
A microfluidic device for real-time monitoring of Bacillus subtilis bacterial spores during germination based on non-specific physicochemical interactions on the nanoscale level†
L.
Zabrocka
b,
K.
Langer
a,
A.
Michalski
b,
J.
Kocik
c and
J. J.
Langer
*a
aFaculty of Chemistry, Laboratory for Materials Physicochemistry and Nanotechnology, A. Mickiewicz University in Poznan, Srem, 63100, Poland. E-mail: orsel@sigmaxi.net
bBiological Threats Identification and Countermeasure Centre of Military Institute of Hygiene and Epidemiology, Pulawy, 24100, Poland
cMilitary Institute of Hygiene and Epidemiology, Warszawa, 01163, Poland
First published on 15th October 2014
Abstract
A microfluidic device for studies on the germination of bacterial spores (e.g. Bacillus subtilis) based on non-specific interactions on the nanoscale is presented. A decrease in the population of spores during germination followed by the appearance of transition forms and an increase in the number of vegetative cells can be registered directly and simultaneously by using the microfluidic device, which is equipped with a conductive polymer layer (polyaniline) in the form of a nano-network. The lab-on-a-chip-type device, operating in a continuous flow regime, allows monitoring of germination of bacterial spores and analysis of the process in detail. The procedure is fast and accurate enough for quantitative real-time monitoring of the main steps of germination, including final transformation of the spores into vegetative cells. All of this is done without the use of biomarkers or any bio-specific materials, such as enzymes, antibodies and aptamers, and is simply based on an analysis of physicochemical interactions on the nanoscale level.
Introduction
Germination of bacterial spores is a complex and still mysterious process1 which results in a breakdown of the structure of the spores and in a concomitant loss of their resistance against physical and biochemical factors. This process, i.e. the conversion of spores into vegetative cells, begins under favourable conditions and is initiated by small molecules (amino acids and nucleosides). After initiation of germination, dormant spores begin to hydrolyse and release monovalent cations, protons and zinc from their core. Also, molecules of dipicolinic acid (DPA) and calcium which is associated with the DPA are released by the spores and replaced with water molecules. This process continues until a sufficient level of hydration is reached, which results in restoring enzyme action and hydrolysis of the spore coat (peptidoglycan) as well as swelling of the germ cell.Only when sufficient nutrients for outgrowth of spore cells are available, small acid-soluble proteins (SASP) associated with spore DNA are degraded, and the vegetative cell escapes from the spore coat.1,2
In recent years, a number of analytical tools for examining the germination of spores have been developed,1e.g. fluorometry,3 flow cytometry,4 laser tweezers Raman spectroscopy,5,6 phase-contrast and fluorescence microscopy,6,7 surface-enhanced Raman scattering (SERS) microscopy,8 and SERS spectroscopy9 as well as an impedance-based method, although not a very accurate one, to detect spores by electrically detecting their germination in real time using a microfluidic biochip.10 Most of these methods are based on advanced (and expensive) instrumentation and complex procedures. Some of the methods mentioned above are very accurate; however, they are limited in that only a few individual cells can be examined, which is not a representative sample, and thus it is difficult to characterise a whole colony of 106–109 (or more) cells when using such a method.
Fluorometry-based methods can use the process of the activation of esterases as a practical parameter to quantitatively measure spore germination since activation of proteases and cortex-lytic enzymes plays a key role during the initial stages of spore germination.
Expression of esterase activity is particularly suitable for measuring the early responses of spores since it displays rapid kinetics measured with diacetyl fluorescein (DAF), a fluorogenic substrate which is hydrolysed nonspecifically by esterases, lipases, and proteases.
Therefore, this method allows monitoring of the germination process directly only in the first 14 minutes after the germinants are added to dormant spores, since there is a 2 minute lag followed by a 2 minute burst of esterase activity during which the activity increases more than 150-fold above the baseline.3
The microfluidic biochip can be used as a method for automatic and rapid electrical detection of germination of viable spores. The device includes special design features that facilitate spore capture and electrodes for impedance measurements. With a detection limit of fewer than 100 spores in a 0.1 nl chamber,10 the spore detection time is similar to that of a conventional method based on the PCR (two hours from heat activation of a suspected organism9). This method, although very advanced and convenient, does not allow for real-time monitoring of spore transformation into vegetative cells.
Surface-enhanced Raman scattering spectroscopy (SERS) has been used to detect the presence of DPAs, i.e. molecules which are released from spores during germination and can be used as a chemical signature for monitoring endospore germination of Bacillus species. Even more fascinating is the fact that only several hundred spores are sufficient to reliably measure the kinetics of germination at different concentrations of the germinant and different temperatures using SERS.9
The downside of this method is the observation of only a single factor, i.e. the release of DPAs from Bacillus spores, as a reference for such a complex germination process.
All methods that can detect spore physiology with limited manipulation and in a small time frame are valuable to public health and safety. Therefore, it is understandable that there is growing social emphasis on developing simple yet reliable techniques that will provide rapid information about a microbial sample without relying on culture techniques.
In this paper we present a new method for simultaneous monitoring of spores and vegetative cells during germination in real time with no such limit and using for testing “reasonable” samples of 105 spores (or more). The method is based on measurements of time-dependent changes in the electrical conductivity of a polyaniline nanofibre network (Fig. 1A); the changes are induced due to the presence of spores and cells interacting directly on conductive polymer nanofibrils. Polyaniline is one of the best materials to use due to its stability and relatively high electrical conductivity13,14 and because it is electroactive and pH-sensitive.
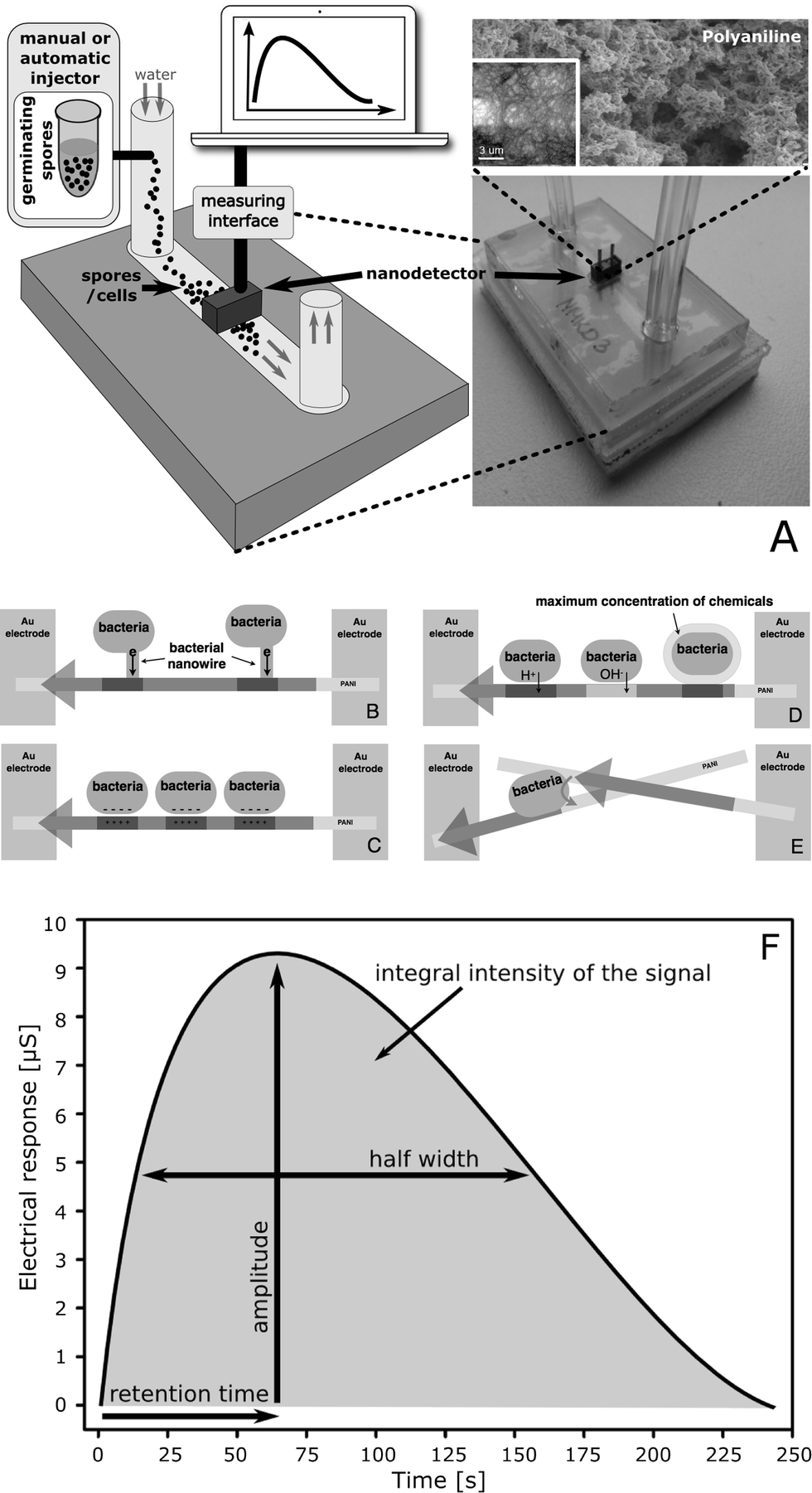 | ||
| Fig. 1 (A) Measurements are performed using a nanodetector which is designed as a lab-on-a-chip device operating in a continuous flow regime. The key component is a free-standing polyaniline (PANI) nanofibre network which is electrochemically formed between two gold micro-electrodes (inset: the initial stage of nanofibre formation in a separate experiment which was done to help visualise the process). Modification of the nano-network's electrical conductivity, induced by the microorganism cells, is a source of the nanodetector's electrical response. The electrical conductivity of polyaniline nanofibres in contact with living cells is modified by (B) the extracellular electron transfer (EET); (C) the presence of a cell surface charge and the FET mechanism; (D) the secretion of chemicals; (E) due to the quantum effects that occur in quasi-one-dimensional nanofibres (high electrical conductivity is possible only along PANI nanofibres and not perpendicularly, through sidewalls). The electrical conductivity is registered as a function of time (F). The result (time profile of the electrical response) is characteristic of different cells. The amplitude, and more precisely the integral intensity of the electrical response, indicates the approximate number of cells in the sample. The retention time depends on the strength of interactions of cells and polyaniline nanofibres. The half-width depends on the number of modes of interactions. | ||
A new nanodetector was designed as a lab-on-a-chip device operating in a continuous flow regime (Fig. 1A), analogously to our previous basic version of a continuous flow nanobiodetector (CFNBD), which has successfully been applied to detect bacterial vegetative cells.11,12 The system, based on polyaniline fibres (antibacterial), is self-cleaning, particularly when it operates in continuous flow mode, so it can be used to analyse multiple and different samples with practically no interference (the baseline is stable between measurements).
After modification and improvement of sensitivity due to construction of a compact, microfluidic, mechanically stable lab-on-a-chip version of the nanodetector and after applying a more accurate measuring interface, we were able to monitor the germination of bacterial spores in detail by using the same single device, which is not possible for other methods of comparable simplicity. When analysing the intensity and shape of the electrical signal registered due to changes in electrical conductivity of the nanofibre network as a function of time (time profile, Fig. 1F), one can obtain information on the number of spores and cells11,12,15 and on the processes in which they are involved. All that is necessary is the injection of a small sample with a suspension of germinating spores (usually below 100 μl and ca. 105 spores) into the detecting device, done every 5 min or more often if necessary. This approach is useful for monitoring the germination process with an accuracy that is better than 5% in a population, i.e. 103 spores or cells, and with a time resolution of process monitoring well below 1 min (in seconds in the case of automatic sampling). These parameters are better than (taking into account the representative character of the samples) or comparable to other methods which are applied in the case of complex systems, e.g. to analyse a mixture of spores and vegetative cells prepared in vitro or formed naturally during germination (please see the section Comparison with other methods). The ability to trace the transformation of spores into vegetative cells is essential not only for science (e.g. microbiology) but it is also of great importance from a practical point of view, e.g. for health, biotechnology, security and defence.
Results and discussion
The basic, standard version of a continuous flow nanobiodetector (CFNBD) was successfully tested only against bacterial vegetative cells.11,12,15 This and similar systems have never been used before to detect spores, and particularly to monitor the germination of bacterial spores, which is much more challenging as an analysis of a complex and dynamic process. Thus, both the device and the method described here are unique.Mechanism of detection
Modification of the electrical conductivity of the nano-network, induced by the spores and cells, is a source of the electrical response of the nanodetector. The detection mechanism is far from a routine approach. It is based on the non-specific (physicochemical) interactions of cells and spores which, when they touch the surface of the conducting polymer nanofibres, leads to a modification of the electrical conductivity of individual nanofibres and, consequently, of the whole network. These include direct charge injection, reversible chemical modifications of the nanofibrils, coulomb interactions leading to field-effect transistor (FET)-type features, and quantum effects associated with the reduced dimensionality of individual nanofibrils (Fig. 1B–E).The electric field generated by ions or the surface charge of cells is able to locally modify the charge carrier density in the nanofibres, which above the percolation threshold leads to an increase in the electrical conductivity due to the FET-type mechanism. Similarly, direct charge injection from the cells influences the electrical properties of the polyaniline nano-network. Quantum effects result in limiting of the electrical conductivity only to one dimension, i.e. along the main axis of the nanofibres. Cells attaching to the surface of the nanofibres (even for a very short period of time) are responsible for breaking the limit, which causes short-circuiting between the fibrils and influences the electrical conductivity of the whole network. Finally, chemical compounds released by cells in the closest vicinity of the nanofibres are active against the conducting polymer, such as polyaniline, which is electro-active and pH sensitive (obviously other polymers of similar properties can also be used but have not yet been tested in our laboratory). Local chemical modifications induced by the cells result in a change in the electrical conductivity of the nanofibres above the percolation threshold.
Owing to all of these effects, a considerable increase in the electrical conductivity is measured directly for the whole nano-network. The response is proportional to the density of the modifications, i.e. the number of cells. Because there are no stable links between the cells and the nanofibrils the processes are dynamic; that is why the response of the nanodetector, which is working in a continuous flow regime, is time dependent.
Obtaining the signal of germinating spores
Injection of a small sample (100 μl, approximately 105 spores) into a narrow channel of the lab-on-a-chip device, where a continuously flowing stream of water transports the sample into the detecting area of the NBD unit, results in the generation of a dynamic response that is registered as an electrical signal as a function of time (Fig. 1F). The time profile of the response is characteristic of different types of cells, including spores and vegetative cells. This is caused by considerable differences in the size and morphology of the cells, in the surface structure and in all biochemical processes. The retention time of the sensor's electrical response depends on the strength of the interactions between cells and the polyaniline nanofibres. The effects measured are related to cell morphology and cell size, to the cell wall structure, cell surface hydrophobicity and the electrostatic surface charge of the cell. The half-width of the sensor's electrical response depends on the number of modes of interactions, which is related to the shape of the micro-organism, the presence of protrusions, tendrils, cilia on the outer surface of the bacteria, and on the symmetry of the electric charge distribution at the surface of the cells.Germination of bacterial spores is associated with a breakdown of their structure, followed by dramatic changes in their chemical and physical properties – particularly of the surface layer, which can easily be detected and identified with the aid of a nanodetector due to the differences in the interactions of each type of cell (spores vs. transition forms vs. vegetative cell).
The data collected during spore germination are used to calculate the integral intensity, retention time and half-width of the registered signal (time profile). The integral intensity and signal amplitude are used to estimate the number of spores or vegetative cells in the sample analysed. The retention time, which is sensitive to the mobility of the cells, defines the type of analysed material (spores/transition forms/vegetative cells); the half-width is associated with interactions, including the influence of morphology of the analysed cells (shape and size). Thus, analysing the shape of the time profiles (Fig. 1F) allows us to distinguish between suspensions of spores, vegetative cells and their mixture, which also provides quantitative information about a mixture's composition.
Real-time monitoring of spore germination
The germination process is performed according to a common procedure3 and monitored instantly by taking small samples (e.g. 100 μl or less) every 5 min for injection into the lab-on-a-chip NBD system (Fig. 1A). The graph of the integral intensity of the total signal measured versus the time of germination is sigmoid (Fig. 2A), which is typical of a transformation from one stage, i.e. the suspension of spores only, into the other, i.e. the suspension of vegetative cells, with a threshold at time 15 min after heat shock (65 °C for 30 min in the presence of the germinates L-alanine and inosine). This corresponds to the dynamic transformation of Bacillus subtilis spores into vegetative cells. Analysing in detail the time profile of the signals, their half-width (Fig. 2B) and the retention time (Fig. 2C), it is possible to obtain direct insight into the interaction of spores and cells and the polyaniline nanofibrils. As can be concluded from the graphs (Fig. 2A–C), the spores and vegetative cells interact with the polyaniline fibrils in a different way. The half-width of the signal measured for the cells (created from the germinating spores of B. subtilis) is more than three times greater (68 seconds) in comparison to that of the spores of the bacteria (22 seconds). Similarly, the values of the retention time, measured at 10 and 60 min after administration of the germinates (mainly for the transition forms and vegetative cells, respectively), are 2.5 and 2.2 times longer compared to those for the spores of B. subtilis ATCC 6633. This is because of the differences in their surface properties, morphology and biological activity. Control experiments (calibration) using purified spores and vegetative cells, both having a defined population, showed that the electrical response of the nanodetector was proportional to the number of spores and vegetative cells in the sample being examined. However, the signal for vegetative cells was stronger; that is why we observed increasing integral intensity of the total signal measured with the time of germination (Fig. 2A). This can be explained by taking into account the difference in the morphology of the spores and cells and their different ability to interact with the nanofibrils. The conducting polymer (polyaniline) is in the form of positively charged macromolecules (poly-cation), which strongly interact with any negatively charged structures. Thus, having a polar-negatively charged surface, the vegetative cells are much more effective than spores16 in interactions with polyaniline nanofibrils, thus giving a stronger response of the NBD. In both cases the signal that was registered was proportional to the number of spores or cells and in particular when taking into account its integral intensity. This has been proved in a series of calibration experiments. We were able to obtain even more information by analysing the time profile of the signals, i.e. the half-width and retention time (Fig. 2B, C), which gives direct insight into interactions between spores (or cells) and nanofibrils. One can clearly find that vegetative cells interact more strongly than spores, as expected when taking into account the difference in their surface properties, morphology and biological activity.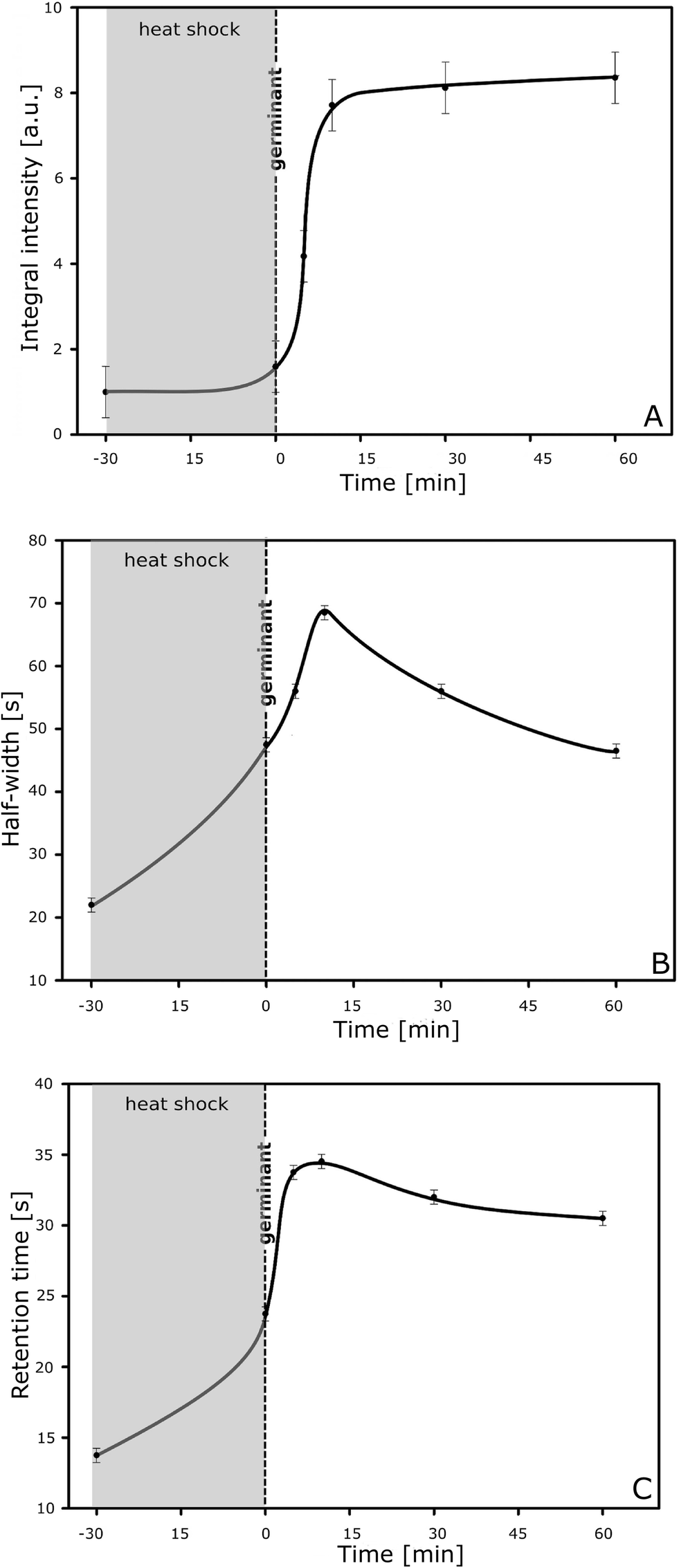 | ||
| Fig. 2 Graphs of (A) integral intensity, (B) half-width and (C) retention time for the signal measured during the whole germination experiment. | ||
In fact, maximum interaction is observed for spores in the transition form 0–15 min after the heat shock. The signal of the transition form is even stronger than that generated by vegetative cells; thus a low population of the transition form in the steady state (below 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ml−1) can easily be detected. Interactions involving spore transition forms are the most intensive and effective. This is due to their unique surface structure.17 Adhesion of such a form is stronger; thus, it leads to a longer retention time and a broader signal (of a greater half-width) for spore transition forms than in any other case (Fig. 2B). Modification of the electrical conductivity of nanofibrils is stronger owing to the interaction of the charged surface of the transformed spores and also because of the direct chemical influence of “biomarkers” produced by the germinating spores,2 which can act on the nanofibrils at the highest possible concentration due to close contact on the nanoscale level.
000 ml−1) can easily be detected. Interactions involving spore transition forms are the most intensive and effective. This is due to their unique surface structure.17 Adhesion of such a form is stronger; thus, it leads to a longer retention time and a broader signal (of a greater half-width) for spore transition forms than in any other case (Fig. 2B). Modification of the electrical conductivity of nanofibrils is stronger owing to the interaction of the charged surface of the transformed spores and also because of the direct chemical influence of “biomarkers” produced by the germinating spores,2 which can act on the nanofibrils at the highest possible concentration due to close contact on the nanoscale level.
Defining specific signals for spores, vegetative cells and the transitional form
When analysing the amplitude of signals at the appropriate retention time (the amplitude is proportional to the number of spores or cells), i.e. 14 s for spores, 30 s for cells and 35 s for the transition form (the values defined from the plot of the RT vs. the duration time of the process, Fig. 2B), one can find information on the dynamics of the main processes induced during germination, including the formation of a transition form of spores before its final transformation into vegetative cells. The results are summarised in Fig. 3. The transition form of spores appears during heat shock, reaching a maximum concentration just after the thermal treatment within the next 15 min. By analysing the data collected in detail we can obtain more information about kinetics. Germination is a complex process which is composed of two main successive stages: [I] annihilation of spores (S) and formation of transition forms (TF) and [II] transformation of transition forms into vegetative cells (C):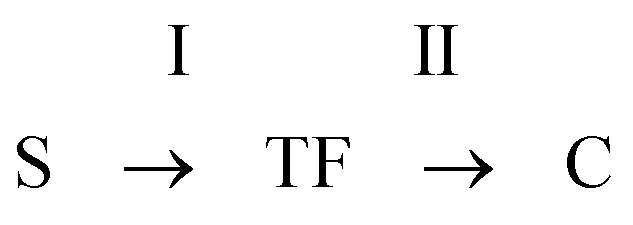
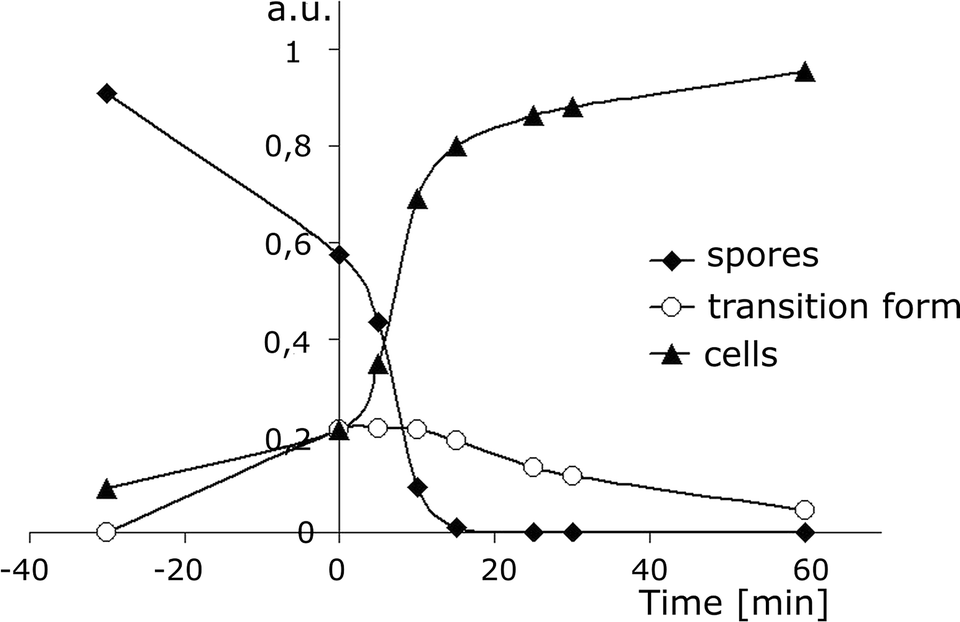 | ||
| Fig. 3 Changes in the population of spores (PS, diamonds), transition forms (PTF, circles) and vegetative cells (PC, triangles) vs. the time of germination, measured as the amplitude of signals at retention times of 14 s, 30 s and 35 s, respectively (Fig. 2B). The values are normalised by 1 according to formula (4), where PS0 = 1 [a.u.]. By analysing these data it is possible to obtain information on the kinetics of the germination process, including the formation of transition forms of spores before their final transformation into vegetative cells. | ||
The process is described by a well-known formula:
| dPS/dt = k1PS | (1) |
| dPC/dt = k2PTF | (2) |
| dPTF/dt = k1PS − k2PTF | (3) |
| PS + PTF + PC = PS0, | (4) |
Based on calibration (an amplitude of 1 a.u. corresponds to 2 × 106 CFU per milliliter of spores, or 2 × 105 spores injected) analysis of the experimental data presented in Fig. 3, one can obtain information on the rate of annihilation of spores (1) during heat shock (−30 to 0 min), which is 373 cells s−1 (in 1 ml). The rate increases dramatically up to 1600 cells s−1 (in 1 ml) under the influence of inducers of germination (inosine and L-alanine), just after their addition at time “0” (Fig. 3, Table 1). The rate of changes in the population of transition forms (TF) within 0–10 min is 0 because the rate of the transformation of spores in the TF is equal to that of the formation of cells from the TF. This is a quasi-stationary state. Then, the population of the TF decreases with a rate of 80 cells s−1 (in 1 ml) within a time range of 30–60 min. Formation of vegetative cells is most intensive (1600 cells s−1 in 1 ml) in the time range of 0–10 min. At the final stage the population of cells increases slowly with a rate of 80 cells s−1 (in 1 ml), which is equal to the value observed for the annihilation of the transition form (TF).
| Time range [min] | −30 to 0 | 0 to 10 | 30 to 60 |
|---|---|---|---|
| P S, spores | −373 | −1600 | 0 |
| P TF, transition forms | 237 | 0 | −80 |
| P C, vegetative cells | 237 | 1600 | 80 |
Comparison with other methods
The results presented here are consistent with those obtained in parallel experiments performed in our laboratories using a conventional technique which is based on fluorescence measurements for detecting the activity of a spore-derived esterase as an indicator of germination and a quantitative parameter of its progress. The plot presenting changes in the fluorescence intensity of fluorescein over time, which is related to esterase activity from the germinating spores of B. subtilis ATCC 6633, shows that the spores remain almost dormant in the absence of germination factors (an increase in fluorescence is low). These results correspond to those obtained using the bioluminometry method as reported elsewhere.18 As expected, the addition of inosine and L-alanine changes the situation, as these compounds are well-known inducers of the germination of spores. They influence spore surface receptors which promote awakening from the dormant state. Then esterase biosynthesis leads to increased transformation of FDA-fluorescein and to more intense fluorescence. The rate of increase in the fluorescence is 0.5 a.u. s−1 at the beginning of the process (0–400 s after adding the germinants) and increases to 1.25 a.u. s−1 within the next 600–800 s (Fig. 4). This corresponds exactly to the time range where the most highly dynamic processes are observed with the use of a nanodetector (Fig. 3).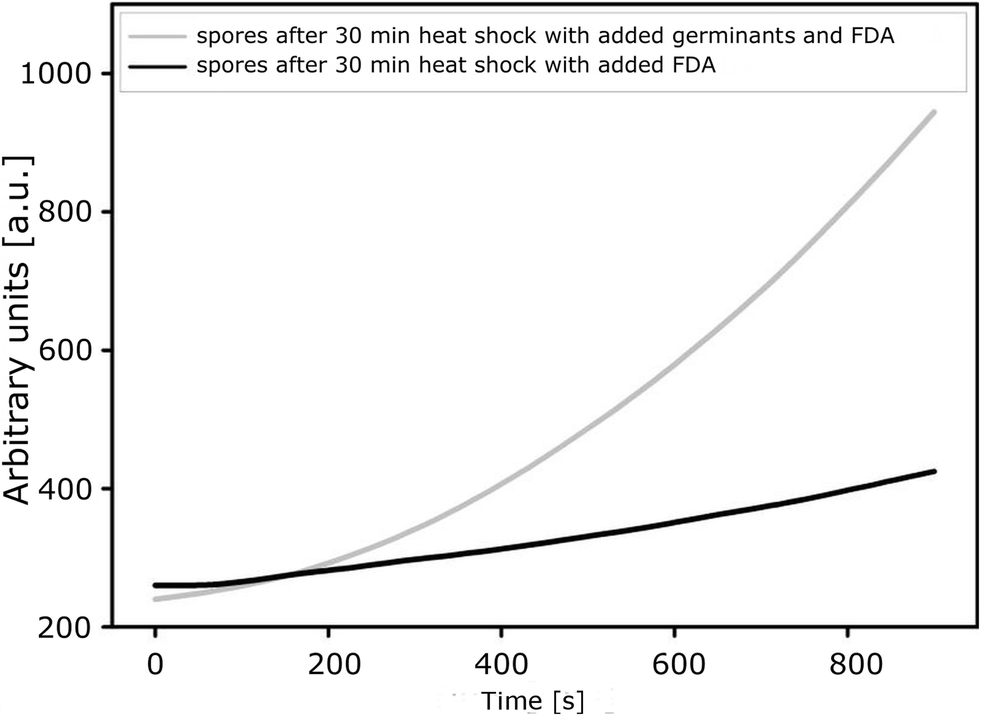 | ||
| Fig. 4 Changes in the fluorescence intensity of fluorescein (generated from the biomarker FDA-fluorescein) during germination of B. subtilis ATCC 6633 with and without germinants. | ||
Experiments performed by Guiwen Wang et al. at the Yong-qing Li laboratory,19 in which Raman spectroscopy and differential interference contrast (DIC) microscopy were used to analyse the kinetics of L-alanine-induced germination of wild-type B. subtilis spores, confirm the time intervals of the germination phases obtained with our method. A clearly visible change of DIC images between 10 and 20 minutes after addition of L-alanine corresponds to the most dynamic changes observed with our method (Fig. 3). This is concomitant with the completion of release of dipicolinic acid (DPA) by the spores, as confirmed by Raman spectroscopy with LTRS.19 Pandey et al. examined the heterogeneity and germination of B. subtilis spores with the help of a phase-contrast microscope. They found that most of the analysed B. subtilis germinated within 3–5 minutes.7 The 3–5 minute interval in the germination of Bacillus spores corresponds to the most dynamic changes and the maximum population of spore transition forms, which is clearly visible when using our method (Fig. 2B and 3).
The method presented here is adequate for the real-time monitoring of transformation of spores into vegetative cells (the germination process) by simultaneous observation of spores, spore transition forms and vegetative cells in a small but representative sample (105 spores). This is achieved by using one, single nanodetector which has been designed as an easy-to-handle lab-on-a-chip unit.
By analysing various parameters of the time profile of the electrical response, i.e. the amplitude, the half-width and the retention time, which are specific for the analysed spores and vegetative cells, it is also possible to detect the presence of highly metabolically active transition forms of spores. The signals, obtained from the nanobiodetector between the 5th and the 20th minute after administration of a germinant, are dominated by a highly active form of B. subtilis which is neither a spore nor a vegetative cell. The maximum activity of the transition form falls on the 45th minute of the experiment, i.e. exactly 15 minutes after exposing the spores (thermally activated) to come into contact with L-alanine and inosine, which are used as the chemical activators of germination. Prior to that, the sample was dominated by spores of B. subtilis, whose number decreased due to the progress of thermal activation. Hence, the amplitude of the signal registered at the retention time, which has been identified as a characteristic of spores, was reduced by about half during 30 minutes of heating. The results are fully consistent with previous, independent studies of the process of transformation sequences of bacteria of the genus Bacillus.7 Additionally, what is worth noting is that the compliance detail also applies to the time scale.
Experimental
The detecting system and procedure
The mechanism of the device is basically similar to that of a standard continuous flow system, CFNBD, which has been successfully applied for bacterial detection. However, now the detector is transformed to a compact lab-on-a-chip system (NBD) supported by a new, more accurate analytical procedure and data processing to reach improved time resolution (in seconds) and sensitivity (0.001 μS), both of which are necessary to monitor the germination of spores.The detecting device consists of two gold electrodes as simple bars (2 mm × 0.5 mm × 0.001 mm) which are deposited on a plastic substrate with the use of a Quorum Technologies Q150R sputter coater. The electrodes are in good contact with the polyaniline nanofibrils formed over a narrow gap (5–10 μm) between the Au microelectrodes so that the current flows through the dense conducting network.
A free-standing polyaniline nanofibril network (Fig. 1A) was prepared by direct synthesis onto Au microelectrodes by electrochemical oxidation of aniline hydrochloride (10% in water, pH ~1) at a potential of 0.8–1.2 V vs. Ag/AgCl with a controlled charge flow (using a Potentiostat Autolab PGSTAT). The morphology of the PANI nanofibrils was examined with a scanning electron microscope (ZEISS EVO 40). The average length was 3–5 μm (maximum above 10 um) and the thickness of single fibrils was about 100 nm or less (Fig. 1A).
To limit the ion current, gold microelectrodes were protected by a self-assembled monomolecular insulating layer formed of benzylthiol (after deposition of PANI nanofibrils). A solution of benzylthiol in methanol (10 mg ml−1) was used. The reaction proceeded for 180 s at room temperature, and then the electrodes were carefully washed with methanol and water. The insulating monomolecular layer considerably improved the electrical response of the nanodetector in the presence of the microorganisms by a significant reduction of the ion current.
The detecting unit was then assembled inside a micro-fluidic platform which consisted of three elements of poly(methyl methacrylate) (32 mm × 50 mm). The outermost element consisted of two inlets for silicone tubing with an internal diameter of 0.2 mm that are 32 mm apart (Fig. 1A). In between there was a hole (3 mm × 6.5 mm) to insert the polyaniline detecting unit which was sealed with a silicone seal. In the middle element a microfluidic channel was formed (3 mm × 3 mm × 32 mm, or 3 mm × 100 μm × 32 mm) that connected both inlets. All of the elements were joined using Tesa sticky tape (double-sided and waterproof).
The system was equipped with a peristaltic pump (GILSON MINIPULS 3), a measuring interface (e.g. ELMETRON CC-505 conductivity meter, which is a tool that is safe for a nanodetector, effective and convenient, despite being used in a non-traditional manner to measure electrical conductivity and temperature at the same time and with very good accuracy) and a PC. Bi-distilled (BiD) or sterile deionised water was used as a medium to transport the samples analysed through the detecting area (flow rate about 1 ml min−1). Generally, a small volume (100 μl or less) of the suspension of germinating spores of bacteria in water was injected every 5 min into the detecting system through a septum. Then the electrical conductivity was measured as a function of time. Changes in the electrical conductivity of nanofibres caused by the presence of spores and cells were plotted as a function of time (a time profile of the electrical response) and analysed. Parameters of the time profile, such as retention time (RT) and half-width (HW), were characteristic of different microorganisms, and the intensity (or amplitude) of the signal was proportional to the number of cells. By measuring the amplitude at the appropriate RT one can obtain information on the components of a complex mixture of microorganisms.
For monitoring the germination of the spores of B. subtilis ATCC 6633 with the NBD system, samples of 100 μl of suspension containing about 2 × 106 spores ml−1 were injected with a microsyringe. The average number of spores injected was about 105; however, the device was able to work efficiently also in the case of a lower and higher number of spores, from 104 to 109. After injection, the electrical conductivity of the nanofibril network was measured with an accuracy of 0.001 μS and registered as a function of time within 300 s (every 1 s with an accuracy of 0.1 s) as the time profile of the electrical response of the NBD. Samples of 100 μl (about 105 spores) were collected and injected into the device through the septum according to the following scheme: at the beginning of the experiment (time: 30 min), at the end of heat shock (time: 0 min), and then at 5 min, 10 min, 15 min, 25 min, 30 min and 60 min after heat shock. This is reasonable because the process is most dynamic at the time of 0–10 min after heat shock, although another sampling time is also possible.
In order to compare the results, measurements were performed with:
- a washed suspension of intact spores,
- water solutions of L-alanine and inosine (blank test),
- a spore suspension after heat shock, and
- a spore suspension after heat shock with the addition of L-alanine and adenosine (0, 5, 10, 15, 25, 30 and 60 minutes after the germinators were added).
The measurements were done in triplicate.
Bacterial cell preparation
Growth and preparation of spores
B. subtilis ATCC 6633 was grown on a standard agar plate with 5% sheep blood (MAST DIAGNOSTICA) at 37 °C (aerobic conditions). After 24 h, single colonies were transferred to flasks (100 ml) with a TSB medium (50 ml). Liquid cultures were grown in a shaking incubator (24 h, 37 °C, 115 rpm). Afterwards, 10 ml of the liquid culture was transferred to a 1-litre Roux bottle containing 200 cm3 of sporulating agar (Sporulating Agar AK, BTL). Roux bottles were incubated at 37 °C for 24 h and subsequently for 4 weeks at room temperature. After 28 days, 10–25 ml of sterile distilled water was added to each bottle (depending on the density of the suspension) and incubated for 1 h at 37 °C. Then the bottles were washed very carefully with gentle shaking (additionally, 10–20 sterile medium-sized (3–4 mm) glass beads were added to intensify the washing). This step was repeated if a significant amount of material was left on the agar surface. Then the obtained suspension was filtered with a gauze (triple-folded) and centrifuged (10000 RCF, 20 min, 4 °C). The supernatant was removed and the pellet was suspended in 10 ml of sterile saline (in this step, additionally 10–20 sterile medium-sized glass beads were added to intensify the mixing). The procedure was repeated. Then the pellet was suspended in 10 ml of 65% isopropanol and incubated for 3 h at 20 °C in order to destroy any remaining vegetative cells. Afterwards the suspension was centrifuged (10000 RCF, 20 min, 4 °C), the supernatant was removed and the pellet was suspended in 10 ml of sterile deionised water at 4 °C. This step was repeated five times. The quality of the spores was determined by microscopic examination to confirm a lack of debris from cell lysis and rods. After washing, the pellet containing spores was suspended in sterile deionised water. The number of spores (CFU ml−1) was estimated by 10-fold serial dilutions (triplicate) which were plated on nutrient agar Petri dishes. After 24 h, plates with a countable number of colonies were selected, colonies were counted and the final concentration was calculated.
Growth and preparation of vegetative cells
B. subtilis ATCC 6633 was grown at 37 °C in a nutrient broth (BTL) with shaking (24 h and 4 h, 115 rpm). Vegetative cells were washed four times with cold (4 °C) deionised sterile water and then suspended in cold sterile deionised water. The quality of the vegetative cells was examined by microscopic observation, looking for the absence of spores. Fresh vegetative cells were prepared each time when required. The concentration of the vegetative cells was estimated by preparing serial log dilutions (the method described above for spores) and expressed as colony-forming units (CFU) per milliliter.Parallel conventional monitoring of spore germination
In order to compare the results, spore germination was tested in parallel at the same time by using a method on the macroscale.3 Spores derived from esterase activity were used as an indicator and a quantitative parameter of germination progress. For this purpose the spores were washed four times with cold (4 °C) deionised sterile water by centrifugation and were subsequently resuspended in water. The final pellet was suspended in cold (4 °C) sterile water at a concentration of about 2 × 106 CFU ml−1. The suspension of spores (1 ml) was incubated at 65 °C for 30 min for induction of germination. To boost germination by receptor interactions, after heat shock the solution of L-alanine and inosine (Calbiochem) was added at a concentration of 10 mM and 2 mM, respectively. Non-fluorescent fluorescein diacetate (FDA) (Sigma-Aldrich), which is a substrate for the cellular esterase enzyme, was used as a germination indicator. The germination process stimulated the enzyme release from waking spores and thus started enzymatic hydrolysis of the FDA. The product of hydrolysis was fluorescein, which is a well-known fluorescent compound. The concentration of the fluorescent product was measured using the F-4500 HITACHI fluorescence spectrophotometer. Solid FDA (Sigma-Aldrich) was dissolved in acetone (POCh) to reach a concentration of 2 mg ml−1 and stored as a stock solution at −20 °C. Prior to each test the FDA was diluted to a concentration of 0.5 mg ml−1, and 10 μl was added to 1 ml of each medium or each media component (FDA final concentration was 0.005 mg ml−1). Measurements were conducted in quartz cuvettes containing 1 ml of spore suspension (about 2 × 106 spores ml−1) in 0.1 M phosphate buffer (pH 7.6). For the measurements the following conditions were applied: excitation wavelength 488 nm, emission wavelength 526 nm, time-scan period 15 minutes, time accuracy 1 second, photomultiplier voltage 750 V, EX/EM slit 10 nm. The results are presented in Fig. 4.Conclusions
The new lab-on-a-chip-type device with a free-standing polyaniline nano-network allows monitoring of the germination process in real time. The method is based on non-specific physicochemical interactions of cells and spores with conducting polymer nanofibrils, which results in a modification of the electrical conductivity of individual nanofibrils and, consequently, of the whole network. This generates a dynamic-type electrical response which is characteristic of different types of cells, including spores and vegetative cells. This is caused by the considerable differences in the size and morphology of spores and vegetative cells as well as in the surface structure and biochemistry of all processes influencing modification of the electrical conductivity of polyaniline. All of these differences in interactions between each type of cell and the polyaniline conducting network, spores, transition forms and vegetative cells can be detected with the aid of the device described here. Our results are fully consistent with previous independent studies of the germination process of bacteria of the Bacillus genus. The method, based on non-specific physicochemical interactions on the nanoscale, is universal and could be useful for advanced studies of the kinetics and the mechanism of other microbiological processes. In a microfluidic version, it is a fast analytical technique, accurate enough for many applications in microbiology, biotechnology, health, security and defence, where spores are in use (proven sensitivity is of 104 spores injected, and the range of application exceeds a value of 1010). Other possible applications are briefly presented in the ESI.†Acknowledgements
The work was supported by the DARPA (USA) within project no. N10PC20099 and in part by the Srem Borough Council.Notes and references
- E. Abel-Santos, Bacterial Spores: Current Research and Applications, Caister Academic Press, UK, 2012 Search PubMed.
- P. Setlow, Curr. Opin. Microbiol., 2003, 6, 550 CrossRef CAS PubMed.
- L. Ferencko, M. A. Cote and B. Rotman, Biochem. Biophys. Res. Commun., 2004, 3, 854 CrossRef PubMed.
- C. Laflamme, J. Ho, M. Veillette, M. C. de Latrémoille, D. Verreault, A. Mériaux and C. Duchaine, Arch. Microbiol., 2005, 2, 107 CrossRef PubMed.
- D. Chen, S. S. Huang and Y. Q. Li, Anal. Chem., 2006, 19, 6936 CrossRef PubMed.
- L. Kong, P. Zhang, G. Wang, J. Yu, P. Setlow and Y. Q. Li, Nat. Protoc., 2011, 5, 625 CrossRef PubMed.
- R. Pandey, A. Ter Beek, N. O. Vischer, J. P. Smelt, S. Brul and E. M. Manders, PLoS One, 2013, 3, e58972 Search PubMed.
- D. D. Jr Evanoff, J. Heckel, T. P. Caldwell, K. A. Christensen and G. Chumanov, J. Am. Chem. Soc., 2006, 39, 12618 CrossRef PubMed.
- J. K. Daniels, T. P. Caldwell, K. A. Christensen and G. Chumanov, Anal. Chem., 2006, 5, 1724 CrossRef PubMed.
- Y. S. Liu, T. M. Walter, W. J. Chang, K. S. Lim, L. Yang, S. W. Lee, A. Aronson and R. Bashir, Lab Chip, 2007, 5, 603 RSC.
- J. J. Langer and K. Langer, Rev. Adv. Mater. Sci., 2005, 10, 434 CAS.
- K. Langer, P. Barczyński, K. Baksalary, M. Filipiak, S. Golczak and J. J. Langer, Microchim. Acta, 2007, 159, 201 CrossRef CAS.
- S. Pal, E. C. Alocilja and F. P. Downes, Biosens. Bioelectron., 2007, 9–10, 2329 CrossRef PubMed.
- Y. Liu, S. Chakrabartty and E. C. Alocilja, Nanotechnology, 2007, 18, 424017 CrossRef PubMed.
- J. J. Langer, P. Barczyński, J. Warchoł, K. H. Bartkowiak and K. Langer, Biosens. Bioelectron., 2009, 9, 2947 CrossRef PubMed.
- E. M. Sonnenfeld, T. J. Beveridge, A. L. Koch and R. J. Doyle, J. Bacteriol., 1985, 3, 1167 Search PubMed.
- A. Driks, Microbiol. Mol. Biol. Rev., 1999, 1, 1 Search PubMed.
- A. Bielawska-Drozd, Applied Science and Analysis, ASA Newsletter, (ASA Inc.), 2005, 2, 18 Search PubMed.
- G. Wang, P. Zhang, P. Setlow and Y.-Q. Li, Appl. Environ. Microbiol., 2011, 10, 3368 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc01009d |
| This journal is © The Royal Society of Chemistry 2015 |