
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cell proliferation and migration inside single cell arrays†
Mayuree
Chanasakulniyom
,
Andrew
Glidle
and
Jonathan M.
Cooper
*
The Division of Biomedical Engineering, School of Engineering, The University of Glasgow, G12 8LT Glasgow, UK. E-mail: jon.cooper@glasgow.ac.uk; Fax: +44 (0)141 330 6002; Tel: +44 (0)141 330 4931
First published on 16th October 2014
Abstract
Cell proliferation and migration are fundamental processes in determining cell and tissue behaviour. In this study we show the design and fabrication of a new single cell microfluidic structure, called a “vertically integrated array” or “VIA” trap to explore quantitative functional assays including single cell attachment, proliferation and migration studies. The chip can be used in a continuous (flow-through) manner, with a continuous supply of new media, as well as in a quiescent mode. We show the fabrication of the device, together with the flow characteristics inside the network of channels and the single cell traps. The flow patterns inside the device not only facilitate cell trapping, but also protect the cells from mechanical flow-induced stress. MDA-MB-231 human breast cancer cells were used to study attachment and detachment during the cell cycle as well as explore the influences of the chemokine SDF-1 (enabling the quantification of the role of chemokine gradients both on pseudopod formation and directional cell migration).
Introduction
Single cell microarrays have provided a simple method for the interrogation of individual cells, which are physically retained and isolated by physical boundaries, known as traps.1–3 Such arrays are regarded as an improvement of plastic multiwell plates for single cell analysis, as they provide easy registration (so that many single cells can be readily located, observed and revisited). The array also enables complex flows to be established so multi-parameter measurements can be made. For example, the unique arrangements of flow enables concentration gradients of chemicals to be established across the device, such that the behaviour of each cell can be quantified in response to different doses of stimulant.4Typically such arrays have been made from PDMS and glass, functionalised either with extracellular matrix (ECM)5 or specific antibodies/cell-adhesive ligands.6 Although such devices are considered to be straightforward for high-throughput trapping and single-cell studies, they still have some limitations in use. The side walls of the traps not only impose restrictions on cell shape, but they also have a high surface area around the cell, which can result in a reduction in cell growth7 and a restriction in cell–cell communication. Thus, although attempts have been made to describe these structures in terms of the cell's natural environment, the reality is that the cell may be under stress, in situ.8
Photodefinable silicon elastomer (PDSE) is a photopatternable, spin-on polymer. The chemical composition of the PDSE is that of a silicone resin dissolved in a plasticising silicone matrix.9,10 PDSE has previously been extensively used for electronics and optical applications9,11–13 as its properties (which include low stress, low modulus, low temperature curing, low shrinkage, good moisture resistance, good dielectric properties, high thermal stability and high transparency) are of benefit to those applications. It has, however, also found various applications in biological studies, not least as it has demonstrated biocompatibility with various cell lines,14 providing an easy way to develop complex 2D and 3D structures. In this respect, the polymer has been shown to adhere well both to glass, as well as other elastomeric polymers such as polydimethylsiloxane (PDMS). In this study we use PDSE, PDMS and other photopatternable resist layers (such as AZ4562 photoresist) to create multilayer constructs providing a new geometry for the study of the chemokines and the response of individual cells.
Stromal cell-derived factor (SDF-1), also known as CXC chemokine ligand-12 is a small cytokine belonging to the CXC chemokine family. It binds exclusively to its receptor CXCR4, expressed on many hematopoietic cells such as CD34+ hematopoietic stem cells, T-lymphocytes, B-lymphocytes and neutrophils. It is also known as a co-receptor for HIV entry to the cell.15 CXCR4 is also expressed in various types of cancer, including those present in breast cancer.16–20 The expression of CXCR4 is undetectable in normal breast, ovarian, prostate epithelial cells. However, it is significantly up-regulated in cancer cells (it is the most common chemokine receptor expressed in most cancer cells).21–23
The interactions of SDF-1 and CXCR4 have been shown to play a critical role in regulating the metastatic destination of breast cancer cells.24 SDF1 has also been shown to increase the invasiveness and migration of breast cancer cells when present as concentration gradients (i.e. the cell responds to the presence of a change in concentration of the chemokine).24–26 As SDF-1 is highly expressed in lymph nodes, bone marrow, lung and liver, it may therefore account for the migration of breast cancer cells to these sites.22,24,27,28 Indeed, reduction of the CXCR4 expression or using CXCR4 antagonists can effectively inhibit the metastasis of breast cancer cells,22,29,30 indicating that the interaction between SDF-1 and CXCR4 is crucial for cancer metastasis.
In this paper, we are interested in the creation of a new chip structure that addresses the limitations of existing trapping geometries (which, it has been argued, can restrict cell growth and cell communication). To demonstrate the efficacy of the new geometry, we explore adhesion, proliferation and migration assays in the study of MDA-MB-231 in the presence of concentration gradients of SDF-1α.
Experimental
Fabrication
Vertically integrated array (VIA) cell traps are composed of three layers (Fig. 1) and were fabricated by photolithography (see ESI† for schematics of the fabrication and the experimental set-up). The upper polymeric layer contained a channel for the introduction of cells, and was linked via a middle layer, which serves to organise single cells, to a lower layer, for the outflow of culture media (Fig. 1A–C). The final device, as shown in the micrographs in Fig. 1D, E comprised a bottom layer with 100 μm circular cavities (for cell proliferation) with each “chamber” linked together by 20 × 80 × 6 μm “VIA” channels.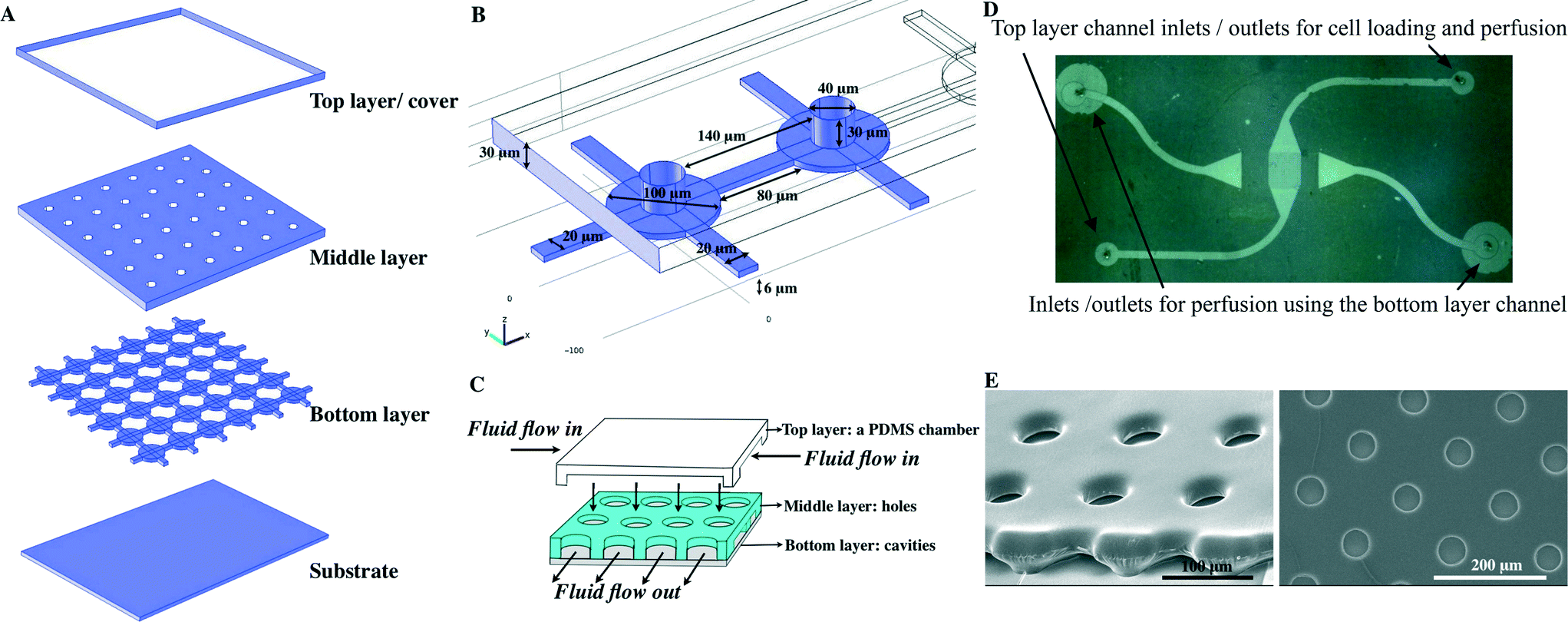 | ||
| Fig. 1 Illustration of the multi-layered VIA device (A) composed of three layers (the upper layer comprises a PDMS chamber, the middle layer has an array of circular holes and the bottom layer has 100 μm circular cavities, linked together by 20 × 80 × 6 μm channels). The substrate is typically a glass microscope slide or coverslip (the latter for ease of cell observation). (B) The detail of the geometry of the VIA devices. (C) The cartoon shows composition of the devices and flow direction. (D) Top view of the VIA array comprising fluid inlets and outlets and an observation area (centre) with an array of holes. (E) SEM images of the VIA devices (viewed from the side (left) and from the top view (right)). | ||
The middle layer has an array of circular holes (40 μm in diameter) for cell trapping, aligned directly above the cavities. The array of holes is used to funnel the individual cells into the cavities of the bottom layer. The structures of both the bottom layer and middle layer were fabricated from a combination of PDSE and AZ4562 photoresist (see ESI†). The top layer was a PDMS chamber used for cell loading and perfusion with cell culture medium.
Computational model
Computational fluid dynamics (CFD) was used to predict the fluid flow and re-circulation within the VIA device using COMSOL Multiphysics 4.0. The culture medium was assumed to have the same properties as water, with a density of 1000 kg m−3 and a viscosity of 0.001 Pa s. The steady-state Navier–Stokes equation for incompressible fluids was used with a no slip boundary condition for walls. A Reynolds number Re ≤ 5.38 × 10−5 was calculated at typical inflow velocities of 1 × 10−4 m s−1 (~0.5 μl min−1) confirming laminar flow characteristics.Cell culture
MDA-MB-231 cells, obtained from ATCC, were cultured in L-15 medium (Leibovitz-15) supplemented with 10% of fetal bovine serum, 10 units of penicillin and 10 μg of streptomycin. Cells were cultured at 37 °C, and were harvested by trypsinization with 0.25% (w/v) trypsin–0.53 mM EDTA solution.Cell loading and perfusion
After fabrication, before loading cells, individual devices were treated by incubating with 0.1 mg ml−1 of fibronectin solution for 30 min before rinsing with PBS. This was followed by priming the device using complete culture media (supplemented with 10% FBS) for 1–2 h.Cell loading was performed by injecting a cell suspension into the top right, top layer inlet port of Fig. 1D using PEEK tubing (100 μm inner diameter) fitted to a gas-tight syringe (Hamilton) and microsyringe pump (Harvard Apparatus). Various cell densities were used to explore their influence on the microwell loading characteristics and the optimal one in terms of single cell occupancy and good overall filling was found to be 3 × 106 cell ml−1 (in culture media) The procedure was as follows: a flow rate of 0.5 μl min−1 was used for the initial introduction of the cells to prevent them sedimenting in the PEEK tubing; this was then reduced to 0.1 μl min−1 after the cells had entered the device (to reduce mechanical shear stress and potential cell damage during loading). The suspended cells were flowed with the flow rate of 0.1 μl min−1 for 30 s after which the flow was paused for 30 s. This procedure was repeated twice to achieve a maximum loading (estimated as 85%). Cells were perfused with culture media containing either 100 ng ml−1 of SDF-1α (Abcam) or free from added SDF-1α. After this was completed, the cell loading syringe was replaced, and SDF-1α containing media was introduced through the top left, top layer inlet (Fig. 1D). At the same time, SDF-1α free media being introduced through the bottom right, top layer inlet (Fig. 1D). For both inlets, the fluid flow rate was maintained at 0.05 μl min−1. Where the two fluid streams meet (in the centre of the microhole patterned region), a steep gradient of SDF-1α was generated by the diffusion of SDF-1α from the SDF-1α media, into the SDF-1α media. As in other studies, after a period of approximately 20 min, the spatial extent of this gradient reaches a steady state as a consequence of the continual removal and replenishment of material by the constantly flowing streams.31 Importantly, the reliability and pulse free nature of the syringe pumps used ensured that this gradient remained stable during the course of the experiments.
Microscopy and image analysis
Cell migration was observed using time-lapse fluorescence microscopy (Axio Observer, Carl Zeiss) under the control of Axiovision software. Long-term cell culture was facilitated by enclosing the microscope within a plexiglass box, maintained at 37 °C by circulating warm air generated by a heating unit (Tempcontrol 37-2 digital 2-channel, Pecon). Images were taken every 15 min for 20 hours.The MDA-MB-231 cell migration toward SDF-1α was analysed using ImageJ32 with two additional plug-in modules, namely Manual tracking (to collect movements of individual cells) and Chemotaxis tool (to analyse the records of cell movement off-line). Cells of interest were selected visually and image sequences were analysed frame by frame.
Results and discussion
Fluid flow characteristics
3D simulations were used to predict the flow characteristics inside the single cell array. In Fig. 2 the velocity profile is shown for a device in which fluid flows in through one of the top layer ports of Fig. 1D. The outlets are the other top layer port and both bottom layer ports. (In the simulation shown the inlet velocity is 1 × 10−4 m s−1, corresponding to a flow rate of 0.5 μl min−1).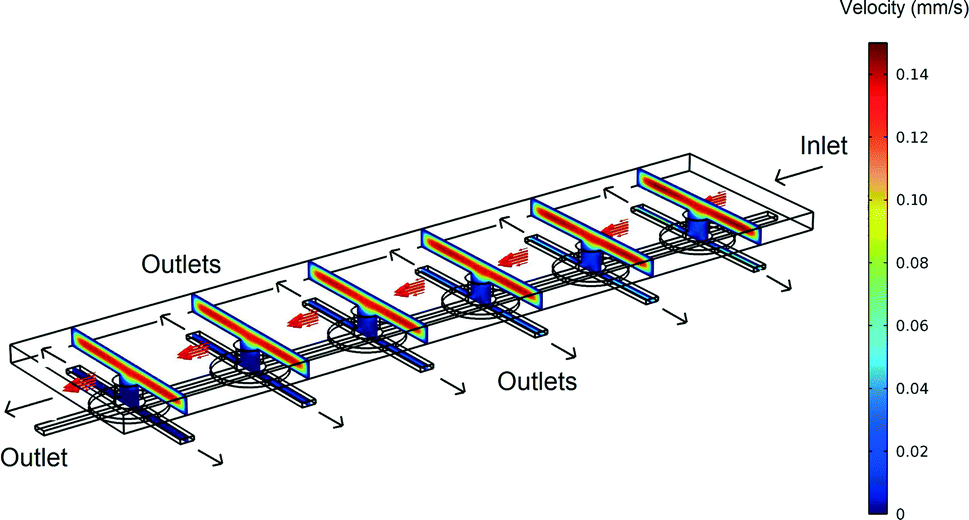 | ||
| Fig. 2 An overview of the velocity contours for a part of the VIA device obtained from 3D CFD simulations. | ||
The velocity contours show that the flow rate inside the VIA is two orders of magnitude lower than the velocity in the top chamber, giving a greatly reduced shear stress (the velocity in the middle of the top layer chamber and the bottom layer linker are approximately 1 × 10−4 m s−1 and 4 × 10−6 m s−1, respectively). Calculations indicate that for an inlet flow rate of 1 × 10−4 m s−1, the shear stress experienced by a cell adhered to the base of the bottom channel in the device would be ~0.0002 Pa. This value is negligible compared to the 0.2 Pa that has been shown to influence T-lymphocyte migration in solutions containing SDF-1α.33 In that study it was shown that cells moved in the direction opposing the shear stress. Thus in studies such as that here, where we wish to explore the influence of chemical gradients alone, it is important to be able to eliminate possible influences from fluid flow, whilst still maintaining a means to deliver fresh nutrients and chemokines. Note, in this device, if desired, after seeding, the adhered cells can be subjected to high shear stress flows by using the bottom layer ports as inlets together with a high inlet flow rate.
As shown in Fig. 3, the majority of the fluid flow (streamlines) are in the top layer. This, combined with the top layer having a larger cross-section area than the bottom layer outlets, make it relatively easy to establish gradients of chemicals across the device (in x,y).
 | ||
| Fig. 3 Shows the CFD generated flow profiles across the device (in cross-section), demonstrating that cells within 5–10 μm of the bottom surface experience little shear-stress. Dimensions in microns, shear stress colour scale from 0 Pa (blue) to 0.04 Pa (orange). | ||
Cell loading, attachment and cell division in VIAs
Cell attachment to a surface such as glass or a suitably treated plastic is also essential for cell growth.34 Attachment is involved in DNA synthesis and growth. If cells remain in suspension, their growth and DNA synthesis will virtually cease.34,35 Attachment is also essential for the anchorage-dependent cells to pass through the restriction point and enter the mitosis phase.36 In this work, the substrates were treated by incubating in 0.1 mg ml−1 of fibronectin solution for 30 min before rinsing with PBS and then priming the devices with complete culture media (supplemented with 10% FBS) for 1–2 h, before loading cells.It was found that when using a loading density of 3 × 106 cells ml−1 and flow rate of 0.1 μl min−1 for 30 s, approximately 60% of the of the chambers occupied were occupied by single cells and ~50% of all of the available chambers were occupied by one or more cells. On repeating the procedure, the overall occupancy of cell loading increases to ~85% (Fig. 4). These numbers are comparable with those obtained using open microhole devices37 with an open hole of the same size as the top layer open hole here. However, as suggested by the data of ref. 37, the structure of the device employed here means that the single cell occupancy is proportionately higher than would be the case if top layer hole opening were the same size as the bottom layer cavity. Importantly, the larger sized bottom layer cavity enables the study of division and migration of cells that are within a semi-encapsulated volume defined in three dimensions.
 | ||
| Fig. 4 Cell loading in a VIA devices showing both single and two cell occupancy after at a time of 1 min after commencement of loading at 0.1 μl min−1. For ~60 s loading time, although the overall microwell occupancy increases, the relative proportion of single to two cell occupancy decreases. | ||
As stated, cell attachment and proliferation inside the VIA devices was observed using time-lapse microscopy. Fig. 5(a–c) is representative of results and illustrates the attachment of MDA-MB-231 cells in three different positions under cell medium perfusion (at 0.05 μl min−1). Attachment occurred between 2.5–5 hours after loading (the process of attachment itself is important for the study of cell responses towards a stimulus, as the cells need to attach to the surface in order to migrate, and forms the basis of routine assays, per se).
 | ||
| Fig. 5 Cell attachment inside the VIA devices under the cell culture medium perfusion rate of 50 nl min−1. Each panel represents cell attachment in individual positions; panels (A) and (B) occurred at 2.5 hours after cell loading while the attachment in panel (C) occurs about 5 hours after loading. Scale bar is 50 μm. | ||
Cell division in the VIA devices, Fig. 6, shows how the cell round-up and lose attachment to the surface. Cells take approximately 1 hour to divide after detachment from the surface. This behaviour and its duration corresponds to the typical timings for eukaryotic cells to complete the M (mitosis) phase where it is known cells become less adhesive and have the rounded morphology due to disassembly of focal adhesions.36,38,39 After mitosis, the cells started to spread again.
 | ||
| Fig. 6 Cell proliferation inside the VIA devices. The MDA-MB-231 cells were introduced into the VIA devices and the proliferation was observed every 15 min under cell culture medium perfusion. Scale bar is 50 μm. | ||
Attachment and detachment during the cell cycle is critical to cell proliferation, and the low fluid flow rates in the VIA serve to protect the dividing and daughter cells well from shear stress, see e.g.Fig. 3. Cell adhesion in Fig. 5 and cell division in Fig. 6 indicate that the natural environment in the device does not have a significant influence on cell survival, nor does it produce effects that lead to a quiescent cell state.
Cell migration toward SDF-1α
To study cell responses toward SDF-1α, MDA-MB-231 cells were perfused using two inlet streams, one containing culture media, the other with media containing 100 ng ml−1 SDF-1α at flow rates of 0.05 μl min−1. This generated a steep gradient of SDF-1α. For the experiment shown in Fig. 7, the uppermost port of the top layer (Fig. 1D) contained the media + SDF-1α solution, and the lowermost port of the top layer contained media alone. Both ports of the bottom layer served as outlets. The gradient generated by this flow pattern could be evaluated and quantified by flowing fluorescein labelled dextran (10 kDa) through the inlet and outlet ports using the same conditions as for the SDF-1α experiments (Fig. 7E–F) (in general this was done after each experiment). Fluorescence measurements showed that the gradient was established within ~20 min of commencing the flow as expected for mass transport based on diffusion and laminar flow, at these flow rates. Measurements with fluorescent dextran were performed on each device fabricated so that small variations in flow characteristics due to fabrication imperfections could be taken into account (ESI† Fig. S3 shows a typical concentration profile obtained from a fluorescent image). Cell migration was observed using time-lapse microscopy by taking images every 15 min for 20 hours.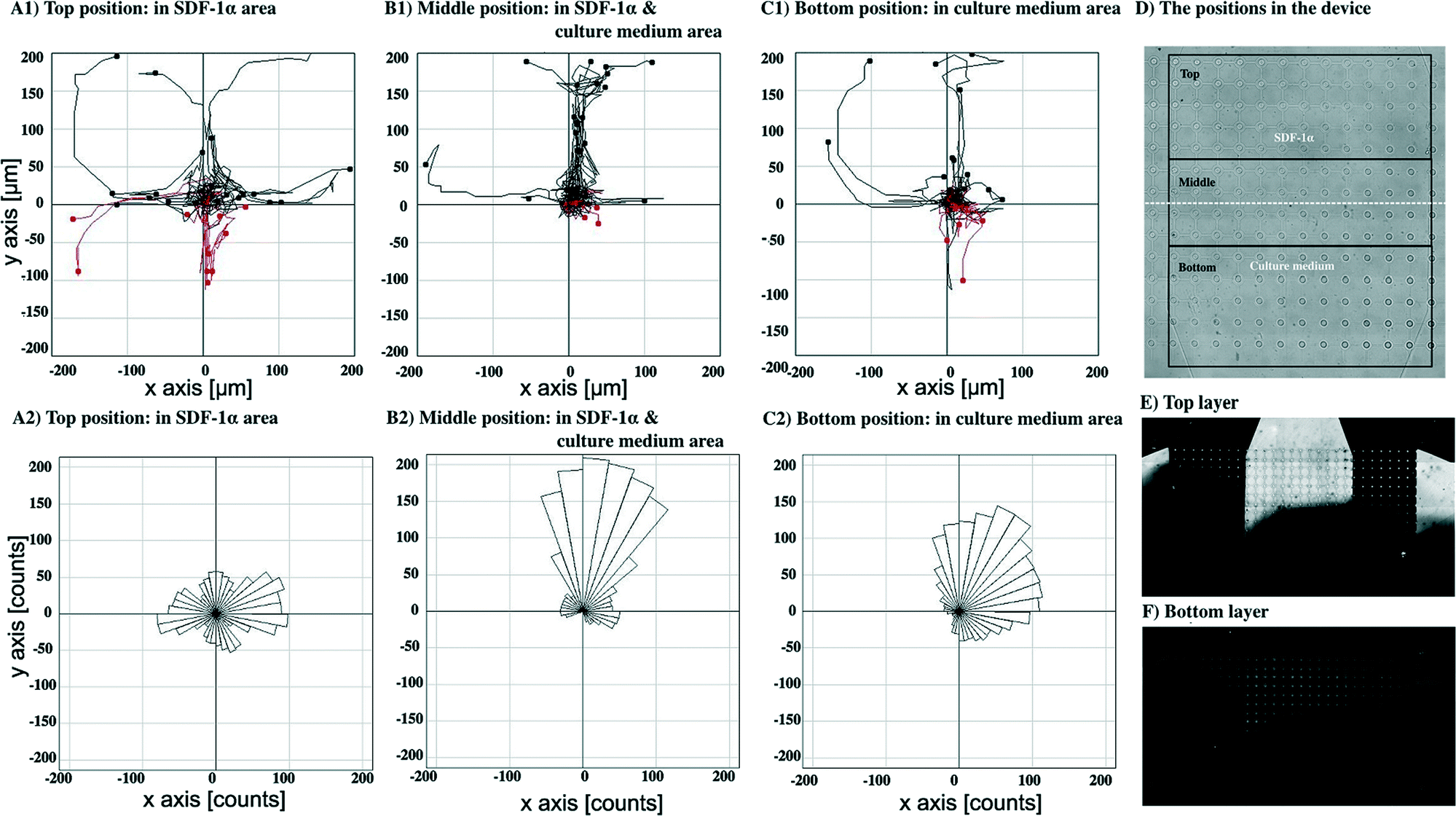 | ||
| Fig. 7 Cell migration towards an SDF-1α gradient by introducing the culture medium through the top layer inlets with flow out through the lower outlets. Cell paths during 20 hours of observation and the final positions of cells at the end time point from top (A1), middle (B1) and bottom (C1). Cells moving upward and downward are shown in black and red, respectively. The directions of pseudopods from top (A2), middle (B2) and bottom (C2) of the device. An overview of the positions in the device (D). Distribution of fluorescein-dextran (MW. 10 kDa) in the top layer (E) and bottom layer (F) of the device using the same perfusion condition as cell culture medium, to illustrate both lateral and vertical concentration gradients. Bright area corresponds to the fluorescein-dextran, the dark area to plain culture medium. | ||
Fig. 7A1, B1 and C1 show cell paths during 20 hours with the final positions of cells at the end time point from the top, middle and bottom position in the device (defined as in Fig. 7D), respectively (n = 30 cells). Cell paths and final cell positions are shown relative to their starting point, as the origin (0,0).
Fig. 7A1 shows migration pathways in a constant concentration of SDF-1α (no gradient) whereas Fig. 7C1 shows the migration in a region of very low SDF-1α concentration (<5 ng ml−1). Fig. 7B1 shows that the greatest number of migration paths are in the region of the steepest concentration gradient.
Fig. 7A2, B2 and C2 show the trajectories of growth of pseudopods analysed from the top, middle and bottom position in the device, respectively. In the middle, Fig. 7B2, and the bottom, Fig. 7C2, the distribution of the pseudopod direction was biased toward the direction of the SDF-1α source, whereas the distribution of pseudopod directions in the top position is random, Fig. 7A2.
To confirm that this directional bias was due to the SDF-1α gradient, the flow direction of the SDF-1α was switched by introducing culture media either containing 100 ng ml−1 of SDF-1α or without the SDF-1α through the bottom layer inlets at the same flow rate, 0.05 μl min−1, Fig. 8. The cell paths and the final position of cells in the middle position having both SDF-1α and cell culture medium were recorded, Fig. 8B1. Again, the controls were observations recorded in regions of uniform SDF-1α concentration, Fig. 8A1 – where cells move randomly. In regions of low SDF-1α concentration, Fig. 8C1 cells again move toward the higher concentration of SDF-1α. Similarly, the distribution of pseudopod directions was biased toward the direction of the SDF-1α source for cells in both this region and the region of the steepest concentration gradient, Fig. 8B2. In contrast, a random distribution of pseudopod directions was observed from the cells in the uniform SDF-1α region, Fig. 8A2.
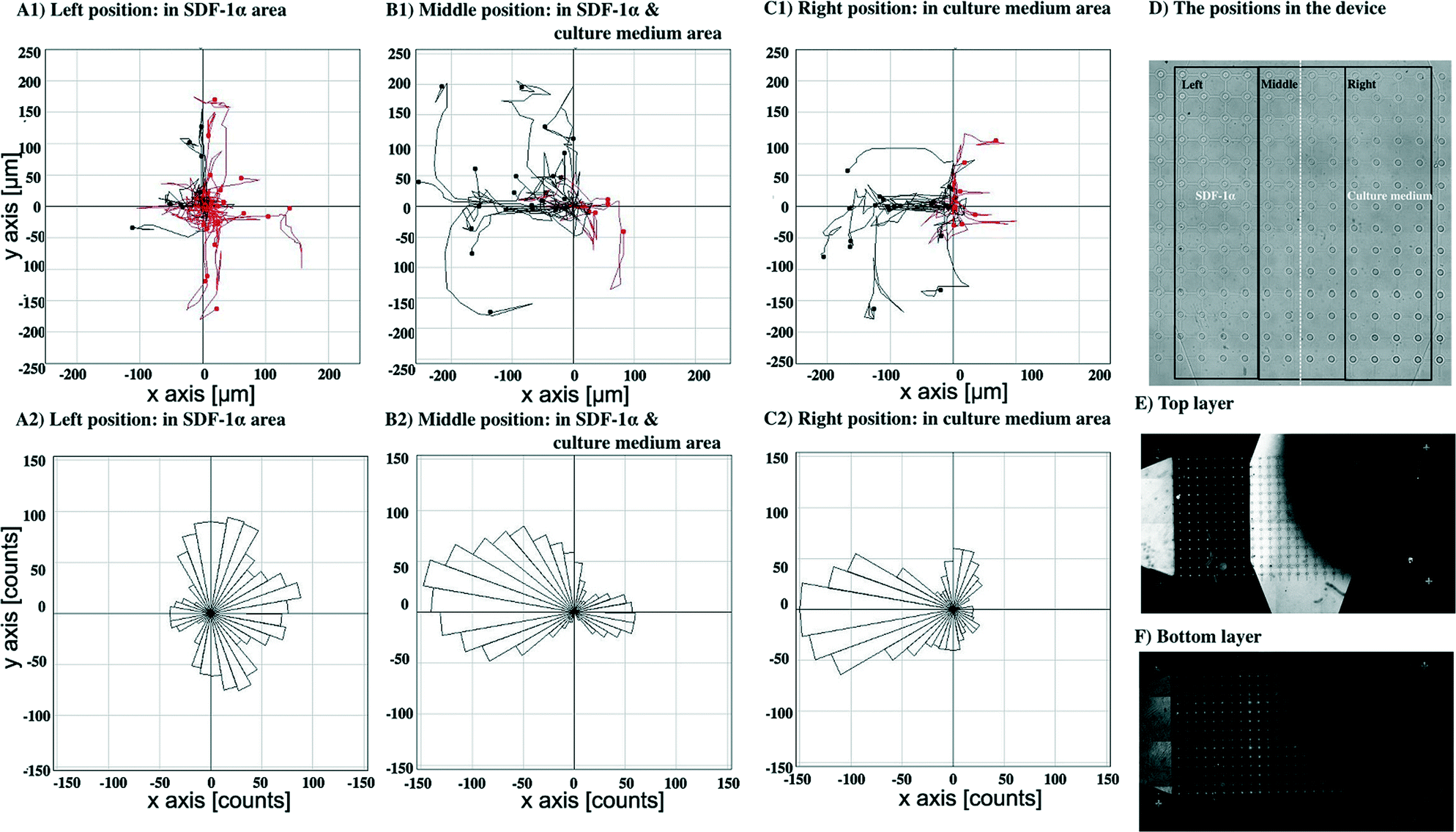 | ||
| Fig. 8 Cell migration towards SDF-1α gradient by introducing the culture medium through the bottom inlets and flowed out via the top layer outlets. Cell paths during 20 hours of observation and the final positions of cells at the end time point from left (A1), middle (B1) and right (C1) positions. Cells moving leftward and rightward are shown in black and red, respectively. The directions of pseudopods from left (A2), middle (B2) and right (C2) positions. An overview of the positions in the device (D). Distribution of fluorescein-dextran (MW. 10 kDa) in the top layer (E) and bottom layer (F) of the device using the same perfusion condition as cell culture medium. Bright area corresponds to the fluorescein-dextran, the dark area to plain culture medium. | ||
An average chemotaxis index40 was calculated for each of the six groupings of cells in Fig. 7 and 8 by evaluating the cosine of the angle that each cell moves with respect to the average direction of the particular group in which the cell is (from the rose diagrams, it can be seen that this average direction is qualitatively similar to the average direction of the fluorescein gradients shown in Fig. 7E and 8E). These chemotaxis index calculations show that the chemotaxis index is low (~0.05) for cells in regions of high SDF-1α concentration (~100 ng ml−1, Fig. 7A and 8A), consistent with a hypothesis that when the cells are surrounded by a moderate or high concentration of SDF-1α, they do not respond to gradients in that concentration. In contrast, for the cells in the regions of steepest gradient (the central parts of the flow field, Fig. 7B and 8B, SDF-1α concentration 25–75 ng ml−1), the migration is strongly directed towards regions of higher SDF-1α concentration (chemotaxis indices of 0.75 and 0.65 respectively).
As a consequence of the VIA traps being arranged on a well defined, regular array, it is easy to compare in more detail the chemotactic movements of individual cells with the concentration profile of SDF-1α. Thus, as Fig. 9 shows, the migration of the cells is clearly towards the higher concentration of SDF-1α (i.e. in the direction right to left, Fig. 9). Furthermore, the extent of movement is generally largest in the region where the gradient is steepest i.e. along the diagonal, top left to bottom right. It is noted that the numbers of cells that experience a particular gradient in any given experiment with a single VIA device are relatively small, but, as a consequence of the well defined, regular array format, chemotactic responses from experiments performed on a series of different devices can be reliably grouped together to improve the statistical quality of the data.
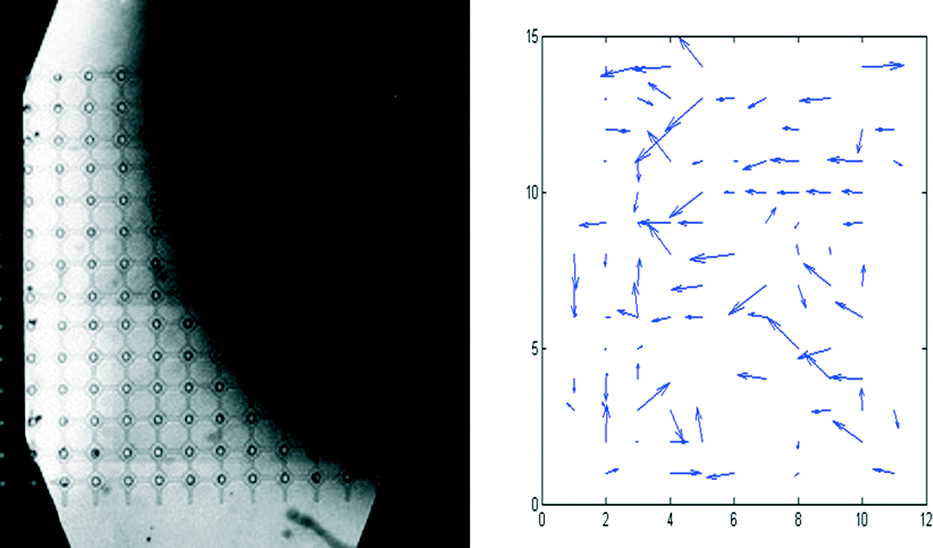 | ||
| Fig. 9 Left: distribution of fluorescein-dextran (MW. 10 kDa, a proxy for SDF-1α) in the top layer of the device of Fig. 8 (the bright area corresponds to the fluorescein-dextran region, the dark area corresponds to the media only region). Right: displacement of individual cells during the course of the 20 h experiment of Fig. 8. | ||
Finally, it is also seen that in areas where the SDF-1α concentration is low (0–25 ng ml−1, Fig. 7C and 8C and top right of Fig. 9), but nevertheless there is still a small gradient in concentration (see ESI† Fig. S3), the cells migrate towards the higher SDF-1α concentration (chemotaxis indices of 0.6 and 0.65 respectively). These results are consistent with the observations that many eukaryotic cells are able to interpret differences in concentration of a chemo-attractant, which may be as little as 2% differences over the length of the cell,41 and the results here support this as a mechanism, however this migration along a concentration gradient may only occur when the stimulant concentration is below a certain threshold value.
In order to migrate, cells will produce pseudopods in response to chemo-attractive signals and which ultimately guide them toward chemoattractants.42Fig. 10 shows such events, namely that the cell extends its pseudopods to sense the surroundings and it maintains only the pseudopod toward the direction of the SDF-1α gradient. This result together with the results above (Fig. 7 and 8) indicate the efficacy of the VIA device in the study of MDA-MB-231 migration in the presence of SDF-1α gradients.
 | ||
| Fig. 10 Extension of cell pseudopods. The culture media containing SDF-1α was introduced through the bottom left inlets. Extension of pseudopods (arrow 1 and arrow 2) within a gradient. The pseudopod (arrow 2) is directed toward a region of higher SDF-1α concentration and remains, whereas other pseudopods (e.g. arrow 1) retract. | ||
Conclusions
This paper demonstrates the use of multilayer VIA devices for cell proliferation and migration studies. The geometry and the flow characteristics inside the array facilitate cell-trapping, serving to reduce shear stress caused by high fluid flow. The array also provides suitable environments to study cell migration toward stimuli under perfusion conditions. Further applications of the array may benefit from the modularity of the design of the different layers, which can accommodate different geometries for different cell types or more complex stimulation conditions. For example, the influence of subchannel dimensions could be readily used to explore spatial constraints on cell migration. Finally, it is noted that the development of the device architecture described here significantly moves forward the array methods that we have used previously43,44 to perform traditional assays.Acknowledgements
The authors are grateful to The Royal Thai Government Science and Technology Scholarship for support of the work. Aspects of the work were also supported by the University of Glasgow, School of Engineering and the EPSRC, reference EP/I017887/1. We also thank the staff of the James Watt Nanofabrication Centre (JWNC) at the University of Glasgow for their help in aspects of equipment maintenance and calibration in our fabrication facilities. The authors also thank Dr. Julien Reboud for critically reading the manuscript.Notes and references
- S. Faley, K. Seale, J. Hughey, D. K. Schaffer, S. Vancompernolle, B. Mckinney, F. Baudenbacher, D. Unutmaz and J. P. Wikswo, Lab Chip, 2008, 8, 1700–1712 RSC.
- S. L. Faley, M. Copland, D. Wlodkowic, W. Kolch, K. T. Seale, J. P. Wikswo and J. M. Cooper, Lab Chip, 2009, 9, 2659–2664 RSC.
- D. Di Carlo, L. Y. Wu and L. P. Lee, Lab Chip, 2006, 6, 1445–1449 RSC.
- K. Chung, C. A. Rivet, M. L. Kemp and H. Lu, Anal. Chem., 2011, 83(18), 7044–7052 CrossRef CAS PubMed.
- I. Kurth, K. Franke, T. Pompe, M. Bornhäuser and C. Werner, Integr. Biol., 2009, 1, 427–434 RSC.
- A. Revzin, K. Sekine, A. Sin, R. G. Tompkins and M. Toner, Lab Chip, 2005, 5, 30 RSC.
- R. Singhvi, A. Kumar, G. P. Lopez, G. N. Stephanopoulos, D. I. Wang, G. M. Whitesides and D. E. Ingber, Science, 1994, 264, 696 CAS.
- T. Xu, W. Yue, C.-W. Li, X. Yao, G. Cai and M. Yang, Lab Chip, 2010, 10, 2271–2278 RSC.
- B. R. Harkness, G. B. Gardner, J. S. Alger, M. R. Cummings, J. Princing, Y. Lee, H. Meynen, M. Gonzales, B. Vandevelde, M. Vanden Bulcke, C. Winters and E. Beyne, SPIE, ed. J. L. Sturtevant, 2004, vol. 5376, pp. 517–524 Search PubMed.
- P. Jothimuthu, A. Carroll, A. A. S. Bhagat, G. Lin, J. E. Mark and I. Papautsky, J. Micromech. Microeng., 2009, 19, 045024 CrossRef.
- G. Gardner, B. Harkness, E. Ohare, H. Meynen, M. V. Bulcke, M. Gonzalez and E. Beyne, Electronic Components and Technology Conference, 2004. Proceedings. 54th, 2004, vol. 1, pp. 170–174 Search PubMed.
- H. Meynen, M. Vanden Bulcke, M. Gonzalez, B. Harkness, G. Gardner, J. Sudbury-Holtschlag, B. Vandevelde, C. Winters and E. Beyne, Microelectron. Eng., 2004, 76, 212–218 CrossRef CAS PubMed.
- F. Y. Shih, B. R. Harkness, G. B. Gardner, J. S. Alger, M. R. Cummings, J. L. Princing, H. Meynen, H. A. Nguyen and W. W. Flack, 5th International Conference on Electronic Packaging Technology, Proceedings, 2003, pp. 316–320 Search PubMed.
- S. P. Desai, B. M. Taff and J. Voldman, Langmuir, 2008, 24, 575–581 CrossRef CAS PubMed.
- G. Alkhatib, Curr. Opin. HIV AIDS, 2009, 4, 96 CrossRef PubMed.
- B. Furusato, A. Mohamed, M. Uhlén and J. S. Rhim, Pathol. Int., 2010, 60, 497–505 CrossRef CAS PubMed.
- M. Kucia, R. Reca, K. Miekus, J. Wanzeck, W. Wojakowski, A. Janowska-Wieczorek, J. Ratajczak and M. Z. Ratajczak, Stem Cells, 2005, 23, 879–894 CrossRef CAS PubMed.
- M. Z. Ratajczak, E. Zuba-Surma, M. Kucia, R. Reca, W. Wojakowski and J. Ratajczak, Leukemia, 2006, 20, 1915–1924 CrossRef CAS PubMed.
- Y.-X. Sun, J. Wang, C. E. Shelburne, D. E. Lopatin, A. M. Chinnaiyan, M. A. Rubin, K. J. Pienta and R. S. Taichman, J. Cell. Biochem., 2003, 89, 462–473 CrossRef CAS PubMed.
- B. A. Teicher and S. P. Fricker, Clin. Cancer Res., 2010, 16, 2927–2931 CrossRef CAS PubMed.
- F. Balkwill, Nat. Rev. Cancer, 2004, 4, 540–550 CrossRef CAS PubMed.
- A. Müller, B. Homey, H. Soto, N. Ge, D. Catron, M. E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S. N. Wagner, J. L. Barrera, A. Mohar, E. Verástegui and A. Zlotnik, Nature, 2001, 410, 50–56 CrossRef PubMed.
- A. Zlotnik, A. M. Burkhardt and B. Homey, Nat. Rev. Immunol., 2011, 11, 597–606 CrossRef CAS PubMed.
- A. Z. Fernandis, A. Prasad, H. Band, R. Klösel and R. K. Ganju, Oncogene, 2004, 23, 157–167 CrossRef CAS PubMed.
- J. D. Holland, M. Kochetkova, C. Akekawatchai, M. Dottore, A. Lopez and S. R. McColl, Cancer Res., 2006, 66, 4117–4124 CrossRef CAS PubMed.
- H. Kang, G. Watkins, C. Parr, A. Douglas-Jones, R. E. Mansel and W. G. Jiang, Breast Cancer Res., 2005, 7, R402–R410 CrossRef CAS PubMed.
- S. Ali and G. Lazennec, Cancer Metastasis Rev., 2007, 26, 401–420 CrossRef CAS PubMed.
- A. Zlotnik, Int. J. Cancer, 2006, 119, 2026–2029 CrossRef CAS PubMed.
- Y. Chen, G. Stamatoyannopoulos and C.-Z. Song, Cancer Res., 2003, 63, 4801–4804 CAS.
- M. C. P. Smith, K. E. Luker, J. R. Garbow, J. L. Prior, E. Jackson, D. Piwnica-Worms and G. D. Luker, Cancer Res., 2004, 64, 8604–8612 CrossRef CAS PubMed.
- B. Li, Y. Qiu, A. Glidle, D. McIlvenna, Q. Luo, J. Cooper, H.-C. Shi and H. Yin, Anal. Chem., 2014, 86(6), 3131–3137 CrossRef CAS PubMed.
- E. Meijering, O. Dzyubachyk and I. Smal, Methods Enzymol., 2012, 504, 183–200 Search PubMed.
- M. P. Valignat, O. Theodoly, A. Gucciardi, Nancy Hogg and A. C. Lellouch, Biophys. J., 2013, 104(2), 322–331 CrossRef CAS PubMed.
- M. Stoker, C. O'Neill, S. Berryman and V. Waxman, Int. J. Cancer, 1968, 3, 683–693 CrossRef CAS.
- J. Folkman and A. Moscona, Nature, 1978, 273, 345–349 CrossRef CAS.
- Y. Yamakita, G. Totsukawa, S. Yamashiro, D. Fry, X. Zhang, S. K. Hanks and F. Matsumura, J. Cell Biol., 1999, 144, 315–324 CrossRef CAS.
- J. R. Rettig and A. Folch, Anal. Chem., 2005, 77, 5628–5634 CrossRef CAS PubMed.
- M. Thery and M. Bornens, Curr. Opin. Cell Biol., 2006, 18, 648–657 CrossRef CAS PubMed.
- K. K. Suzuki and K. K. Takahashi, J. Cell. Physiol., 2003, 197, 297–305 CrossRef CAS PubMed.
- C. P. McCann, P. W. Kriebe, C. A. Parent and W. Losert, J. Cell Sci., 2010, 123, 1724–1731 CrossRef CAS PubMed.
- J. S. King and R. H. Insall, Trends Cell Biol., 2009, 19, 523–530 CrossRef CAS PubMed.
- P. K. Mattila and P. Lappalainen, Nat. Rev. Mol. Cell Biol., 2008, 9, 446–454 CrossRef CAS PubMed.
- D. Wlodkowic, S. Faley, M. Zagnoni, J. P. Wikswo and J. M. Cooper, Anal. Chem., 2009, 81(13), 5517–5523 CrossRef CAS PubMed.
- S. Faley, M. Copland, J. Reboud and J. M. Cooper, Integr. Biol., 2012, 4, 368–373 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fabrication of VIA trap and experiment set-up. See DOI: 10.1039/c4lc00774c |
| This journal is © The Royal Society of Chemistry 2015 |