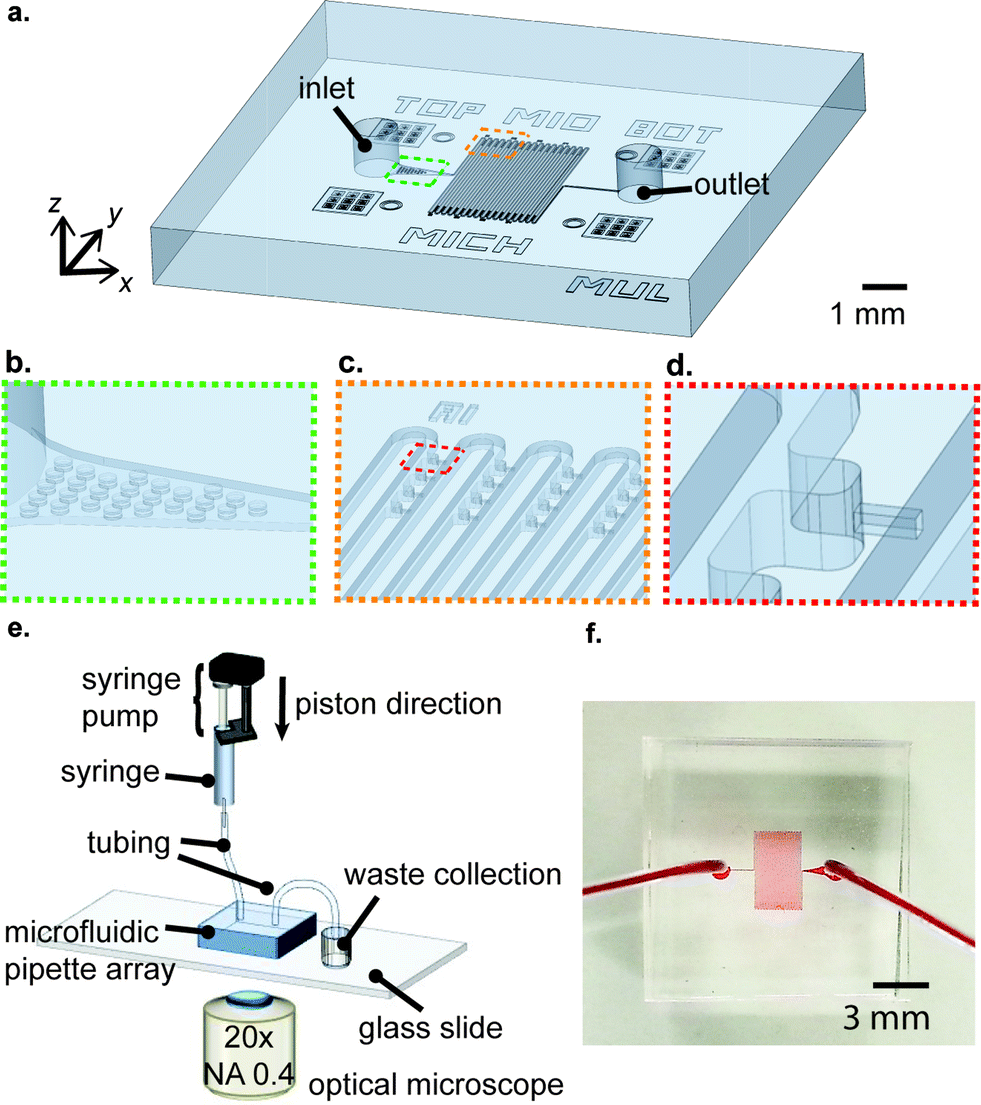
The infrastructure of the μFPA device is shown in Fig. 1(a). The device is composed of a main meandering microfluidic channel and a pair of inlet/outlet. Next to the inlet, there is an array of posts, which serve to block aggregated cell clumps or large debris, preventing them from entering the main microfluidic channel (Fig. 1(b)). The trapping/aspiration arrays are positioned near the turnings of the main microfluidic channel. This arrangement enhances trapping efficiency and maximizes the magnitude of pressure exerted on the trapped cells (Fig. 1(c)). In our current implementation, the device has 16 columns and each column has 4 trapping/aspiration chambers at each side, yielding 128 trapping/aspiration chambers in total. The aspiration pipette was constructed along the centerline of the trapping chamber such that the trapped cells were levitated from the channel bottom upon aspiration (Fig. 1(d)). For the operation of our μFPA device, the inlet was connected to a syringe pump (Fusion 400, Chemyx) filled with a cell suspension. The outlet was connected to a waste collection. The μFPA device was mounted under an optical microscope for observation of cell deformation (Fig. 1(e)). A picture of our μFPA device is illustrated in Fig. 1(f).
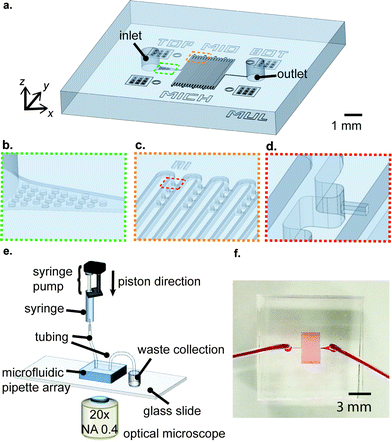 |
| | Fig. 1 Overview and operation of the μFPA device. (a) Overall schematic, (b) filtering unit, (c) aspiration chamber array, and (d) single aspiration pipette unit. (e) Experimental setup for device operation and (f) a picture of the device. | |
Device fabrication
The microfabrication process of the microfluidic micropipette device was based on the polydimethylsiloxane (PDMS) soft-lithography technique. The microfluidic device was composed of two pieces of PDMS substrates which were aligned and bonded together. The PDMS substrates were molded by SU-8 patterned silicon wafers. The silicon mold for the top substrate was made by double-layer SU-8 patterning, in which the first patterned layer defines the cross section of the micropipettes, trapping structures and microfluidic channel, while the second layer defines the trapping structures and the main microfluidic channel. The silicon mold for the bottom substrate was made by single-layer SU-8 patterning, which defines trapping structures and the main microfluidic channel. This fabrication method allowed us to manufacture symmetric features. A photomask with a resolution of 8 μm was manufactured by inkjet printing of transparency which was produced by CAD/Art Services (Bandon, OR, USA). The feature of the SU-8 pattern of the bottom silicon wafer resembled the mirror image of the second SU-8 layer patterned on the top wafer. To start with, both silicon wafers were dehydrated by hotplate baking at 150 °C for 5 min to promote photoresist adhesion. In patterning of the first layer of the top substrate, SU-8 2010 was spin coated on the silicon wafer at 5000 rpm, which gave a thickness of 8 μm. For the second layer, SU-8 2010 was applied with a spinning speed of 2000 rpm and gave a total thickness of 15 μm. After patterning and development of the first SU-8 layer, the silicon wafer was hard-baked at 150 °C for 30 minutes to ensure that the SU-8 pattern is fully cured before the application of the second layer. For the bottom substrate, a single layer of SU-8 2010 was patterned with a thickness of 15 μm under a spinning speed of 1500 rpm. The thickness of SU-8 patterns on both wafers was measured with a profilometer. After silanization with trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma-Aldrich) in a desiccator, both silicon molds were casted with PDMS (Sylgard-184) with a mixing ratio of 10:1 (base:curing agent). Two PDMS substrates were then cured at 60 °C overnight and de-molded from the wafers afterwards. Inlet/outlet holes of 1 mm diameter were punched on the top PDMS substrate. Top and bottom PDMS substrates were aligned and bonded under a customized alignment platform under an optical microscope. A schematic summary for the fabrication process can be found in the ESI† Fig. S1.
Simulation
The fluid flow of our μFPA device was numerically studied using COMSOL 4.4 (COMSOL Multiphysics). A three-dimensional model was built which represents the repeating unit of the microfluidic device to save computational power. The velocity and pressure fields were computed using the laminar flow module. The problem is modeled as incompressible flow, including the inertial term. Water is selected as the material property for the entire domain. No slip boundary conditions were imposed to all walls except for inlet and outlet. Laminar inflow conditions were imposed with a zero entrance length in the channel inlet for different flow rates. For the channel outlet, constant pressure (P = 0) was imposed with backflow suppression. In the cell trapping simulation, particle tracing for fluid flow is coupled with the laminar flow module for the time-dependent study. Stokes' drag law was imposed and a cell radius of 10 μm was used.
Preparation of cell lines
HeLa cells were maintained in growth medium consisting of high-glucose Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Thermo Scientific), 50 units ml−1 penicillin, and 50 units ml−1 streptomycin (Invitrogen). MCF-10A cells were cultured in growth medium (1:1 Ham's F-12:DMEM with 2 mM L-glutamine, Gibco, Grand Island, NY) supplemented with 5% horse serum (Invitrogen), 2.5 μg ml−1 Fungizone (Invitrogen), 5 μg mL−1 gentamicin (Invitrogen), 10 μg ml−1 insulin (Sigma-Aldrich, St. Louis, MO), 0.5 μg ml−1 hydrocortisone (Sigma-Aldrich), 0.02 μg ml−1 epidermal growth factor (Sigma-Aldrich), and 0.1 μg ml−1 cholera toxin (Sigma-Aldrich). MDA-MB-231 cells were cultured in growth medium (RPMI 1640 with 2 mM L-glutamine (Gibco)) supplemented with 10% fetal bovine serum, 50 units ml−1 penicillin, 50 units ml−1 streptomycin (Invitrogen), 2.5 μg ml−1 Fungizone (Invitrogen), 5 μg mL−1 gentamicin (Invitrogen). Fresh 0.25% (for MCF-10A) or 0.05% (for HeLa and MDA-MB-231) trypsin–EDTA in phosphate buffered saline (PBS) was used to detach cells for preparing the cell suspension. For experiments involved with latrunculin-A, MCF-10A cells were incubated in latrunculin-A and medium for 20 minutes prior to loading into the device.
Expression of bacterial MscL in mammalian cells
Details of the MscL expression in mammalian cells have been described previously.26 Briefly, MscL WT and its mutant MscL G22S (with lower activation threshold) constructs were cloned into a tetracycline (tet)-regulatable adenovirus vector (pADtet) using seamless cloning. Human embryonic kidney 293 (HEK293) Cre4 cells were transfected with pADtet-MscL-WT or pADtet-MscL-G22S constructs to generate adenoviruses. For experiments involving MscL expression, retinal pigment epithelial (RPE) cells, which were maintained in DMEM/F12 supplemented with 10% fetal bovine serum, were used. Adenoviruses containing MscL WT or MscL G22S with an encoded (tet)-regulatable promoter (pADtet-MscL-WT or pADtet-MscL-G22S) were co-infected with tetracycline transactivator (tTA) adenovirus in RPE cells for 12–16 hours prior to aspiration experiments. All MscL constructs were verified by DNA sequencing and the successful expression of MscL on the cell membrane of RPE cells was confirmed by Western blot analysis.
Image acquisition and processing to study cell deformation in micropipette aspiration
The μFPA device was mounted on an optical microscope (Nikon, Ti Eclipse) for image acquisition under a 20× objective. The field of view was about 0.5 mm and the optical resolution was about 650 nm. This field of view allowed us to observe 16 aspiration chambers simultaneously without the use of the motorized x–y stage. The images were captured at 20 s intervals with exposure times of 10 ms in bright field and 200 ms in fluorescence mode. A customized Matlab program was developed to automatically analyze cell deformation and fluorescence dye uptake during the aspiration experiment. Briefly, the program first aligned video frames to compensate for possible drifting during image acquisition. After applying a Wiener filter and a binarization process, the positions of the pipette mouth and the leading edge of the aspirating cell were recognized. The protrusion lengths of the cells were determined. The program was also able to estimate the radius of cells by using a curving fitting algorithm. This Matlab program can process most cells, except for images that were slightly defocused or the cell boundary was very faint. Images which were not analyzable in the Matlab program were manually analyzed in ImageJ (http://imagej.nih.gov/ij/). A homebuilt micropipette aspiration system with a graduated manometer was used for conventional micropipette aspiration experiments. Glass micropipettes with inner diameters of ~10 μm were filled with 0.2% bovine serum albumin (BSA) in PBS in order to allow smooth movement of the cell membrane inside the glass micropipette. Negative pressure in the micropipette tip was generated by aspirating water from the main manometer reservoir and increased gradually in −100 Pa increments. Nikon Advanced Modulation Contrast optics (NAMC) mounted on a Nikon Ti-S microscope and CoolSnap MYO CCD camera (Photometrics, Tucson, AZ) were used to acquire live-cell phase contrast images and analyzed manually in ImageJ.
Results and discussion
Cell loading
The cell loading mechanism of our μFPA device is similar to a hydrodynamic trapping scheme for microarray applications, which was first reported by Tan and Takeuchi.27 This trapping mechanism has been recently adopted for on-chip cell culture and sorting applications.28–30 To better explain this trapping concept, we will simplify our illustration to one repeating unit of our microarray structure (Fig. 2(a)). Near the trapping chamber, the microfluidic channel is branched into two paths, the main microfluidic channel and the aspiration pipette channel. Our design is based on the rationale to impose a higher flow resistance in the main microfluidic channel than that in the aspiration pipette such that the majority of the fluid will flow into the aspiration pipette. Flow resistance is a function of cross section and path length. The cross-sectional area of our aspiration pipette is smaller than that of the main microfluidic channel. Thus, we need to have a much longer path length to increase the flow resistance. The volume flow rate ratio between path 1, the aspiration pipette and path 2, the main microfluidic channel is represented as:| | 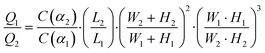 | (1) |
where Q1 is the volume flow rate through the aspiration pipette channel and Q2 is the volume flow rate through the main microfluidic channel; L1, W1, H1, are the length, width and height of the aspiration pipette, respectively; and L2, W2, H2, are the path length, width and height of the main microfluidic channel, respectively. C(α) is the laminar friction constant, which is a function of the cross section aspect ratio, α. α1 and α2 are defined by the smaller value of height/width or width/height, which are both equal to one for square cross sections of both the aspiration pipette and the main microfluidic channels. Given the low velocity in typical microfluidic flows, the characteristic Stokes' number of cells flowing across the trapping chamber is on the order of 0.001. Thus, the cells will flow along the streamline of the velocity flow field and the cells' inertia is negligible. After a cell is trapped in the first aspiration chamber, it greatly increases its flow resistance. Given the square cross section of the aspiration pipette, we can roughly assume that the trapped cell completely blocks the fluid flow. Other cells will flow along the main microfluidic flow instead of entering the trapping chamber and the pressure drop across the aspiration pipette will be fully exerted on the trapped cells. Based on Darcy–Weisbach equation, the pressure difference is represented as:| | 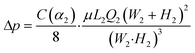 | (2) |
where C(α2) = 56.91 for α2 = 1. Δp = p2 − p1, and μ is the dynamic viscosity of the fluid (0.001 Pa s for water at 20 °C). After cells are trapped in the aspiration pipette channels, Q2 takes the value of the volume flow rate set by the syringe pump by continuity of incompressible flow. We have analyzed three designs with parameters summarized in Table 1. We selected device 3 as the ultimate design parameters since it gave an optimized trapping efficiency with a volume flow rate ratio of Q1/Q2 = 1.954. Furthermore, it provided a significant pressure difference of several kilopascals across the aspiration pipette channel for pipette aspiration under a typical volume flow rate range of 0–0.75 μl min−1. The side length of the mouth of the aspiration channel is designed to be 8 μm so that the cell sized larger than 10 μm will be robustly trapped in the aspiration chamber. The channel length of the aspiration channel is designed to be 25 μm to provide a sufficient protrusion length of the trapped cell for aspiration. During cell loading, the microfluidic channel is operated with a flow rate of 0.1 μl min−1 with pulse durations of 10 s until cells filled up the regions of interest. Trapping structures closer to the inlet were filled first compared to the downstream columns of trapping structures. This flow velocity minimizes the chance of cell clogging by sedimentation and had minimal perturbation to the trapped cells. We used a cell suspension of 1.0 × 106 cells ml−1, corresponding to an average cell separation of about 1 mm within the main microfluidic channel. This arrangement minimized the effect of clumping and possible pressure fluctuation during aspiration experiments. After the cells were trapped in the aspiration chambers, an idle time of two minutes was given before ramping up the volume flow rate again for aspiration experiments. This idle time allowed the recovery of cells to their original states before data collection. We noted that cell sedimentation may occur during this idle time. However, since the microfluidic device was operating in a relatively high flow rate of 1 mm s−1 even for the lowest volume flow rate of 0.05 μl min−1, we observed that fluid flow was able to re-suspend the settled cell during aspiration experiments. A demonstration of cell trapping under a loading volume flow rate of 0.1 μl min−1 is illustrated by numerical simulations (Fig. 2b). The particle flowing close to the side wall followed the streamline and entered the trapping/aspiration chamber. A typical cell loading result in the μFPA device is shown in Fig. 2c, with an illustration of the corresponding pressure difference at individual trapping chamber. Note that we have taken this small pressure difference into account in all aspiration experiments in subsequent calculations. We also noticed that any empty chamber will lead to lower pressure differences exerted on other aspiration chambers during cell aspiration experiments. Under normal operations, we ensured a 100% of trapping efficiency (including conditions where a gas bubble or debris was trapped in the pipette channel). This condition is not difficult to achieve given the high volume rate, Q1/Q2 ratio.
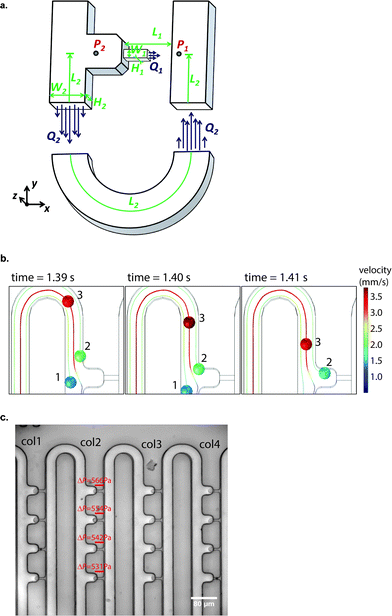 |
| | Fig. 2 Illustrations of cell trapping in the μFPA device. (a) Modeling of an aspiration chamber of cell trapping mechanism. Q: Volumetric flow rate; P: pressure; L, W, H: length, width, and height of the microchannel, respectively. (b) Demonstration of cell trapping in numerical simulations (the color on both the particles and the streamlines indicates the magnitude of velocity), (c) cell loading of single HeLa cells in different columns of trapping structures (col 1 closest to the inlet) in the μFPA device. The pressure difference at individual trapping chamber was illustrated at a loading volume flow rate of 0.1 μl min−1. | |
Table 1 Geometric parameters from different designs of the μFPA device
|
|
W
1, μm |
L
1, μm |
W
2, μm |
L
2, μm |
H
1, μm |
H
2, μm |
Q
1/Q2 |
Δp, Pa (Q = 0.75 μl min−1) |
| Device 1 |
8 |
25 |
60 |
5708 |
8 |
60 |
0.072 |
157 |
| Device 2 |
8 |
25 |
60 |
5661 |
8 |
60 |
1.145 |
2486 |
| Device 3 |
8 |
25 |
30 |
9661 |
8 |
30 |
1.954 |
4243 |
Cell aspiration
In general, mechanical properties of cells can be studied using two different mechanical models. In simplified pictures, the mechanical behavior of cells can either be modeled as a drop of liquid enclosed by a membrane or a piece of elastic solid. Micropipette aspiration is a versatile technique which can provide measurements of both behaviors. In the liquid-drop model, the cell is deformed with a constant volume and the deformation is attributed to a change of cortical tension of the cell membrane–actin composite material. The cortical tension, which is a sum of lipid bilayer tension and the tension from the underlying actin–myosin cortex, is assumed to be homogeneous and at equilibrium during pipette aspiration and can be calculated from the Young–Laplace equation:| | 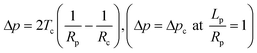 | (3) |
where Tc is the cortical tension as defined above, Lp is the protrusion length of the trailing edge of the cell into the pipette, Rc is the radius of the cell outside the pipette and RP is the hydraulic radius of the aspiration pipette, which can be found as follows:| | 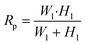 | (4) |
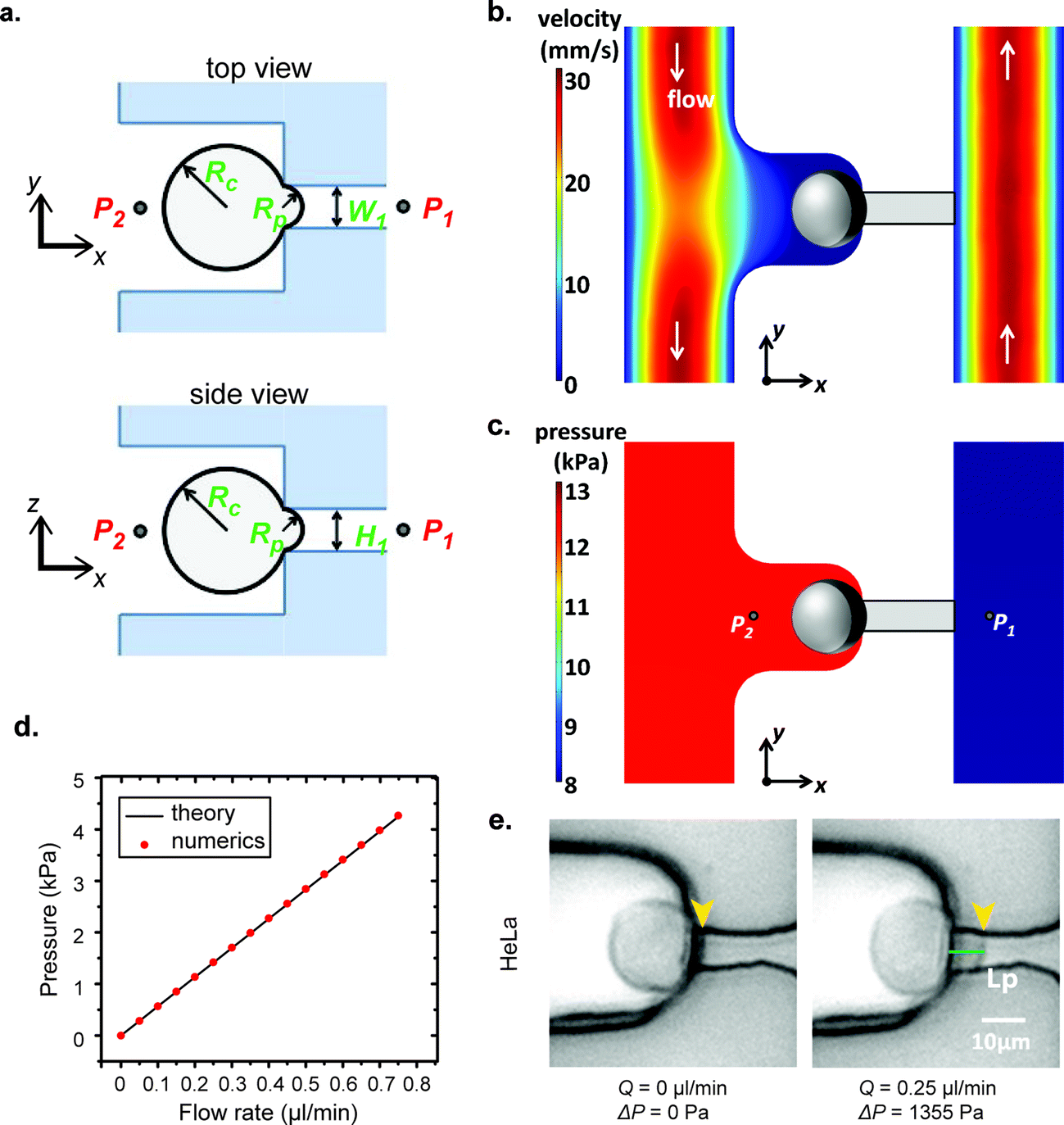
In our μFPA device, the cross section of the aspiration pipette channel is a square. Thus, the hydraulic radius is equivalent to half the side length. This square cross section ensures that the cells aspirated fully enclose the mouth of the pipette to prevent pressure leakage. The pressure difference is exerted entirely on the trapped cells along the axis of the aspiration pipette. This configuration also minimizes the friction and dragging force on the cell membrane with pipette side walls in contrast to a traditional cylindrical micropipette. The sectional views of our aspiration pipette unit are shown in Fig. 3a. Under a volume flow rate of 0.75 μl min−1 (which is the maximum operation flow rate of our device), the velocity field distribution in one aspiration unit is shown in Fig. 3b. We set this flow rate as our maximum working threshold and did not encounter any leakage problem when the device was operated below this flow rate. The cells trapped in chambers were isolated from the shear flow of the main microfluidic channel. The average shear stress exerted on a spherical trapped cell in our μFPA device was 0.072 dyn cm−2 (ESI† Fig. S2). This value is at least 200 times lower than the threshold to activate shear stress-activated ion channels.31,32 For all of the cells under aspiration, we did not observe any event of shear-induced rotational motion or cell deformation. The corresponding pressure distribution is shown in Fig. 3c. Given the large L2/L1 ratio in our design, the pressure distribution closed to the aspiration region is reasonably constant spatially. We ignored minor losses due to bends, expansions in the aspiration units. The pressure difference across the trapped cell calculated from eqn (2), in which a straight channel was assumed, is plotted against different volume flow rates and the comparison with numerical simulation results is shown in Fig. 3d. These two results showed a discrepancy of 0.05% in maximum flow rate, revealing that we can reliably use eqn (2) to estimate the pressure difference upon pipette aspiration. The pressure difference is solely determined by the volume flow rate set by the syringe pump. Since a syringe pump was used directly to generate aspirating pressure difference, the resolution of pressure difference is limited by the peristalsis of the syringe pump where the fluctuation of a syringe pump may cause pressure fluctuation.33 The syringe pump we used can exert a linear force of 50 lbs and has a step resolution of 0.016 μm. We also used a small syringe (0.5 ml) with a diameter size of 3.26 mm. This arrangement can minimize the effect of peristalsis for liquid pumping. We operated our microfluidic device at a low Reynolds number (Re < 1). The viscous effect and laminar nature provided stability on the pressure field.
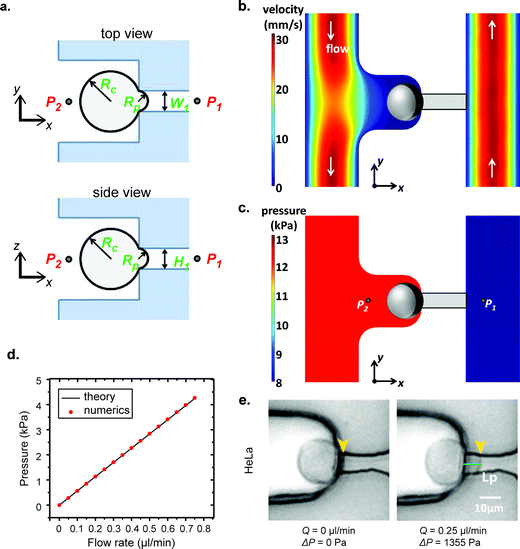 |
| | Fig. 3 Illustrations of cell aspiration. (a) Sketch of an aspiration chamber to determine the cortical tension of the single cell. P: Pressure; W, H: width and height of aspiration micropipette, respectively; Rc, Rp: radii of cell and aspiration micropipette, respectively. (b) Numerical simulations of velocity field and (c) pressure distribution. (d) Comparison of pressure difference across the microfluidic pipette versus the flow rate between theory and numerical simulation results. (e) Demonstration of pipette aspiration of the HeLa cell in a μFPA device. | |
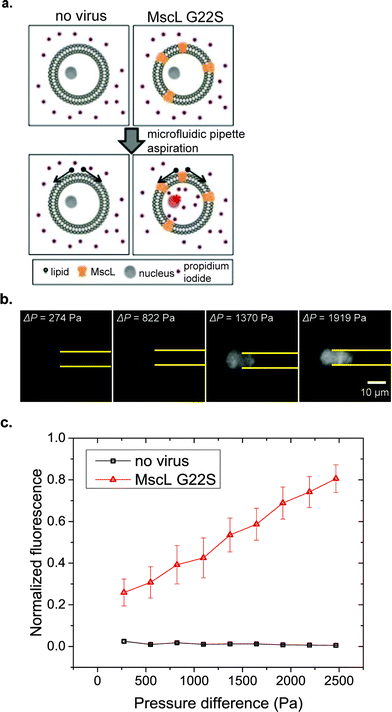
Adapted from the theoretical analysis of Theret et al., a cell can be modeled as a homogeneous elastic solid.34 We can study the stiffness of a single cell as follows:
| | 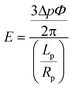 | (5) |
where
E is the Young's modulus,
Φ is a constant which is determined by the geometry of the micropipette, which typically takes a value of 2.1. During a pipette aspiration measurement, the volume flow rate increased from 0.05 μl min
−1 to 0.75 μl min
−1 at step increases of 0.05 μl min
−1. There were a total of 16 intervals. At each interval, the flow rate was held constant for 2 minutes and we observed that the velocity flow field stabilized within 10 s for a flow rate increment at each interval by examining the motion of 2 μm fluorescent beads in our set up (not shown). Thus, one complete measurement took 32 minutes. As a demonstration, a HeLa cell under aspiration is shown in
Fig. 3e. At a flow rate of 0.25 μl min
−1, a pressure difference of ~1.36 kPa was exerted and a clear cell protrusion length of a HeLa cell into the micropipette was observed. To determine the value of the Young's modulus, the advancing linear portion of the protrusion curves of each cell under aspiration was fitted with a linear regression.
6,12 The slope of the fitted line was used to calculate the Young's modulus of each cell according to
eqn (5).
Mechanophenotyping of breast cancer cells
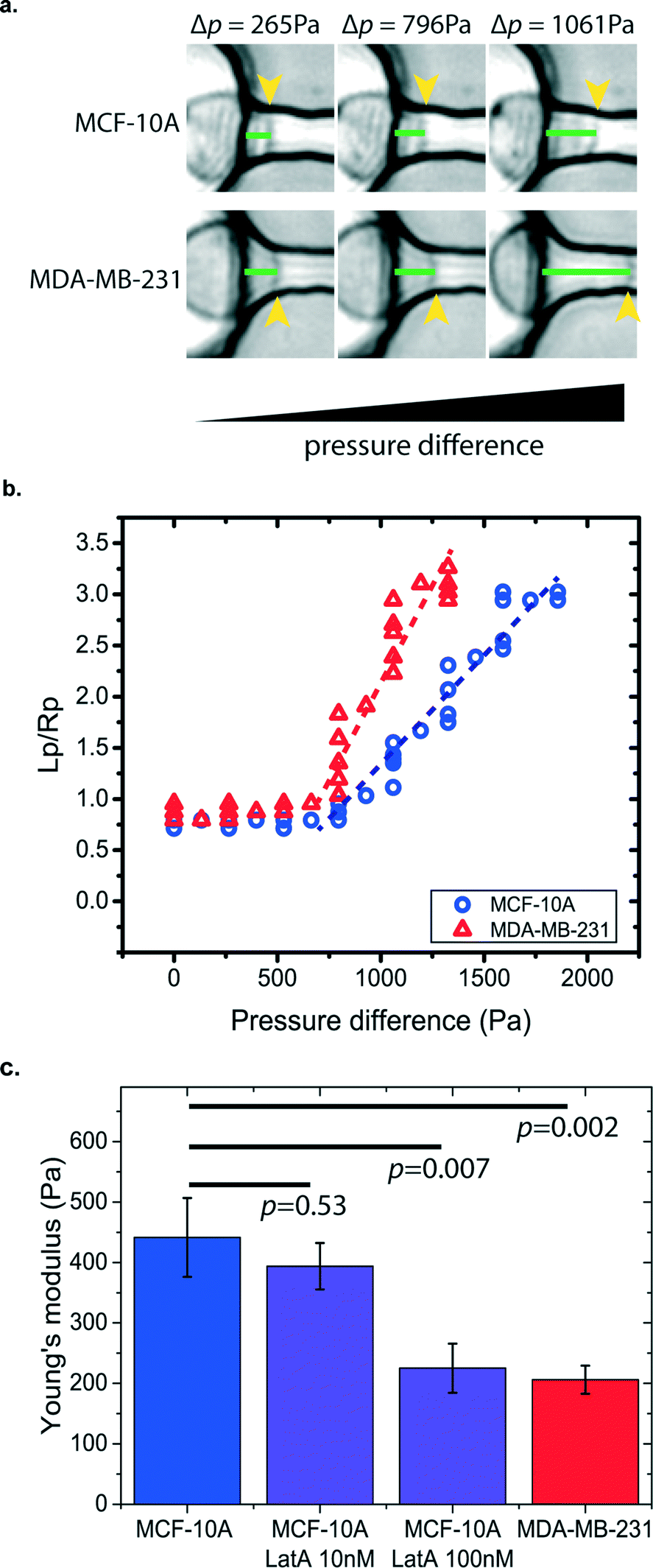
Cancer cells have different mechanical properties from normal cells.35–37 The progression of human cancer alters the structures and dynamics of the cell cytoskeleton. Some cancer cells have been found to have higher deformability in order to transmigrate through the basement membrane to enter the bloodstream and spread cancer to other organs.35,38 Thus, by measuring cell stiffness, we can evaluate the metastatic potential of cancer cells. Other than the electrical-based flow cytometry method for cell deformation studies,39 micropipette aspiration provides a simple, direct, and label-free approach to measure cancer cell stiffness. However, the traditional micropipette aspiration system is limited by its very low testing throughputs since measurement is administered to one cell at a time.40 This hinders its potential to extract statistically significant data from a heterogeneous cell population for clinical diagnostic applications. Our μFPA device provides an array-based platform for cell mechanical measurement of cancer cells in a parallel manner. As a proof-of-principle demonstration, we have used our μFPA device to measure human breast cell lines of non-tumorigenic cells, MCF-10A, in comparison with the metastatic cancer cell line, MDA-MB-231. The preparation of the cell suspension with the optimized concentration is described in the materials and methods section. Given the same pressure difference exerted across the aspiration pipette channel, the two cell lines showed different cell deformability and protrusion lengths as shown in Fig. 4a and b. Based on eqn (5), the Young's moduli of the MCF-10A and MDA-MB-231 were calculated to be 441.4 ± 65.3 Pa and 206.2 ± 23.1 Pa (mean ± s.e.), respectively (Fig. 4c). The measured values of these two cell populations were statistically different, and this result is comparable with previous measurements by atomic force microscopy41 as well as measurements done using conventional micropipette aspiration (ESI† Fig. S3). We believed that the ~10% difference between our measurements is due to changes in physiological conditions of cells in traditional micropipette aspiration, which typically took 2 hours for measurement of 10 cells.
 |
| | Fig. 4 Mechanical characterization of human breast non-tumorigenic (MCF-10A) and metastatic cancer (MDA-MB-231) cells. (a) Demonstration of different protrusion lengths under increasing applied aspiration pressures, (b) one representative plot of protrusion length and pipette radius ratio with an applied pressure difference for a single MCF-10A and MDA-MB-231 cell. (c) Determination of the Young's modulus (mean ± s.e.) using the μFPA device. Dotted lines are fits of the advancing linear region through the data points. MCF-10A: 441.4 ± 65.3 Pa, n = 32; MCF-10A incubated with 10 nM LatA: 393.7 ± 38.3 Pa, n = 12; MCF-10A incubated with 100 nM LatA: 225.2 ± 40.6 Pa, n = 10 and MDA-MB-231: 206.2 ± 23.1 Pa, n = 21. Error bars represent the standard error of the mean, and p values are also indicated on the graph from a Student's t-test. | |
Since cell stiffness depends on the integrity of the actin cytoskeleton, we investigated the sensitivity of our μFPA devices by measuring cell stiffness using low dosages of latrunculin-A to MCF-10A cells. Latrunculin-A disrupts the microfilament organization in the cell cortex and hence reduces cell stiffness.42 Typical cell biology experiments use latrunculin-A in the micromolar range to inhibit actin-based cell migration.43 Our device is able to detect small changes in cell deformability in the 10s and 100s nanomolar range, where treatment of MCF-10A cells with 10 nM or 100 nM latrunculin-A yielded Young's moduli of 393.7 ± 38.3 Pa and 225.2 ± 40.6 Pa, respectively. The ability to measure changes in cell stiffness at these low levels of latrunculin-A demonstrates the sensitivity of our device.
Mechanical gating of mechanosensitive channels
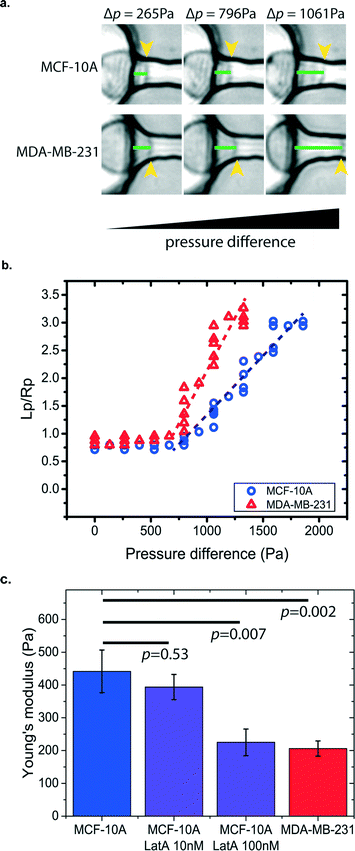
Mechanical perturbations are recognized to regulate diverse cellular processes.44 Mechanical forces can be transduced into biochemical signals in cells through mechanosensitive (MS) channels on the cell membrane. Some MS channels are known to be gated purely by lipid bilayer tension.45,46 The lipid bilayer is directly coupled with the actomyosin cortex and it is recognized that the cortical tension represents 90% of the composite membrane–actin cortex tension.47 Thus, by altering cortical tension, micropipette aspiration can also alter membrane tension.
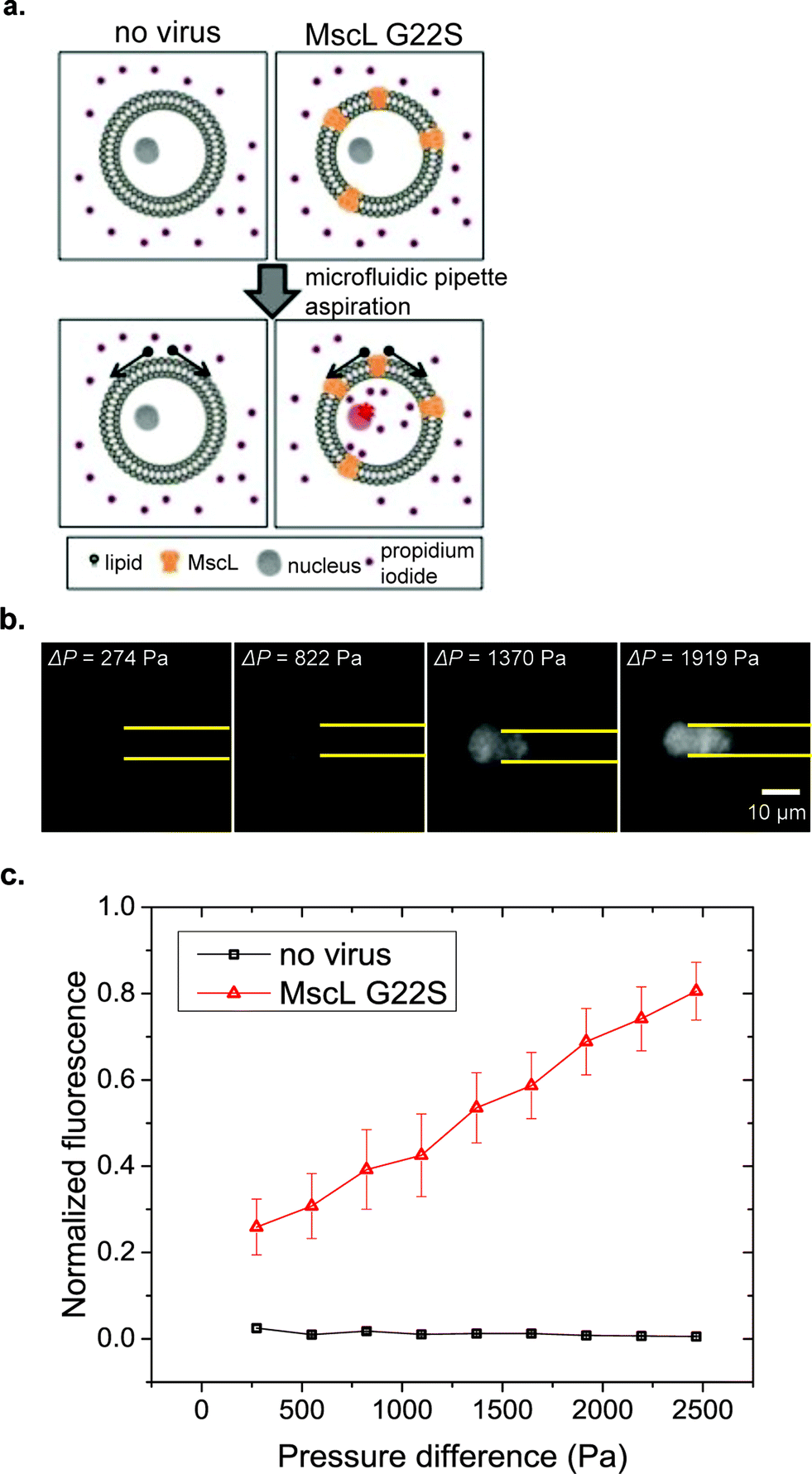
MscL is a bacterial MS channel that has recently been reconstituted in mammalian cells.26,48 Mechanical gating properties of MscL can be studied through the uptake of a small membrane impermeable stain, propidium iodide (PI), which fluoresces upon binding to DNA and RNA molecules as shown in Fig. 5a. Two MscL constructs were used in our experiments, wild-type MscL and a G22S mutant MscL that has a lower gating threshold.49 Compared with other flow-based cytometers for cell deformation measurement, our μFPA device provides the advantage of trapping cells at fixed positions that allows simultaneous monitoring of cell deformations and fluorescent dye uptake over time. In our μFPA device, cells under aspiration are levitated from the top and bottom channel walls. This ensures an accurate measurement of cortical tension according to eqn (3). Using RPE cells expressing MscL, uptake of PI was observed in correlation with the magnitude of cell deformation under micropipette aspiration shown in Fig. 5b. The tension required to activate MscL-G22S-infected cells was estimated to be 4.5 mN m−1. We were unable to determine the activation threshold of MscL-WT-infected cells with the span of pressure difference we studied. A portion of RPE cells escaped from the aspiration pipette at high magnitudes of pressure difference over 2500 Pa, roughly corresponding to a cortical tension of 9.0 mN m−1. This value is lower than the threshold required to activate MscL-WT in mammalian cells, which is about 12.0 mN m−1.50 We can possibly improve the stability of cell trapping and increase the cortical tension exerted on cells for aspiration by the design of the micropipette channel with smaller cross section. Nonetheless, MscL G22S-expressing cells clearly had PI uptake at lower cortical tension (Fig. 5c). Our results here demonstrated the application of our novel μFPA device for single-cell mechanotransduction studies of MS channels.
 |
| | Fig. 5 Mechanical gating of MscL-expressing RPE cells. (a) Illustration of MscL gating by exertion of tension by pipette aspiration, (b) demonstration of propidium iodide (PI) influx into the MscL-G22S-expressing RPE cells with increasing applied aspiration pressure in correlation with cell deformation, (c) plot of normalized fluorescence of PI in RPE cells versus pressure difference. No virus: n = 9 and MscL G22S: n = 8. Error bars represent the standard error of the mean. The error bars for the case of no virus is too small to be seen in the plots. | |
Conclusion
Compared with other advanced measurement techniques in cell mechanics studies, such as atomic force microscopy or optical tweezer systems, which involve sophisticated electronic, optical and mechanical components to operate, the micropipette aspiration system provides a simple and direct approach to measure mechanical properties of single cells. Despite its simplicity, micropipette aspiration is still a widely adopted method in many recent advanced cell mechanics studies on bleb growth,47 cytoskeletal cortex dynamics,51 and mechanosensing.52 Traditional micropipette aspiration systems are limited by its measurement accuracy and throughput. In this work, we have developed a microfluidic device, namely μFPA, which takes advantage of the laminar and stable nature of microfluidic flow, to conduct pipette aspiration in a parallel manner. Using only a syringe pump, the device is able to autonomously trap cells in designated aspiration chambers and make quantitative measurement of mechanical properties of single cells by observation of their deformation under an optical microscope. With the aid of theoretical modeling and numerical simulations, we have designed the μFPA device such that the aspiration pressure exerted on trapped cells is directly proportional to the flow rate set by the syringe pump. This aspiration pressure is exerted along the axis of the pipette and cells under aspiration are free from shear stress. We currently operate the device in a single-use manner. This is mainly due to the possibility of clogging at the filtering unit near the microfluidic channel inlet due to successive cell loading and unloading.
As a demonstration, we have applied our μFPA to measure the Young's modulus of human breast non-tumorigenic (MCF-10A) and metastatic cancer (MDA-MB-231) cells. Our μFPA device measured statistically significant Young's moduli to differentiate MCF-10A from MDA-MB-231 cells. Our μFPA device provides an alternative to cell migration assays which takes hours rather than minutes for cancer cell diagnostics.53 It also has potential to be further developed as a versatile test bed for rapid drug screening and discovery for molecular cancer therapy based on cell mechanophenotyping. We have also applied our μFPA device to study mechanical gating properties of MscL-expressing RPE cells. The uptake of a small fluorescence stain, PI, was measured with increasing applied cortical tension. The activation tension in MscL-G22S-infected RPE cells was found to be 4.5 mN m−1. The ability to apply dynamic mechanical loading using the μFPA device could open up opportunities for more detailed analysis of cellular mechanotransduction pathways. Altogether, the μFPA device presented here has versatile uses that will enable research in cell mechanics and mechanotransduction.
Acknowledgements
This work is supported by the NIH Director's New Innovator Award (DP2 HL117748-01). We appreciate the assistance from Victoria Murray and Johanna Heureaux in generating MscL adenoviruses and on MscL expression. We thank Johanna Heureaux, Kwun Yin Ho and Christopher Coyne for their help in the traditional micropipette aspiration experiments. The breast cell lines were kindly provided by Dr. Sofia Merajver at the University of Michigan. We also acknowledge the technical support from the Lurie Nanofabrication Center (LNF) at the University of Michigan.
Reference
- G. Bao and S. Suresh, Nat. Mater., 2003, 2, 715–725 CrossRef CAS PubMed.
- M. J. Rosenbluth, W. A. Lam and D. A. Fletcher, Biophys. J., 2006, 90, 2994–3003 CrossRef CAS PubMed.
- N. Wang and D. E. Ingber, Biochem. Cell Biol., 1995, 73, 327–335 CrossRef CAS PubMed.
- Z. Z. Fan, Y. B. Sun, D. Chen, D. Tay, W. Q. Chen, C. X. Deng and J. P. Fu, Sci. Rep., 2013, 3 Search PubMed.
- H. Zhang and K. K. Liu, J. R. Soc., Interface, 2008, 5, 671–690 CrossRef CAS PubMed.
- R. M. Hochmuth, J. Biomech., 2000, 33, 15–22 CrossRef CAS PubMed.
- T. Das, T. K. Maiti and S. Chakraborty, Lab Chip, 2008, 8, 1308–1318 RSC.
- Y. Shao, X. Y. Tan, R. Novitski, M. Muqaddam, P. List, L. Williamson, J. P. Fu and A. P. Liu, Rev. Sci. Instrum., 2013, 84 Search PubMed.
- S. M. Block, Nature, 1992, 360, 493–495 CrossRef CAS PubMed.
- D. Psaltis, S. R. Quake and C. H. Yang, Nature, 2006, 442, 381–386 CrossRef CAS PubMed.
- X. D. Fan and I. M. White, Nat. Photonics, 2011, 5, 591–597 CrossRef CAS PubMed.
- E. Shojaei-Baghini, Y. Zheng and Y. Sun, Ann. Biomed. Eng., 2013, 41, 1208–1216 CrossRef PubMed.
- J. S. Dudani, D. R. Gossett, H. T. K. Tse and D. Di Carlo, Lab Chip, 2013, 13, 3728–3734 RSC.
- D. R. Gossett, H. T. K. Tse, S. A. Lee, Y. Ying, A. G. Lindgren, O. O. Yang, J. Y. Rao, A. T. Clark and D. Di Carlo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7630–7635 CrossRef CAS PubMed.
- J. P. Shelby, J. White, K. Ganesan, P. K. Rathod and D. T. Chiu, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14618–14622 CrossRef CAS PubMed.
- M. J. Rosenbluth, W. A. Lam and D. A. Fletcher, Lab Chip, 2008, 8, 1062–1070 RSC.
- H. W. Hou, Q. S. Li, G. Y. H. Lee, A. P. Kumar, C. N. Ong and C. T. Lim, Biomed. Microdevices, 2009, 11, 557–564 CrossRef CAS PubMed.
- A. Adamo, A. Sharei, L. Adamo, B. Lee, S. Mao and K. F. Jensen, Anal. Chem., 2012, 84, 6438–6443 CrossRef CAS PubMed.
- S. Byun, S. Son, D. Amodei, N. Cermak, J. Shaw, J. H. Kang, V. C. Hecht, M. M. Winslow, T. Jacks, P. Mallick and S. R. Manalis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7580–7585 CrossRef CAS PubMed.
- M. Mak and D. Erickson, Integr. Biol., 2013, 5, 1374–1384 RSC.
- M. Mak, C. A. Reinhart-King and D. Erickson, Lab Chip, 2013, 13, 340–348 RSC.
- Q. Guo, S. Park and H. S. Ma, Lab Chip, 2012, 12, 2687–2695 RSC.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- J. M. Kwan, Q. Guo, D. L. Kyluik-Price, H. S. Ma and M. D. Scott, Am. J. Hematol., 2013, 88, 682–689 CrossRef CAS PubMed.
- Q. Guo, S. J. Reiling, P. Rohrbach and H. S. Ma, Lab Chip, 2012, 12, 1143–1150 RSC.
- J. Heureaux, D. Chen, V. L. Murray, C. X. Deng and A. P. Liu, Cell. Mol. Bioeng., 2014, 7(3), 307–319 CrossRef PubMed.
- W. H. Tan and S. Takeuchi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1146–1151 CrossRef CAS PubMed.
- B. Deng, X. F. Li, D. Y. Chen, L. D. You, J. B. Wang and J. Chen, Sci. World J., 2014, 2014, 929163 CAS.
- S. Kobel, A. Valero, J. Latt, P. Renaud and M. Lutolf, Lab Chip, 2010, 10, 857–863 RSC.
- J. Kim, J. Erath, A. Rodriguez and C. Yang, Lab Chip, 2014, 14, 2480–2490 RSC.
- S. S. Ranade, Z. Z. Qiu, S. H. Woo, S. S. Hur, S. E. Murthy, S. M. Cahalan, J. Xu, J. Mathur, M. Bandell, B. Coste, Y. S. J. Li, S. Chien and A. Patapoutian, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10347–10352 CrossRef CAS PubMed.
- J. Li, B. Hou, S. Tumova, K. Muraki, A. Bruns, M. J. Ludlow, A. Sedo, A. J. Hyman, L. McKeown, R. S. Young, N. Y. Yuldasheva, Y. Majeed, L. A. Wilson, B. Rode, M. A. Bailey, H. R. Kim, Z. Fu, D. A. Carter, J. Bilton, H. Imrie, P. Ajuh, T. N. Dear, R. M. Cubbon, M. T. Kearney, R. K. Prasad, P. C. Evans, J. F. Ainscough and D. J. Beech, Nature, 2014 DOI:10.1038/nature13701.
- Z. D. Li, S. Y. Mak, A. Sauret and H. C. Shum, Lab Chip, 2014, 14, 744–749 RSC.
- D. P. Theret, M. J. Levesque, M. Sato, R. M. Nerem and L. T. Wheeler, J. Biomech. Eng., 1988, 110, 190–199 CrossRef CAS PubMed.
- V. Swaminathan, K. Mythreye, E. T. O'Brien, A. Berchuck, G. C. Blobe and R. Superfine, Cancer Res., 2011, 71, 5075–5080 CrossRef CAS PubMed.
- W. Xu, R. Mezencev, B. Kim, L. Wang and J. McDonald,
et al. Cell Stiffness Is a Biomarker of the Metastatic Potential of Ovarian Cancer Cells, PLoS One, 2012, 7(10), e46609 CAS.
- P. Katira, M. H. Zaman and R. T. Bonnecaze, Phys. Rev. Lett., 2012, 108 Search PubMed.
- S. Suresh, Acta Mater., 2007, 55, 3989–4014 CrossRef CAS.
- N. Bao, Y. H. Zhan and C. Lu, Anal. Chem., 2008, 80, 7714–7719 CrossRef CAS PubMed.
- E. Shojaei-Baghini, Y. Zheng, M. A. S. Jewett, W. B. Geddie and Y. Sun, Appl. Phys. Lett., 2013, 102 Search PubMed.
- M. Nikkhah, J. S. Strobl, E. M. Schmelz and M. Agah, J. Biomech., 2011, 44, 762–766 CrossRef PubMed.
- R. Vargas-Pinto, H. Gong, A. Vahabikashi and M. Johnson, Biophys. J., 2013, 105, 300–309 CrossRef CAS PubMed.
- K. M. Stroka, H. Y. Jiang, S. H. Chen, Z. Q. Tong, D. Wirtz, S. X. Sun and K. Konstantopoulos, Cell, 2014, 157, 611–623 CrossRef CAS PubMed.
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265–275 CrossRef CAS PubMed.
- E. S. Haswell, R. Phillips and D. C. Rees, Structure, 2011, 19, 1356–1369 CrossRef CAS PubMed.
- A. Anishkin, S. H. Loukin, J. F. Teng and C. Kung, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7898–7905 CrossRef CAS PubMed.
- J. Y. Tinevez, U. Schulze, G. Salbreux, J. Roensch, J. F. Joanny and E. Paluch, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18581–18586 CrossRef CAS PubMed.
- J. F. Doerner, S. Febvay and D. E. Clapham, Nat. Commun., 2012, 3 Search PubMed.
- K. Yoshimura, A. Batiza, M. Schroeder, P. Blount and C. Kung, Biophys. J., 1999, 77, 1960–1972 CrossRef CAS PubMed.
- S. I. Sukharev, W. J. Sigurdson, C. Kung and F. Sachs, J. Gen. Physiol., 1999, 113, 525–539 CrossRef CAS PubMed.
- J. Brugues, B. Maugis, J. Casademunt, P. Nassoy, F. Amblard and P. Sens, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15415–15420 CrossRef CAS PubMed.
- T. Z. Luo, K. Mohan, P. A. Iglesias and D. N. Robinson, Nat. Mater., 2013, 12, 1063–1070 CrossRef PubMed.
- N. Kramer, A. Walzl, C. Unger, M. Rosner, G. Krupitza, M. Hengstschlager and H. Dolznig, Mutat. Res., Rev. Mutat. Res., 2013, 752, 10–24 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc01218f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.