A general method for the separation of amphiphilic surface-active poly(ethylene glycol) mono- and di-esters with long-chain ionic liquid-based biphasic systems†
Liyun
Kong
,
Qiwei
Yang
,
Huabin
Xing
*,
Baogen
Su
,
Zongbi
Bao
,
Zhiguo
Zhang
,
Yiwen
Yang
and
Qilong
Ren
Key Laboratory of Biomass Chemical Engineering of Ministry of Education, Department of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: xinghb@zju.edu.cn; Fax: +86-571-87952375; Tel: +86-571-87952375
First published on 15th October 2013
Abstract
A new method for the effective separation of amphiphilic poly(ethylene glycol) mono- and di-esters with long-chain ionic liquid-based biphasic systems has been developed. These biphasic systems exhibited high selectivity, a large distribution coefficient and a good ability to eliminate the emulsification resulting from the surface active character of those amphiphilic molecules.
Poly(ethylene glycol) (PEG) esters are well known as a class of biodegradable, nontoxic and highly desired “green” amphiphilic surfactants which have been widely applied as solubilisers, emulsifiers and lubricants in pharmaceuticals, cosmetics and other industries.1 In the emerging polymer-based drug delivery systems, PEG functionalized products (PEGylated) are dominant and also the gold standard for other polymer-based products due to their lack of toxicity, biodegradability, absence of antigenicity and immunogenicity, and solubility in water and organic solvents.2 Methods for preparing PEG esters involve ethoxylation, i.e. reaction of ethylene oxide with an acid, esterification between acids and PEG, or transesterification between PEG and simple acid esters.3 However, a mixture of PEG mono- and di-esters (Scheme 1) is usually obtained due to the equal reactivity of the two terminal hydroxyl groups in PEG.4 The PEG diesters with two hydrophobic substituents at the ends of PEG exhibit a lower hydrophilic–lipophilic balance number and worse water affinity compared with the monoesters. Furthermore, no free hydroxyl group exists in diesters for further reactions to form polymer-conjugations with drugs or copolymers with other polymers.5,6 Because of the pronounced differences between PEG mono- and di-esters, there is a strong need to separate PEG monoesters from their mixtures.
 | ||
| Scheme 1 General structure of PEG mono- and di-esters (1), and molecular structures of PEG mono- and di-TS (2), PEG mono- and di-laurates (3), PEG mono- and di-oleates (4), PEG mono- and di-stearates (5). | ||
However, the separation of these amphiphilic molecules has been very challenging due to their structural similarity and surface-active property. Chromatographic technologies using a preparative stationary bed chromatography system or a simulated moving bed (SMB) chromatography system are the most popular methods for the separation of PEG mono- and di-esters, which exhibit problems of low capacity, high organic solvent consumption, a large amount of solid waste emission (adsorbent) and large equipment investment, and thus are not economically and environmentally favourable for large-scale production.4,7,8 Liquid–liquid extraction is a simple and economical separation method, which offers advantages of a large sample output, small quantities of solvents, relatively simple equipment and is easy to scale up. Nevertheless, the attempt to establish a feasible liquid–liquid extraction method for separating PEG esters is strongly limited by the serious emulsification caused by their excellent surface-active character (detailed discussion shown below).
In this work, we report a new extraction method for the separation of PEG mono- and di-esters with long-chain ionic liquid-based biphasic systems. The extractants in these biphasic systems consist of ionic liquids (ILs) and water, both of which are green solvents. This method showed excellent separation performance with a selectivity of PEG monoester to PEG diester up to 796. The utilization of long-chain ILs as extractants also achieved a large extraction capacity by forming micelle or lyotropic liquid crystal structures microscopically. In addition, the emulsification phenomenon was eliminated by using ILs as extractants.
PEG mono- and di-tocopherol succinates (PEG mono- and di-TS, Scheme 1(2)) with PEG having an average molecular weight of 1000 were selected as model objects in this study. PEG mono-TS, a water soluble derivative of natural vitamin E, is an excellent amphiphilic molecule that has been widely applied as a solubiliser, an emulsifier, an absorption enhancer and as a vehicle for lipid-based drug delivery formulations.7,9 This method was also explored in the separation of other PEG mono- and di-esters, such as PEG mono- and di-laurates, PEG mono- and di-oleates and PEG mono- and di-stearates (Scheme 1(3), (4) and (5)).
When the traditional water–ethyl acetate (H2O–EtOAc) biphasic system was used for the separation of PEG mono- and di-TS, a serious emulsification phenomenon was observed (Fig. 1(a)). Setting for 3 days was not long enough for complete phase separation. More than 20 minutes was required to achieve complete phase separation by centrifugation at a speed of 4000 rpm. Furthermore, the distribution coefficient of PEG mono-TS (Dmono-) in the H2O–EtOAc biphasic system was very low (Dmono- = 0.072), which manifested that PEG mono-TS was scarcely extracted by aqueous solution. Inorganic salts are a kind of well-known demulsifying agents. Thus a typical inorganic salt, sodium chloride, was introduced into the biphasic system and a good demulsification effect was achieved (Fig. 1(b)). However, an obvious salting-out effect was observed because Dmono- decreased to 0.016 in the 3 M sodium chloride aqueous solution.
 | ||
| Fig. 1 Phase separation conditions of PEG mono- and di-TS separated by (a) water–ethyl acetate system, setting for 72 h, (b) 3 M sodium chloride aqueous solution–ethyl acetate system, setting for 10 min, and (c) 4 mol% of [C8Py]Br aqueous solution–ethyl acetate system, setting for 10 min. The initial concentration of PEG TS in the ethyl acetate phase was 2 mg mL−1. | ||
ILs, composed of asymmetric organic cations and organic or inorganic anions, are liquid salts at room temperature. ILs have been revealed to have the strong ability to interact with organic molecules through various mechanisms, and the interactions between ILs and solutes can be finely adjusted through the change of ILs’ cations or anions task-specifically.10 Furthermore, ILs can readily form immiscible liquid–liquid biphasic systems with various solvents due to their high cohesive energy.11 Benefiting from the unique physicochemical properties of ILs, good separation efficiencies were achieved with ILs as extractants in the extraction of metal ions,12 organic molecules,13 protein,14 and bioactive compounds,15 and so on. Thus, different from traditional inorganic salts, IL-based ternary biphasic system, IL/water–ethyl acetate (IL/H2O–EtOAc), was expected to achieve effective separation of PEG mono- and di-esters and to eliminate the emulsification problem, simultaneously. In this article, ILs 1-alkyl-3-methylimidazolium chloride ([Cnmim]Cl, n = 2–12) and N-alkyl-pyridine bromide ([CnPy]Br, n = 2–12) were investigated. Complete phase separation was obtained through a setting of 5 minutes without emulsification in all investigated IL/H2O–EtOAc biphasic systems (Fig. 1(c)).
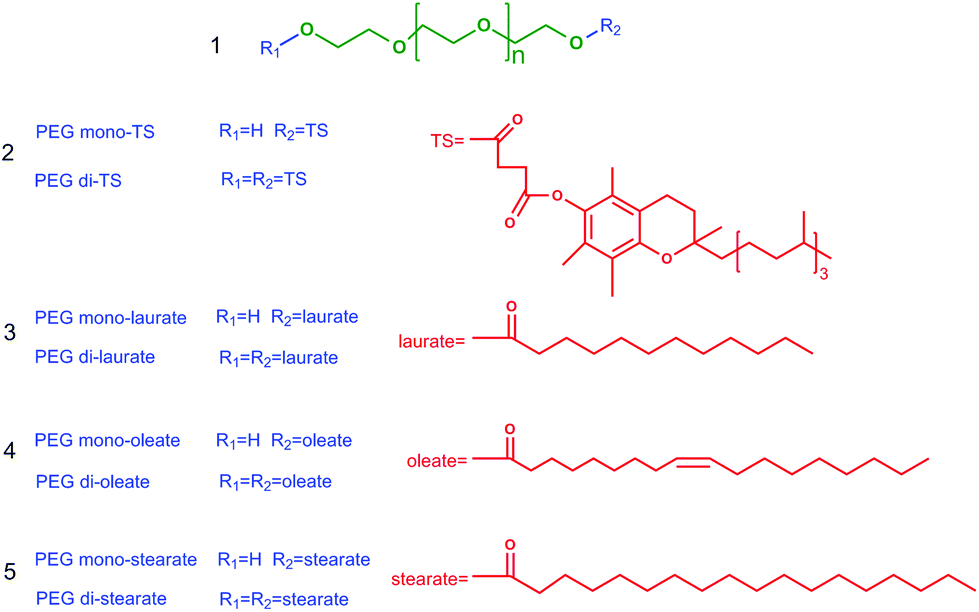
As shown in Fig. 2, the values of Dmono- increased rapidly with the increase of the alkyl chain length of ILs, meaning that ILs with longer alkyl chains had better affinity for PEG mono-TS. The separation performance of ILs with shorter alkyl chains (n = 2, 4) was similar to that of traditional inorganic salts, exhibiting a good demulsification effect, while Dmono- of PEG mono-TS was very low (Dmono- < 0.004 for both [Cnmim]Cl and [CnPy]Br when n = 2 and 4). For [CnPy]Br when n = 6 and 8, Dmono- were 1.91 and 7.75, respectively, which were dozens of times larger than Dmono- in the H2O–EtOAc biphasic system (Fig. 2(b)). At the same time, the selectivities of PEG mono-TS to PEG di-TS were very high, and the values were up to 796 and 23 for [C6Py]Br and [C8Py]Br systems, respectively, revealing the effectiveness of this method as an efficient separation approach. In addition, it was interesting that Dmono- were 8.64 and 15.09 in biphasic systems with [C12mim]Cl and [C12Py]Br, respectively, which were almost twice as high as Dmono- in [C8mim]Cl and [C8Py]Br systems. At the same time, the selectivities of PEG mono-TS to PEG di-TS were still larger than 8 in these biphasic systems. Thus, the majority of PEG mono-TS could be selectively extracted into the aqueous phase with [C12mim]Cl or [C12Py]Br as an extractant.
 | ||
| Fig. 2 The distribution coefficients of PEG mono-TS (Dmono-) and PEG di-TS (Ddi-) in IL/H2O–EtOAc biphasic systems with ILs having different alkyl chain lengths: (a) for [Cnmim]Cl (n = 2–12), (b) for [CnPy]Br (n = 2–12). Experimental conditions: 303 K, the initial concentration of PEG TS in the ethyl acetate phase was 2 mg mL−1, the initial concentration of IL in water was 5 mol%. | ||
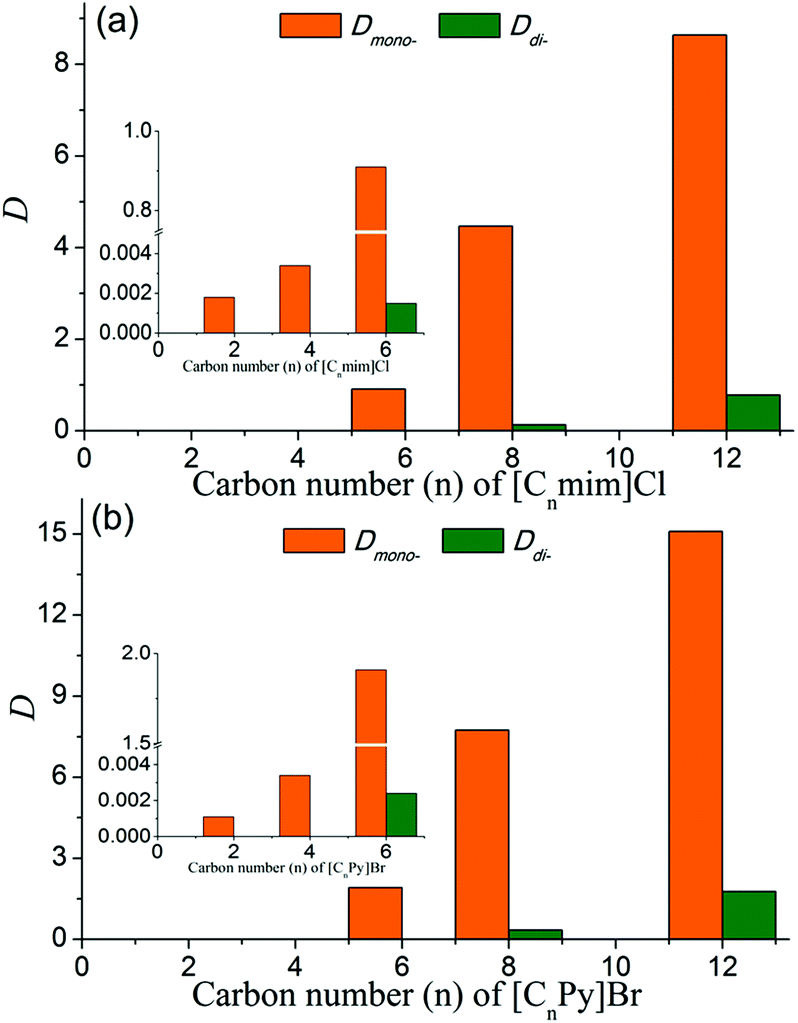
In order to explore the underlying extraction mechanism of the long-chain IL-based biphasic systems, dynamic light scattering (DLS) and polarizing optical micrograph (POM) were employed to investigate the microstructure of the IL-rich aqueous phase. According to the results of DLS, no micelle structure was found in the IL-rich aqueous phase when the carbon number of the alkyl chain of IL was not more than 6 (Fig. 3(a) for [C6mim]Cl). As the carbon number of the alkyl chain increased to 8, i.e. [C8mim]Cl and [C8Py]Br, micelle structures were observed in the IL-rich aqueous phase (Fig. 3(b) and Fig. S1(b)†). POM results indicated that no liquid crystal structures were generated in the aqueous phase of [Cnmim]Cl (n = 6 and 8) and [CnPy]Br (n = 6 and 8); only a dark-background was observed through the POM measurements (data not shown). On increasing the carbon number of the alkyl chain to 12, i.e. [C12mim]Cl and [C12Py]Br, liquid crystal structures were observed (Fig. 3(c) and 3(d)). Although the micelle and lyotropic liquid crystal structures for long-chain ILs in aqueous solution or some nonaqueous solution have been reported previously,16,17 the micelle and lyotropic liquid crystal structures in this ternary system of IL/H2O–EtOAc were first reported. Clearly, with the increase of the alkyl chain length, the aqueous solution changed from isotropic to micelle and liquid crystal structures, and then larger distribution coefficients of PEG mono-TS in the IL-rich phase were obtained.
 | ||
| Fig. 3 DLS images of IL-rich aqueous phase of the IL/H2O–EtOAc systems ([C6mim]Cl (a) and [C8mim]Cl (b)), POM images of the IL-rich aqueous phase with [C12mim]Cl (c), and [C12Py]Br (d). The initial concentrations of ILs were 5 mol%, and the detections were done at 303 K. | ||
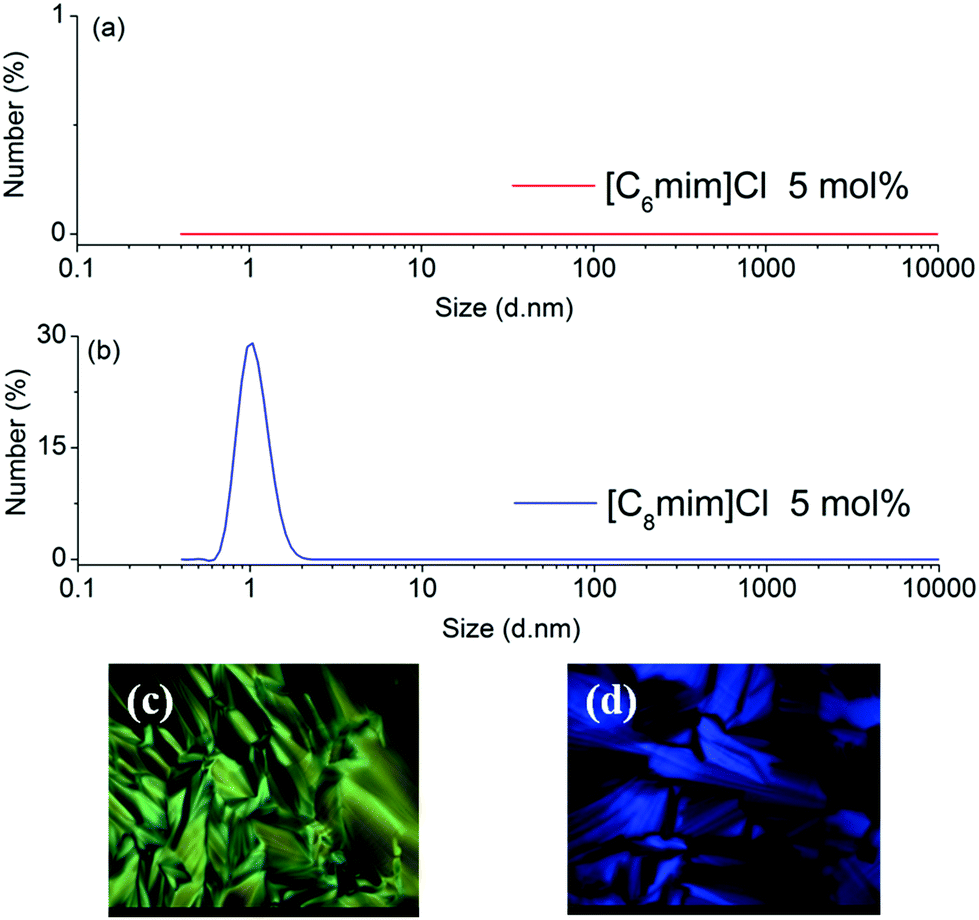
The effects of anions on the extraction performance and the microstructure of the ternary biphasic systems have also been investigated with ILs having the same cation ([C8mim]+) but different anions (Cl−, Br−, [NO3]− and [H2PO4]−) as extractants. As is evident from the results shown in Fig. 4(a), the type of anions had a moderate effect on the distribution coefficients of PEG mono- and di-TS, and both Dmono- and Ddi- increased with the order of [NO3]− > Br− > [H2PO4]− > Cl−. This order was consistent with the increasing order of the size of micelles in the IL-rich aqueous phase (Fig. 4(b)). When the anions vary from Cl− to [NO3]−, the size of micelles changed from 0.75 to 1.30 nm, and Dmono- increased from 5.06 to 8.09. Thus with ILs having the same cation but different anions as extractants, the biphasic systems having larger sized micelles in the IL-rich aqueous phase exhibited higher distribution coefficients.
 | ||
| Fig. 4 The anion effects on the distribution coefficients of PEG mono- and di-TS and the size of micelles in the IL-rich aqueous phase. The cation of the ILs was [C8mim]+, the initial concentration of PEG TS in the ethyl acetate phase was 2 mg mL−1, the initial concentration of ILs in water was 4 mol%, both the extraction experiments and DLS detections were done at 303 K. | ||
The effect of the molar fraction of ILs in the aqueous phase (xIL) on the extraction performance was investigated, and a diluent-swing effect was observed in [C8Py]Br or [C12Py]Br/H2O–EtOAc systems. When the initial [C8Py]Br concentration in water varies from 0.4 to 20 mol%, Dmono- first increased from 0.82 to 8.09, and then decreased to 0.50 (Fig. 5). Maximums of Dmono- and Ddi- appeared when the IL concentration reached 4 mol%. The existence of a maximum meant that there was an optimum content of IL in water for the separation of PEG mono- and di-TS. It is generally appreciated that the problems of large viscosity and considerable cost of ILs hinder the large-scale application and further development of IL-based liquid–liquid extraction. Using mixtures of ILs and polar molecular solvents as the extractants greatly reduces these problems in certain cases. On the one hand ionic liquid usage can be reduced to cut costs, on the other hand and more importantly, it provided an efficient means to isolate the high boiling point solute from ILs, which was a great challenge in the application of ILs. Furthermore, after the successful separation of the solute from the IL solution, the IL can be reused as an extractant to cut cost and a continuous process is possible. First, PEG mono-TS was selectively extracted into the IL-rich phase with 4 mol% [C8Py]Br aqueous solution. Then the IL-rich phase containing PEG mono-TS was concentrated to remove water, and the PEG mono-TS could be facilely recovered by back-extraction with EtOAc, taking advantage of the low Dmono- in the high-concentrated IL phase. Finally, the target product PEG monoester was obtained and the extractant IL could also be recycled.
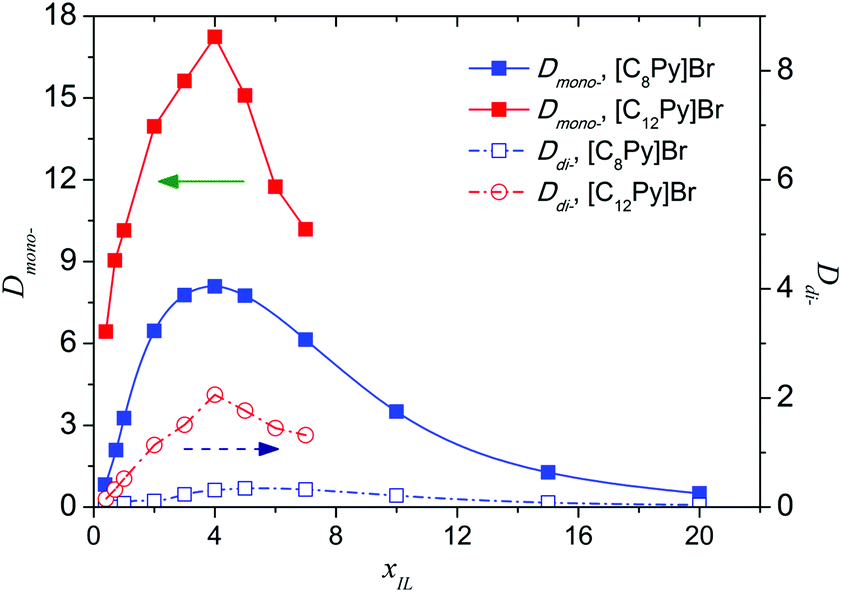 | ||
| Fig. 5 The effect of initial molar fraction of IL in aqueous phase (xIL) on the distribution coefficients of PEG mono-TS (Dmono-) and PEG di-TS (Ddi-) at 303 K. The initial concentration of PEG TS in the EtOAc phase was 2 mg mL−1. | ||
For an extraction process, temperature is an important factor as well. In this paper, the extraction experiments with the [C8Py]Br/H2O–EtOAc system over the temperature range 283 to 313 K were conducted. The results in Fig. 6 revealed a decreasing tendency of Dmono- and Ddi- with the temperature increment. Dmono- decreased from 14.32 to 2.63 when the temperature increased from 283 K to 313 K. Thus extraction at lower temperature can achieve higher distribution coefficient of PEG mono-TS in the extraction phase.
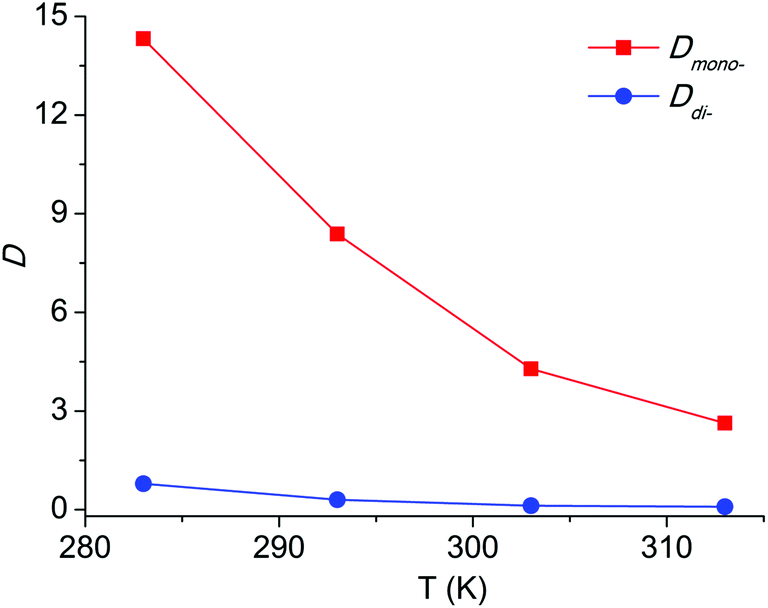 | ||
| Fig. 6 Temperature effect on the distribution coefficients of PEG mono- and di-TS. The initial concentration of PEG TS in the ethyl acetate phase was 2 mg mL−1, the initial concentration of IL in water was 5 mol%. | ||
The long-chain ILs employed in this paper, which actually was a kind of cationic surfactants, behaved excellently in the separation of PEG mono- and di-TS, so we wondered about the performance of other surfactants such as conventional anionic and non-ionic surfactants. Triton X-100 (polyethylene glycol octylphenol ether) and sodium dodecyl sulfate (SDS) were chosen as the representative of non-ionic and anionic surfactants, respectively. The distribution coefficients of PEG mono- and di-TS with Triton X-100 aqueous solution and SDS aqueous solution as extractants were determined under the conditions of 4 mol% surfactants in water initially, 2 mg mL−1 PEG TS in the ethyl acetate phase and at 303 K. However, the non-ionic surfactant Triton X-100 had a large mutual solubility with ethyl acetate, so a higher volume ratio of ethyl acetate to Triton X-100 aqueous solution was required to constitute a biphasic system. A volume ratio up to 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was adopted in the case of Triton X-100, while the volume ratio is just 1:1 in the case of IL-based biphasic systems. The distribution coefficients of PEG mono- and di-TS were very low with Triton X-100 as an extractant and the values were 0.38 and 0.0043, respectively. SDS also showed a larger mutual solubility with ethyl acetate compared with ILs, so more ethyl acetate was also needed to constitute a biphasic system. The 2:1 volume ratio between ethyl acetate and SDS aqueous solution was adopted in this paper. Dmono- and Ddi- were 11.48 and 2.07 for SDS, and the selectivity was 6. While Dmono- and Ddi- obtained under similar conditions (except the equal volume of ethyl acetate and IL aqueous solution) were 17.24 and 2.06 for [C12Py]Br, the selectivity was 8. A better separation performance with higher distribution coefficients and higher selectivity was obtained with the IL-based system. It was probably because the organic cations of ILs exhibited better affinity to the organic solute, and the lower mutual solubility with the ethyl acetate ensured higher selectivity and less solvent consumption. In addition, the structural tunability of ILs made the IL-based biphasic systems more charming for the separation of diversified amphiphilic compounds.
:1 was adopted in the case of Triton X-100, while the volume ratio is just 1:1 in the case of IL-based biphasic systems. The distribution coefficients of PEG mono- and di-TS were very low with Triton X-100 as an extractant and the values were 0.38 and 0.0043, respectively. SDS also showed a larger mutual solubility with ethyl acetate compared with ILs, so more ethyl acetate was also needed to constitute a biphasic system. The 2:1 volume ratio between ethyl acetate and SDS aqueous solution was adopted in this paper. Dmono- and Ddi- were 11.48 and 2.07 for SDS, and the selectivity was 6. While Dmono- and Ddi- obtained under similar conditions (except the equal volume of ethyl acetate and IL aqueous solution) were 17.24 and 2.06 for [C12Py]Br, the selectivity was 8. A better separation performance with higher distribution coefficients and higher selectivity was obtained with the IL-based system. It was probably because the organic cations of ILs exhibited better affinity to the organic solute, and the lower mutual solubility with the ethyl acetate ensured higher selectivity and less solvent consumption. In addition, the structural tunability of ILs made the IL-based biphasic systems more charming for the separation of diversified amphiphilic compounds.
High-purity PEG monoester can be easily obtained by batch crosscurrent extraction or continuous countercurrent extraction. The crosscurrent extraction was conducted with 4 mol% [C6Py]Br aqueous solution as an extractant to extract an equal volume of PEG mono- and di-TS (80.0 wt% of PEG mono-TS in it) solution in EtOAc with a concentration of 18.0 mg mL−1 at 303 K. The IL/H2O phase and the EtOAc phase were collected after the extraction, and then a fresh equal volume of the extractant was added to extract the EtOAc phase collected again (for graphical representation see the ESI†). The collected IL-rich phases were mixed after the two-stage crosscurrent extraction, and the relative purity and recovery of PEG mono-TS were up to 99.9 wt% and 82.0%, respectively. High purity as well as good recovery yield of PEG mono-TS could also be achieved through a countercurrent extraction using [C8Py]Br–H2O as an extractant (for detailed information see the ESI†).
To test the generality of this method, extraction separation of other PEG mono- and di-esters (PEG mono- and di-laurates, PEG mono- and di-oleates and PEG mono- and di-stearates) with different degrees of polymerization of PEG and different types of fatty acids was carried out in H2O–EtOAc systems without or with ILs. A serious emulsification phenomenon was observed in the H2O–EtOAc biphasic system without ILs when the average molecular weight of PEG was larger than 400. And the values of Dmono- were all less than 0.015 in the H2O–EtOAc biphasic system. As we expected, the long-chain IL-based biphasic systems showed excellent separation performance for all investigated PEG mono- and di-esters. Data in Table 1 showed that the selectivities (α) of PEG monoesters to PEG diesters were larger than 13 for all esters investigated, and Dmono- in the IL-rich phase were larger than 1.4. In addition, the values of Dmono- increased with the increase of the polymerization degree of PEG. Thus, the long-chain IL-based biphasic systems can be successfully applied to the separation of other PEG mono- and di-esters, achieving efficient separation and an excellent demulsification effect simultaneously.
| PEG esters | [C8Py]Br/H2O–EtOAc | [C12Py]Br/H2O–EtOAc | ||||
|---|---|---|---|---|---|---|
| D mono- | D di- | α | D mono- | D di- | A | |
| PEG400 laurates | 2.63 | 0.090 | 29 | 3.69 | 0.19 | 19 |
| PEG600 oleates | 4.95 | 0.24 | 21 | 8.82 | 0.53 | 17 |
| PEG1000 oleates | 14.00 | 0.96 | 15 | 23.94 | 1.57 | 15 |
| PEG400 stearates | 1.42 | 0.11 | 13 | 3.22 | 0.21 | 15 |
| PEG1000 stearates | 15.23 | 1.05 | 15 | 25.45 | 1.68 | 15 |
In summary, we have developed a new method for the facile separation of surface-active PEG mono- and di-esters using long-chain IL-based biphasic systems. These long-chain IL-based biphasic systems exhibited a large distribution coefficient and high selectivity as well as a good ability to eliminate the emulsification phenomenon. The success of the batch and continuous extractions demonstrated that this IL-based extraction was efficient and had great potential for the production of high-purity PEG monoester in industrial applications. As we know, the amphiphilic molecules contribute to a large proportion of compounds, and there are theoretically numerous kinds of “designable” ILs that can be tuned to have specific properties. Thus this method opens a door to effectively purify amphiphilic surface-active molecules with various related structures.
This work was supported by the National Natural Science Foundation of China (21222601, 20936005 and 21076175), the Zhejiang Provincial Natural Science Foundation of China (LR13B060001), and the Program for Zhejiang Leading Team of S & T Innovation (2011R50002).
Notes and references
- (a) B. Thierry and H. J. Griesser, J. Mater. Chem., 2012, 22, 8810 RSC; (b) H. Xu, Y. H. Deng, D. W. Chen, W. W. Hong, Y. Lu and X. H. Dong, J. Controlled Release, 2008, 130, 238 CrossRef CAS PubMed; (c) H. Ihre, O. L. P. De Jesus and J. M. J. Frechet, J. Am. Chem. Soc., 2001, 123, 5908 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- K. Kosswig, in Nonionic Surfactants, Surfactant Science Series, ed. N. M. van Os, Marcel Dekker, New York–Basel–Hong Kong, 1998, vol. 72, p. 123 (and references therein) Search PubMed.
- G. J. Papariello, S. Chulkaratana, T. Higuchi, J. E. Martin and V. P. Kuceski, J. Am. Oil Chem. Soc., 1960, 37, 396 CrossRef CAS.
- J. Zhao, Y. Mi and S. S. Feng, Biomaterials, 2013, 34, 3411 CrossRef CAS PubMed.
- Z. P. Zhang, S. H. Lee and S. S. Feng, Biomaterials, 2007, 28, 1889 CrossRef CAS PubMed.
- E. M. Collnot, C. Baldes, M. F. Wempe, J. Hyatt, L. Navarro, K. J. Edgar, U. F. Schaefer and C. M. Lehr, J. Controlled Release, 2006, 111, 35 CrossRef CAS PubMed.
- L. Y. Kong, B. G. Su, Z. B. Bao, H. B. Xing, Y. W. Yang and Q. L. Ren, J. Chromatogr., A, 2011, 1218, 8664 CrossRef CAS PubMed.
- K. Y. Win and S. S. Feng, Biomaterials, 2006, 27, 2285 CrossRef CAS PubMed.
- (a) N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC; (b) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS PubMed.
- (a) X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079 CAS; (b) A. Arce, M. J. Earle, S. P. Katdare, H. Rodriguez and K. R. Seddon, Chem. Commun., 2006, 2548 RSC; (c) M. G. Freire, A. F. M. Claudio, J. M. M. Araujo, J. A. P. Coutinho, I. M. Marrucho, J. N. C. Lopes and L. P. N. Rebelo, Chem. Soc. Rev., 2012, 41, 4966 RSC; (d) M. G. Freire, C. Neves, J. N. C. Lopes, I. M. Marrucho, J. A. P. Coutinho and L. P. N. Rebelo, J. Phys. Chem. B, 2012, 116, 7660 CrossRef CAS PubMed.
- (a) X. Q. Sun, H. M. Luo and S. Dai, Chem. Rev., 2012, 112, 2100 CrossRef CAS PubMed; (b) A. E. Visser, R. P. Swatloski, W. M. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis and R. D. Rogers, Chem. Commun., 2001, 135 RSC; (c) G. T. Wei, Z. S. Yang, C. Y. Lee, H. Y. Yang and C. R. C. Wang, J. Am. Chem. Soc., 2004, 126, 5036 CrossRef CAS PubMed.
- (a) A. Arce, M. J. Earle, H. Rodriguez, K. R. Seddon and A. Soto, Green Chem., 2009, 11, 365 RSC; (b) H. S. Gao, C. Guo, J. M. Xing, J. M. Zhao and H. Z. Liu, Green Chem., 2010, 12, 1220 RSC; (c) A. Bosmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz and P. Wasserscheid, Chem. Commun., 2001, 2494 RSC.
- (a) Z. Y. Li, X. X. Liu, Y. C. Pei, J. J. Wang and M. Y. He, Green Chem., 2012, 14, 2941 RSC; (b) J. F. B. Pereira, A. S. Lima, M. G. Freire and J. A. P. Coutinho, Green Chem., 2010, 12, 1661 RSC; (c) Z. Du, Y. L. Yu and J. H. Wang, Chem.–Eur. J., 2007, 13, 2130 CrossRef CAS PubMed.
- (a) M. G. Freire, C. Neves, I. M. Marrucho, J. N. C. Lopes, L. P. N. Rebelo and J. A. P. Coutinho, Green Chem., 2010, 12, 1715 RSC; (b) Q. W. Yang, H. B. Xing, B. G. Su, K. Yu, Z. B. Bao, Y. W. Yang and Q. L. Ren, Chem.–Eng. J., 2012, 181, 334 CrossRef PubMed; (c) Y. F. Cao, H. B. Xing, Q. W. Yang, B. G. Su, Z. B. Bao, R. H. Zhang, Y. W. Yang and Q. L. Ren, Green Chem., 2012, 14, 2617 RSC.
- (a) M. Blesic, M. H. Marques, N. V. Plechkova, K. R. Seddon, L. P. N. Rebelo and A. Lopes, Green Chem., 2007, 9, 481 RSC; (b) J. Bowers, C. P. Butts, P. J. Martin, M. C. Vergara-Gutierrez and R. K. Heenan, Langmuir, 2004, 20, 2191 CrossRef CAS; (c) O. Rojas, B. Tiersch, C. Rabe, R. Stehle, A. Hoell, B. Arlt and J. Koetz, Langmuir, 2013, 29, 6833 CrossRef CAS PubMed.
- (a) S. Ujiie and Y. Yano, Chem. Commun., 2000, 79 RSC; (b) C. K. Lee, H. W. Huang and I. J. B. Lin, Chem. Commun., 2000, 1911 CAS; (c) T. L. Greaves, D. F. Kennedy, S. T. Mudie and C. J. Drummond, J. Phys. Chem. B, 2010, 114, 10022 CrossRef CAS PubMed; (d) K. Ma, A. A. Shahkhatuni, B. S. Somashekhar, G. A. N. Gowda, Y. Tong, C. L. Khetrapal and R. G. Weiss, Langmuir, 2008, 24, 9843 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, extraction experiments and microstructure characterization are included. See DOI: 10.1039/c3gc41804a |
| This journal is © The Royal Society of Chemistry 2014 |