Sustainable route to methyl-9-hydroxononanoate (polymer precursor) by oxidative cleavage of fatty acid methyl ester from rapeseed oil†
Kévin
Louis
a,
Laurence
Vivier
a,
Jean-Marc
Clacens
a,
Markus
Brandhorst
b,
Jean-Luc
Dubois
c,
Karine De Oliveira
Vigier
*a and
Yannick
Pouilloux
*a
aUniversité de Poitiers – IC2MP – UMR CNRS 7285, 4, rue Michel Brunet, 86022 Poitiers Cedex, France. E-mail: yannick.pouilloux@univ-poitiers.fr; karine.vigier@univ-poitiers.fr; Tel: +33 5 49 45 39 51
bARKEMA FRANCE, Rue Henri Moissan, 69493 Pierre-Bénite, France
cARKEMA FRANCE, 420 Rue d'Estienne d'Orves, 92705 Colombes, France
First published on 4th October 2013
Abstract
Fatty acid methyl esters from rapeseed oil can be converted to monomers for polymer industries (85% methyl-9-hydroxynonanoate) by an oxydoreductive cleavage step in solvent free medium at room temperature, followed by a reduction step. All by-products are valuable and the reactions are performed under mild conditions.
The depletion of the fossil carbon reserve and the global warming concern lead to the use of biomass as a raw material. Biomass and specifically vegetable oils with their long-chain and aliphatic structure can be converted into polymers.1–5 Unsaturated fatty acids contain two functional groups (a carbonyl and several unsaturated bonds) where chemical reactions can be performed. Owing to these properties, fatty acid derivatives contribute to the flexibility, toughness, light stability, biodegradability, and low toxicity of many plastics.6,7 For example, soybean oil is used in making bio-based polyols for polyurethane applications.8,9 Many other vegetable oils are used such as castor oil to produce bio-based epoxy resins,1,10,11 sunflower oil to provide high oleic sunflower oil derivatives12–15 and bio-based nanocomposites from epoxidized linseed oil.16 Vegetable oils can be converted to bio-sourced polymers after oxidation of the FAME (Fatty Acid Methyl Ester) to produce the corresponding aldehyde. After hydrogenation of reductive amination, this aldehyde can be transformed to a monomer for polymer industries (Fig. 1). These products and other derivatives of polyunsaturated vegetable oils have many potential uses (i.e. plasticizers, polyamide plastics, wool treatment, and new coatings).
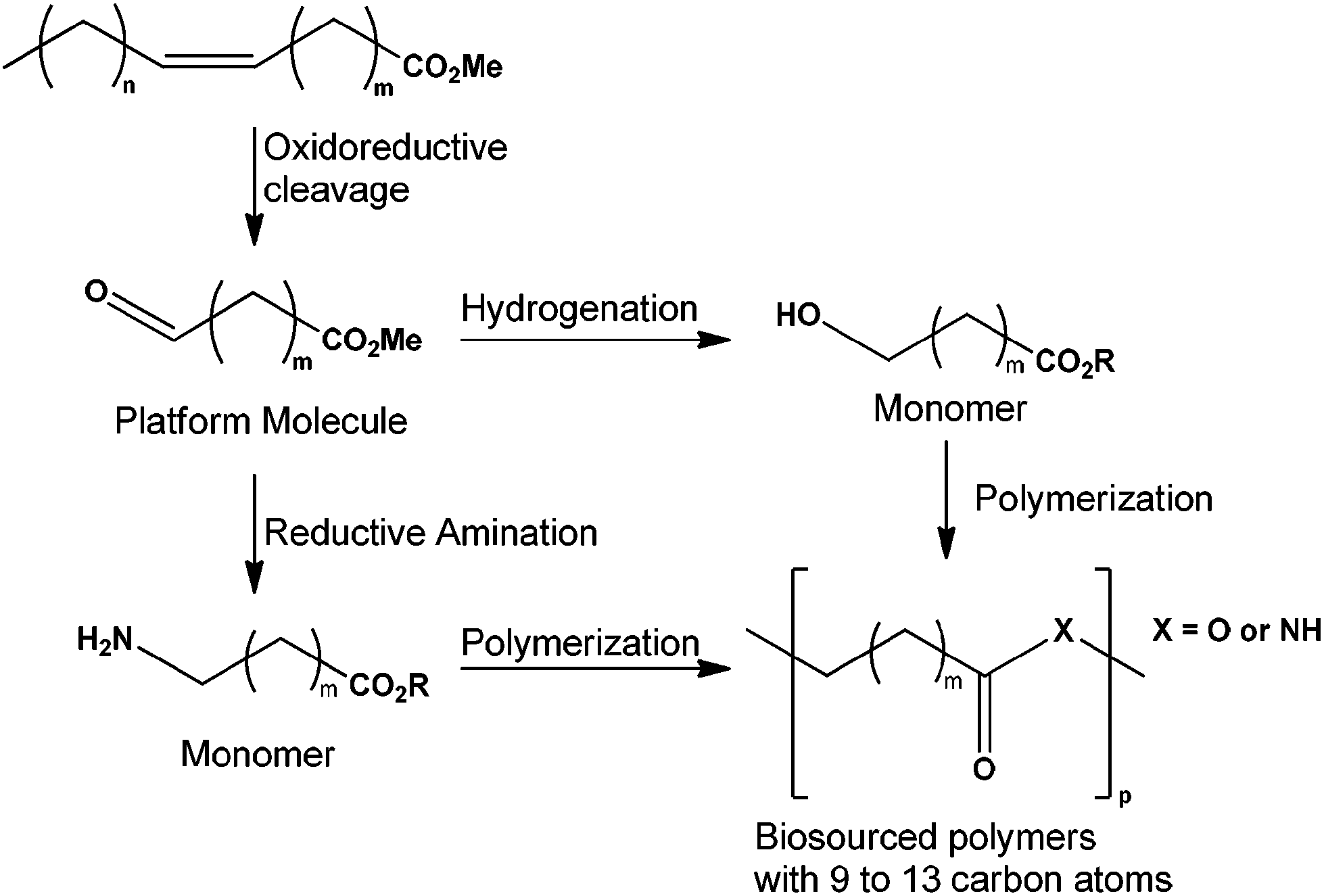 | ||
| Fig. 1 Synthesis of polyamide or polyester from FAME. | ||
Ozonolysis is a convenient and highly effective method for producing a large panel of compounds such as carboxylic acids, ketones, aldehydes and alcohols. Several attempts were made to favor the formation of aldehydes as the main products by controlling the double bond ozonolysis of soybean oil, rapeseed oil and canola oil using different solvents or a mixture of solvents.17–21 Indeed, the nature of the solvent used plays an important role during ozonolysis. Solvents may be classified broadly as participating and non-participating agents. Participating solvents will react chemically with intermediates formed during the ozonolysis reaction.22 For example, it is well-known that when ozonolysis is carried out in a solvent such as alcohol or water, which is a proton donor, this leads to the formation of hydrogen peroxide.23,24 On the other hand, the ozonolysis performed in aprotic solvents such as hydrocarbons (e.g. pentane, hexane) and chlorinated hydrocarbons (e.g. dichloromethane and chloroform) leads to the formation of ozonides.25–29
Nevertheless, all these studies reported in the literature suffer from a lack of selectivity to the desired product due to (i) the reactivity of intermediaries with protic solvents (see below) and (ii) the utilization of reducers which can damage aldehyde function23 (Zn/AcOH) or give rise to more complicated purification steps (Ph3P/Ph3PO).30,31
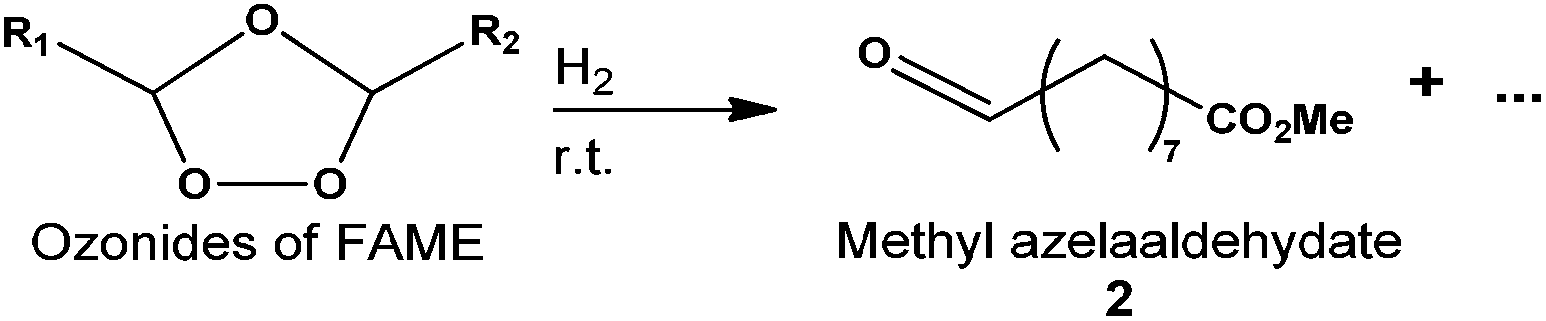
Herein the synthesis of methyl azelaaldehydate was performed from methyl oleate and more largely from FAME of rapeseed oil. The reaction was carried out in the absence of solvent at room temperature in the presence of Pd/C and hydrogen with yield and purity over 90%. The catalyst was recycled and Pd leaching was only 5 ppm. The hydrogenation of methyl azelaaldehydate was investigated, and a 93% yield of the corresponding alcohol (methyl-9-hydroxynonanoate) was obtained.
Oxydoreductive cleavage of methyl oleate and FAME
In a first set of experiments, methyl oleate was used as a model molecule thanks to its mono-unsaturated structure to study the ozonolysis reaction. Ozonolysis was performed through saturation of the solvent by ozone – at low temperature (i.e. −78 °C) – prior to the addition of unsaturated compounds which react instantaneously.32 This procedure led to much experimental damage as explosions due to free radical decomposition of unstable intermediates (i.e. molozonides). In the following experiments, the decomposition was prevented by the addition of ozone into a mixture of methyl oleate and dichloromethane at −78 °C (Table S1†). Under these conditions, a lower amount of ozone reacted instantaneously with the double bond of methyl oleate limiting the amount of unstable molozonides. Moreover, the ozone quantity was reduced to 10%.The selectivity to ozonides was followed by 1H NMR and HPLC analysis at room temperature due to the decomposition of ozonides in the GC injector. Indeed, ozonides are not stable at temperatures higher than 50 °C.19 One should mention that the ozonides were stable at room temperature since no modification or degradation was observed by 1H NMR analysis after several weeks under nitrogen storage (Fig. S1†). The reaction was performed with O3/O2 bubbling (1.6 molar%) in CDCl3 at room temperature. Under these conditions, ozonides were formed with a high selectivity (>99%).
In dichloromethane, independently of the temperature (−78 °C, 0 °C or 25 °C) the conversion of methyl oleate was total and ozonides were selectively produced (Table S1†). These results show that the synthesis of methyl azelaaldehydate can be performed in dichloromethane at room temperature without any drop in selectivity. As shown by HPLC analysis (Fig. 2), molozonide decomposition led to three ozonides which each have two isomers with a cis and trans configuration. The mechanism of the ozonide formation is described in Fig. S2.† The quantification of each ozonide was not possible. However, independently of the reactions conditions, nonanal and methyl azelaaldehydate were always obtained in similar amount. The results are similar to other nonprotic solvents such as pentanes, hexanes and heptanes at room temperature (100% conversion and 99% selectivity to ozonides, Table S1†). One should point out that when the reaction is performed at low temperature (−78 °C and 0 °C), precipitation of ozonides is observed for concentrations higher than 10 g L−1 of methyl oleate. The ozonolysis of methyl oleate at room temperature allows its conversion to ozonides at any concentration. Indeed, at room temperature, methyl oleate ozonides are soluble in solvents and liquids under solvent-free conditions. In order to develop ozonolysis of methyl oleate in an environmentally friendly process, an experiment was performed with direct ozone bubbling (1.6 molar %) in methyl oleate, without solvent, at room temperature. Solvent-free ozonolysis leads to the same results as those under solvent conditions (Table S1†).
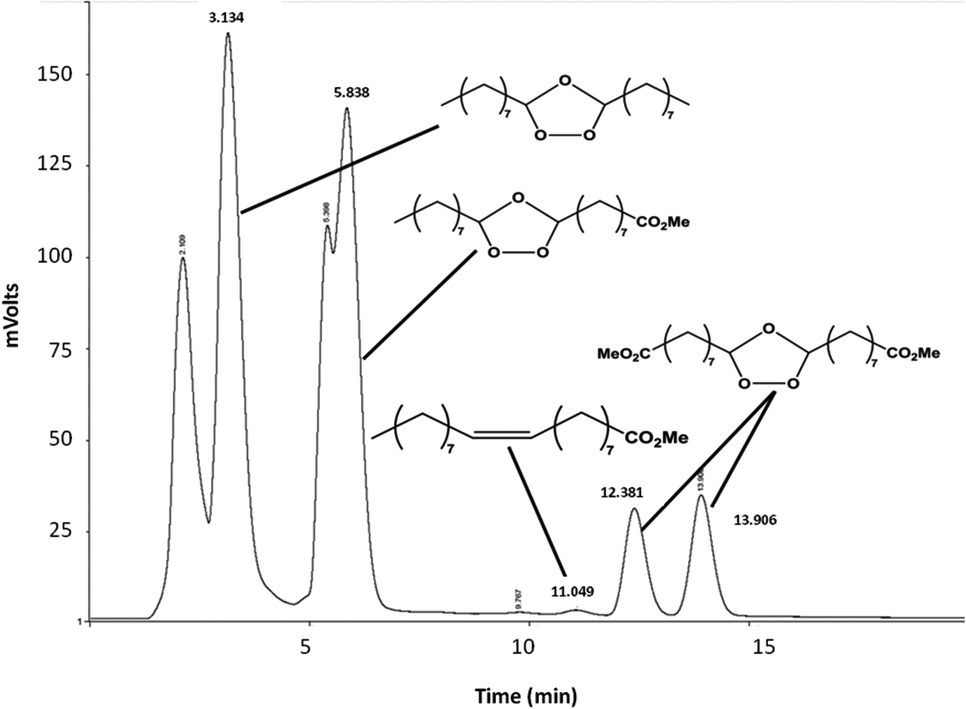 | ||
| Fig. 2 Reversed-phase HPLC chromatogram of the six ozonides of methyl oleate. Conditions: ODS 3 μm, 150 × 2.1 mm, MeOH–H2O (92/8) – 0.25 mL min−1. | ||
This ozonolysis process was applied to FAME from rapeseed oil. Rapeseed oil has a low level of saturated fats (∼10 wt%) along with high levels of monounsaturated (MUFA, ∼60 wt%) and polyunsaturated (PUFA, ∼25 wt%) fatty acids. All of MUFA (mostly methyl oleate) and PUFA (methyl linoleate and α-linolenate) contain carbon–carbon double bonds within their fatty acyl groups which are readily available for chemical/structural modifications. All these UFA led to C-9 aldehyde-ester/acid. Ozonolysis of FAME from rapeseed oil was carried out without solvent at room temperature as previously described (Table S1†). The conversion of unsaturations was total; saturated FAME did not react during ozonolysis and was recovered at the end of the reaction. The molar ratio between ozone and unsaturations was similar to those obtained with methyl oleate.
Next, the selective reduction of ozonides to aldehydes was studied at room temperature to develop a one-pot two-step process. Starting from the reaction media obtained from the ozonolysis reaction, a reducer was added after the elimination of excess O2 by N2 bubbling. The idea was to use a reducer that can form a by-product which can be valorized afterwards. The ozonide reduction was first studied in the presence of DMS, the by-product being DMSO, a capital solvent in organic synthesis (Table 1, entry 1). Unfortunately, the conversion of methyl oleate was only 50% and the yield of aldehydes was 45%. When the reaction was performed for a prolonged time (up to 17 h) the conversion and the yield were not improved. DMS was thus replaced by triphenylphosphine. The conversion of methyl oleate was total and the selectivity to aldehydes was over 90% (Table 1, entry 2). One should note that even with this high activity, this reducer cannot be recovered – as previously mentioned – from the reaction media without using environmentally unfriendly compounds.31 Having all these results in hand, heterogeneous catalysis reduction was investigated for its environmental and economic interest (Table 1). The reduction of methyl oleate ozonides was studied in the presence of a commercial Pd/C catalyst from Sigma Aldrich with 5 wt% Pd at room temperature under atmospheric pressure of H2 (200 mL min−1). Water was formed as a by-product. The conversion (>99%) and the yield (92%) were similar to the ones obtained in the presence of triphenylphosphine (Table 1, entry 3). Moreover, the reaction time was divided by 17 since only 1 hour was necessary to achieve 93% methyl azelaaldehydate in the presence of Pd/C. Conversion and yield were similar under solvent and solvent free conditions. However, the reaction time was increased to 3 h in the absence of solvent due to a lower solubility of hydrogen in ozonides and saturated FAME (Table 1, entry 4) than in the presence of solvent (Table 1, entry 3). The catalyst and products were easily separated by filtration on Whatman™ paper at room temperature. The solvent was removed and methyl azelaaldehydate was purified either on a silica column using petroleum ether–diethyl ether (85/15) as an eluent or by distillation under vacuum (i.e. 8 mmHg) at respectively 49 °C and 90 °C for nonanal and methyl azelaaldehydate. The silica column gave high purity (∼99%) but needed the use of a solvent.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reducer | Amount of reducer | Reaction time (h) | Conv. (%) | Yielda (%) | |
| 2 | 3 | |||||
| Conditions: methyl oleate ozonides (3.4 mmol), CDCl3 (50 mL), room temperature.a Isolated yield.b H2 (200 mL min−1).c No solvent – H2 (200 mL min−1). | ||||||
| 1 | DMS | 4 eq. | 5 | 50 | 45 | 41 |
| 2 | PPh3 | 1.1 eq. | 17 | >99 | 95 | 93 |
| 3 | H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b Pd(5)/C b Pd(5)/C |
10 wt% | 1 | >99 | 93 | 92 |
| 4 | H2c Pd(5)/C |
10 wt% | 3 | >99 | 92 | 90 |
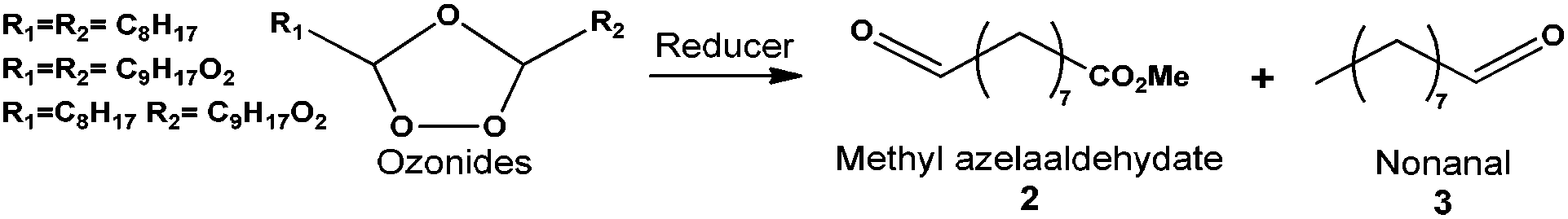
Distillation under vacuum offered a solvent-free purification step with 93% purity of expected aldehyde-ester. Purity of methyl azelaaldehydate as a monomer is a critical point for the functionalization and polymerization steps afterwards (see below). One can note that the by-products of this reaction are water and nonanal. The latter can be valorized in the cosmetic industry for example.
The catalytic reduction was applied to ozonides of FAME from rapeseed oil directly in solvent-free media. This experiment was undertaken at room temperature in the presence of Pd/C as a catalyst under hydrogen bubbling directly in ozonides (Table 2, entry 1). Under these conditions, the catalyst content was related to the ozonide amount taking into account the total conversion of unsaturations in FAME (Table S1†). The yield of methyl azelaaldehydate (91%) was similar to those obtained without solvent with ozonides from methyl oleate (Table 1, entry 4). In order to improve the catalytic process, the content of palladium on activated carbon was kept at 5 wt% and the catalyst content was varied from 5 to 10 wt% (Table 2, entries 1 to 3). As expected, an increase from 5 to 10 wt% led to a decrease in the reaction time from 16.5 to 9.5 h to reach a total conversion of ozonides (Fig. 3). However, all catalysts led to total conversion and notably high yield of 2. Next, studies were performed with 10 wt% Pd(5)/C to have a shorter reaction time.
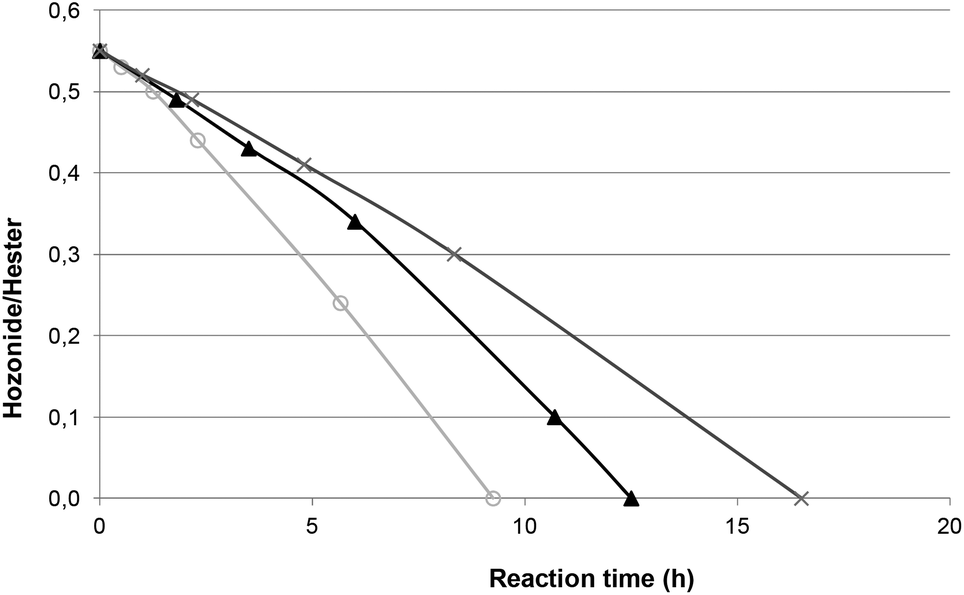 | ||
| Fig. 3 Conversion of ozonides of FAME from rapeseed oil with different amounts of Pd(5)/C, followed by integrating 1H NMR ozonide and ester signals. Conditions: ozonides from FAME (28 mmol), H2 (200 mL min−1), room temperature, catalyst content: (○) 10 wt%, (▲) 7.5 wt%, (×) 5 wt%. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Catalyst amount (wt%) | Reaction timea (h) | Yieldb (%) |
| Conditions: ozonides from FAME (28 mmol) – H2 (200 mL min−1) – room temperature.a Until total conversion of starting material.b Isolated yield.c PH2 = 16 bars. | ||||
| 1 | Pd(5)/C | 10 | 9.5 | 91 |
| 2 | Pd(5)/C | 7.5 | 12.25 | 89 |
| 3 | Pd(5)/C | 5 | 16.5 | 91 |
| 4 | Pd(5)/Cc | 10 | 0.54 | 92 |
The recyclability of the catalyst was then investigated by removing the catalyst at the end of the reaction from the reaction media by filtration. The recovered catalyst was washed with a small amount of dichloromethane and dried under vacuum (150 mbar) at 60 °C for 15 h before another cycle. After 9 runs, the yield of methyl azelaaldehydate remained unchanged showing the stability of the catalyst in the reaction media (Fig. S3†). This result, showing that the reduction step of ozonides can be performed in a continuous flow reaction, is of prime importance.
The leaching of Pd in the reaction media was also studied by ICP analysis. The amount of Pd in the crude mixture (5 ppm) is very low. To ascertain that this leaching is not responsible for the catalyst activity, the solid was removed when the conversion reached 50%. After the removal of the catalyst, hydrogen was introduced into the reaction media as previously described and the reaction was performed for 1 h. No conversion of ozonides was observed showing that the leaching of Pd did not allow the formation of methyl azelaaldehydate. Pd(5)/C has high catalytic properties without leaching in media and gives rise to high yields and selectivity for catalytic reduction of FAME's ozonides. Moreover, this catalyst is recyclable.
In order to improve this process, the amount of hydrogen used in the reduction step was also investigated. Indeed, solvent-free bubbling of hydrogen requires a huge amount (138 eq.) of H2 due to the low solubility of H2 in ozonides compared to the reduction reaction in solvent media. When hydrogen is under pressure, its solubility can be increased. Under a pressure of 16 bars of H2, the conversion and yield obtained were similar to those observed under atmospheric pressure. However, the reaction time was critically decreased (Table 2, entry 4). The catalyst was always highly selective and did not reduce aldehyde function to alcohol. With 16 bars of H2, the quantity of hydrogen was significantly decreased to 20% (1.2 eq.). One can note that a pressure of 16 bars of hydrogen is required to convert totally ozonides to aldehyde esters. Below this pressure of hydrogen the conversion was not total.
Furthermore, this reductive ozonolysis process (i.e. ozonolysis and selective catalytic reduction under pressure) was applied to other unsaturated starting materials (Table 3). Methyl 10-undecenoate gave the C-10 aldehyde ester (Table 3, entry 2) and methyl erucate, the C-13 (Table 3, entry 3). The yields of aldehyde after isolation of the desired products were as good as those from FAME of rapeseed oil. These results show that the ozonolysis reduction process can be applied to several FAMEs.
|
|
||||
|---|---|---|---|---|
| Entry | A | Conv. A (%) | Yield B (%) | |
| R | n | |||
| 1 | C8H17 | 7 | >99 | 92 |
| 2 | H | 8 | >99 | 93 |
| 3 | C8H17 | 11 | >99 | 94 |
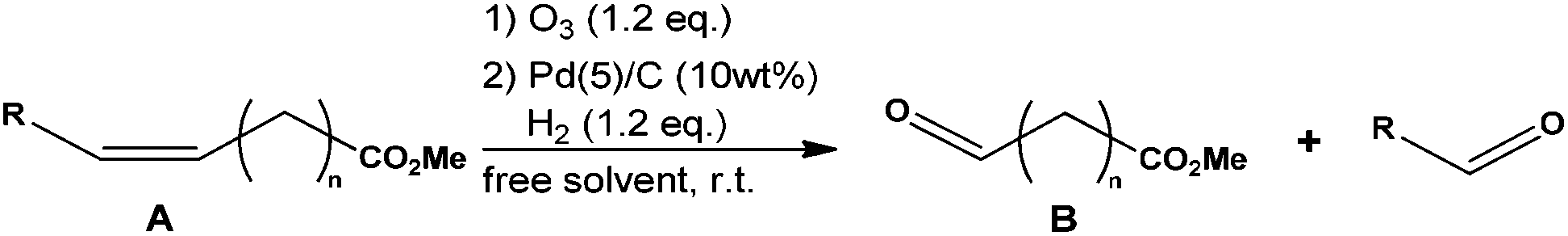
Synthesis of methyl-9-hydroxynonanoate
Methyl azelaaldehydate can be easily produced from renewable starting materials. Moreover, reduction of this aldehyde-ester gives rise to the synthesis of methyl-9-hydroxynonanoate 4. This alcohol-ester can be used in the polymer industry (Fig. 1) to synthesize bio-sourced polyester. The first attempt to produce methyl-9-hydroxynonanoate directly from ozonides of methyl oleate without solvent (or from FAME) was carried out without success. Only reduction to aldehyde ester 2 has been observed when ozonide was used as starting material. We have investigated the use of a solvent in this hydrogenation step. Therefore, the catalytic reduction of pure methyl azelaaldehydate 2 to produce methyl-9-hydroxynonanoate 4 was performed in solvent media under 50 bars pressure of H2 and at 50 °C (Table 4). The reduction was first performed in the presence of Pd/C, and the yield of 4 was 81% after 4 h in n-heptane (Table 4, entry 1). However, conversion was not higher than 72% within this reaction time. The conversion of 2 using Pt(5)/C (Table 4, entry 2) was comparable to Pd(5)/C while conversion was total with the RANEY® nickel catalyst in the same reaction time (Table 4, entry 3). In spite of total conversion with nickel, the yield of methyl-9-hydroxynonanoate was similar for all three different catalysts. In the latter case, mono-methyl azelaic acid was formed as a by-product.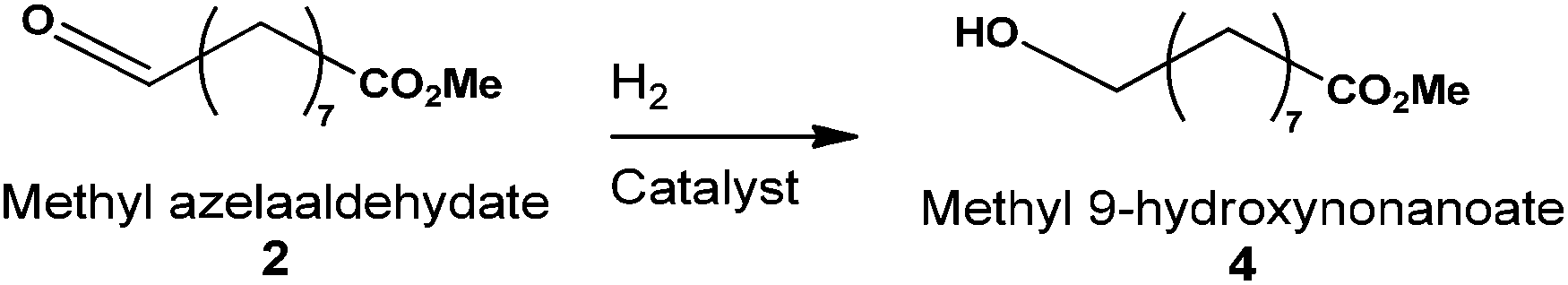
Catalytic reduction of methyl azelaaldehydate was performed in methanol using RANEY® nickel as a catalyst (Table 4, entry 4). Under these conditions, the reaction time was significantly decreased as a result of the difference between the solubility of hydrogen in heptane and methanol. Indeed, the solubility of hydrogen in methanol33 is 4.379 mol m−3 bar−1vs. 1.204 mol m−3 bar−1 in n-heptane.34 Conversion of 2 in both solvents was the same but the yield of methyl-9-hydroxynonanoate increased due to a lower amount of mono-methyl azelaic acid formed. However, in the presence of heptane (Table 4, entry 5), the reaction time to obtain 4 is three times higher than in the presence of methanol, showing that the nature of the solvent is of prime importance in this reaction. In order to perform the synthesis of 4 directly from ozonides of FAME of rapeseed oil, the use of Pd(5)/C to carry out reduction of 2 was studied. Compared to the RANEY® nickel catalyst, conversion of 2 was lower after 1.5 h (Table 4, entry 6) but was total only after 4 hours of reduction reaction (Table 4, entry 7). Under these conditions, methyl-9-hydroxynonanoate was obtained with a 93% yield.
Based on these results, the reduction of ozonides from FAME was carried out in the presence of methanol under 50 bars of H2 at 50 °C and in the presence of Pd(5)/C catalyst. Conversion of ozonides was total after 4 hours. 2 and 4 were obtained with respectively a yield of 91% and 7%. Under these conditions, methyl azelaaldehydate was formed as the main product. From this result, it seems that the impurities are responsible for the low yield of methyl-9-hydroxynonanoate. The reduction of ozonides from FAME to methyl-9-hydroxynonanoate can be performed in the presence of Pd/C, but the impurities obtained after the first reduction to methyl azelaaldehydate have to be removed from the reaction media.
The presence of a polar solvent such as methanol is required to obtain methyl-9-hydroxynonanoate from methyl azelaaldehydate owing to a higher solubility of hydrogen in such a solvent. In this reaction step, the solubility of hydrogen should be high enough to reduce the aldehyde function to alcohol. However, the reaction cannot be performed directly from ozonide reduction probably due to the presence of impurities.
Conclusions
In conclusion aldehyde-esters of various chain lengths were prepared from unsaturated starting materials using a solvent-free catalytic process at room temperature in the presence of a heterogeneous catalyst. The only by-products of this process are oxygen from ozone delivery and water from the reduction step. The catalyst was removed from the reaction media by a simple filtration and was reused at least 9 times. Simple, bifunctional aldehyde-ester was obtained with exceptionally high purity and may serve as an intermediate in numerous polymerization steps. For example, catalytic hydrogenation of aldehyde into the corresponding alcohol has been performed to obtain a bifunctional alcohol-ester monomer. The conversion of methyl azelaaldehydate was total and the yield of methyl-9-hydroxynonanoate was over 90% using RANEY® nickel or Pd on activated carbon catalysts. With this global process, alcohol-ester synthesis can be carried out from renewable feedstock in three steps with a total yield of 85%. Moreover, all by-products are valuable.Experimental
Ozonolysis reaction
Ozone (O3) production was performed using an O1-Generator (Ozone.ch) from pure oxygen gas. An O3/O2 mixture (1.6%, 200 mL min−1) was bubbled directly into unsaturated starting material (10–100 g) at room temperature. The conversion of the unsaturated compound has been followed with HPLC and 1H NMR as non-destructive analysis due to the instability of 1,2,4-trioxolanes when the temperature is over 30 °C. The crude material is directly used in the reduction step without any purification.Reduction to aldehyde ester 2
After elimination of O3/O2 by N2 bubbling, a catalyst was carefully added to the crude materials obtained after ozonolysis. Hydrogen was then introduced at 25 °C both by bubbling (200 mL min−1) and under pressure (16 bars). The products formed were analyzed as previously described and the conversion was determined. The crude material was filtered to remove the catalyst and purified by distillation under vacuum (90 °C at 8 mmHg) or on a silica column (petroleum ether–diethyl ether 85/15). Both give rise to 2 with purity up to 90%.Synthesis of methyl-9-hydroxynonanoate 4
Methyl azelaaldehydate 2 was dissolved in a solvent. A catalyst was added under stirring. The solution was heated at 50 °C under a dinitrogen atmosphere. Hydrogen was then introduced under pressure (50 bars). The conversion of 2 was followed by gas chromatography analysis without treatment while 4 has to be silylated before GC analysis. Silylation was performed by 0.1 mL BSTFA–TMCS 99:1. The crude material was filtered to remove the catalyst and purified using a silica column (heptane–ethyl acetate 80/20), giving rise to 4 with high purity (>99%). Methyl-9-hydroxynonanoate can be kept at room temperature under nitrogen flush without degradation or polymerization.
Analysis of the products
The 1H NMR spectra were recorded using a Bruker Avance III Ultrashield+ 400 mHz spectrometer with CDCl3 as a solvent, and chemical shifts were given in ppm downfield from trimethylsilane (TMS).HPLC was carried out using a reversed-phase ODS, 3 μm, 2.1 × 150 mm column. The mobile phase consisted of 92/8 methanol–water isocratic. The eluent flow rate was 0.25 mL min−1, and the detector was monitored at 210 nm.
Gas chromatographic analysis was performed using a Varian 3800 equipped with an HT5 column, 25 m × 0.32 mm × 0.25 μm, at a nitrogen flow of 1 mL min−1. A split/splitless injector (1%) was heated at 250 °C. For each injection, 0.1 μL of the solution to be analyzed was used. The FID detector was heated to 350 °C using air and at hydrogen flow of respectively 300 and 30 mL min−1.
Acknowledgements
We are grateful to the French National Research Agency for the financial support of the project ANR-GUI-AAP-03.Notes and references
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- S. S. Narine and X. Kong, Vegetable Oils in Production of Polymers and Plastics, John Wiley & Sons, Inc., 2005 Search PubMed.
- J. C. Ronda, G. Lligadas, M. Galià and V. Cádiz, Eur. J. Lipid Sci. Technol., 2011, 113, 46–58 CrossRef CAS.
- Y. Xia, R. L. Quirino and R. C. Larock, J. Renewable Mater., 2013, 1, 3–27 CrossRef CAS.
- R. Shogren, Z. Petrovic, Z. Liu and S. Erhan, J. Polym. Environ., 2004, 12, 173–178 CrossRef CAS.
- S. Pramanik, R. Konwarh, K. Sagar, B. K. Konwar and N. Karak, Prog. Org. Coat., 2013, 76, 689–697 CrossRef CAS PubMed.
- S. Caillol, M. Desroches, G. Boutevin, C. Loubat, R. Auvergne and B. Boutevin, Eur. J. Lipid Sci. Technol., 2012, 114, 1447–1459 CrossRef CAS.
- D. P. Pfister, Y. Xia and R. C. Larock, ChemSusChem, 2011, 4, 703–717 CrossRef CAS PubMed.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5630–5644 CrossRef CAS.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6717–6727 CrossRef CAS.
- L. Montero de Espinosa, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6843–6850 CrossRef CAS.
- L. Montero de Espinosa, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1159–1167 CrossRef CAS.
- L. M. De Espinosa, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4051–4063 CrossRef.
- L. Montero de Espinosa, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 869–878 CrossRef CAS.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Biomacromolecules, 2006, 7, 3521–3526 CrossRef CAS PubMed.
- E. Pryde, D. Anders, H. Teeter and J. Cowan, J. Am. Oil Chem. Soc., 1961, 38, 375–379 CrossRef CAS.
- Z. S. Petrović, W. Zhang and I. Javni, Biomacromolecules, 2005, 6, 713–719 CrossRef PubMed.
- C. S. Fitchett, N. G. Laughton, C. G. Chappell, M. L. Khan, V. Tverezovskiy, J. Tomkinson and P. Fowler, US 2005/0010069 A1, 2005.
- J. Yue and S. Narine, J. Am. Oil Chem. Soc., 2007, 84, 803–807 CrossRef CAS PubMed.
- T. S. Omonov, E. Kharraz and J. M. Curtis, J. Am. Oil Chem. Soc., 2011, 88, 689–705 CrossRef CAS PubMed.
- R. Criegee, Angew. Chem., Int. Ed., 1975, 14, 745–752 CrossRef.
- E. H. Pryde, D. E. Anders, H. M. Teeter and J. C. Cowan, J. Org. Chem., 1960, 25, 618–621 CrossRef CAS.
- D. Anders, E. Pryde and J. Cowan, J. Am. Oil Chem. Soc., 1965, 42, 236–243 CrossRef CAS.
- H. Edwards, Lipids, 1966, 1, 1–5 CrossRef CAS PubMed.
- O. Lorenz and C. R. Parks, J. Org. Chem., 1965, 30, 1976–1981 CrossRef CAS.
- O. Privett and C. Nickell, J. Am. Oil Chem. Soc., 1962, 39, 414–419 CrossRef CAS.
- O. S. Privett and E. C. Nickell, J. Lipid Res., 1963, 4, 208–211 CAS.
- E. Nickell and O. Privett, Lipids, 1966, 1, 166–170 CrossRef CAS PubMed.
- M. Sasaoka, D. Suzuki and T. Shiroi, US 5770729, 1998.
- Y. S. Hon and K. C. Wu, Tetrahedron, 2003, 59, 493–498 CrossRef CAS.
- S. Ramachandran, P. V. Rao and D. G. Cornwell, J. Lipid Res., 1968, 9, 137–139 CAS.
- K. Radhakrishnan, P. A. Ramachandran, P. H. Brahme and R. V. Chaudhari, J. Chem. Eng. Data, 1983, 28, 1–4 CrossRef CAS.
- S. K. Lachowicz, D. M. Newitt and K. E. Weale, Trans. Faraday Soc., 1955, 51, 1198–1205 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc41491d |
| This journal is © The Royal Society of Chemistry 2014 |