Copper-catalyzed homocoupling of ketoxime carboxylates for synthesis of symmetrical pyrroles†
Longfei
Ran
,
Zhi-Hui
Ren
,
Yao-Yu
Wang
and
Zheng-Hui
Guan
*
Department of Chemistry & Materials Science, Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, Northwest University, Xi'an 710069, P. R. China. E-mail: guanzhh@nwu.edu.cn
First published on 14th October 2013
Abstract
A novel and efficient copper-catalyzed homocoupling of ketoxime carboxylates has been developed for the synthesis of symmetrical pyrroles. This reaction tolerates a wide range of functional groups and provides a synthetically useful process to synthesize valuable symmetrical pyrroles under mild conditions.
Pyrroles are one of the most important heterocycles which are prevalent in numerous natural products, pharmaceuticals and functionalized materials.1 Particularly, large numbers of symmetrical pyrroles have recently been discovered to have remarkable bioactivity and fluorescence, such as COX-2 isoenzyme inhibitors (a), DNA minor groove recognition reagent DB884 (b), bio-antioxidant (c), fluorophores TPPy (d) and NPANPy (e) (Fig. 1).2 Over the past decades, tremendous efforts have been devoted to the synthesis of various substituted pyrroles.3 Apart from the classical protocols,4 many new strategies, such as cycloaddition reactions,5 multicomponent reactions,6 dehydrogenative cyclization reactions,7 coupling of enamides with alkynes,8 and oxidative cyclization of N-allylimines9 have been developed for the synthesis of pyrroles. However, most of these methods are focused on versatile unsymmetrical substituted pyrroles, general protocols for the synthesis of valuable symmetrical pyrroles are rarely developed.10
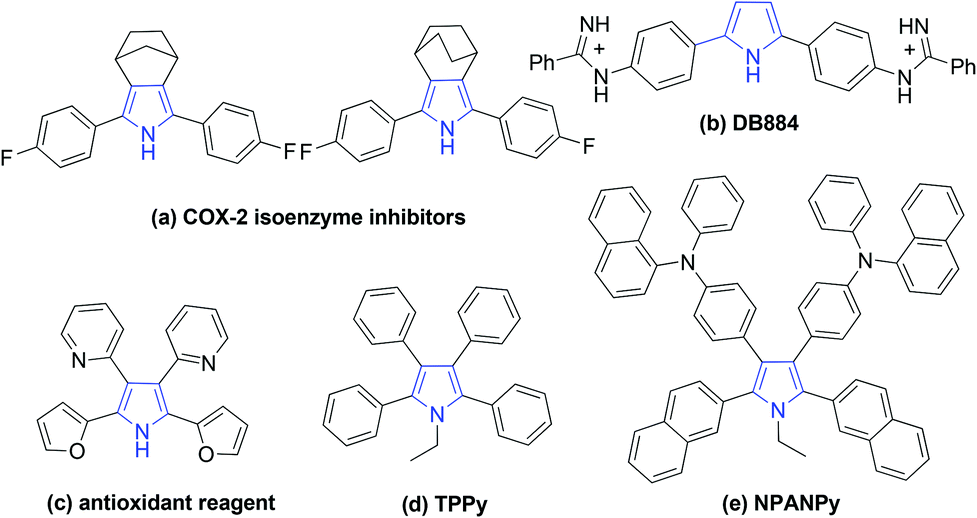 | ||
| Fig. 1 Bioactive or fluorescent symmetrical pyrroles. | ||
Oximes and their derivatives are versatile synthetic building blocks.11 They are well-known fruitful candidates for Beckmann rearrangement reactions in amide preparation,12 and dehydrations to produce nitriles.13 Recently, reductive acylation of ketoximes has been developed as a practical method for the synthesis of enamides.14 Meanwhile, much attention has been paid to the coupling reactions of oxime carboxylates, because the N–O bond cleavage of oxime carboxylates could be used as an internal oxidant to make reactions proceed under redox-neutral conditions. Thus, transition-metal catalyzed coupling of oxime carboxylates with aldehydes, alkynes and organoboronic acids have emerged for the synthesis of aza-heterocycles.15 In connection with our interest in oximes14a,b,15a and green synthesis of pyrroles,5 we have developed a novel and efficient copper-catalyzed homocoupling of ketoxime carboxylates for the synthesis of valuable symmetric pyrroles.
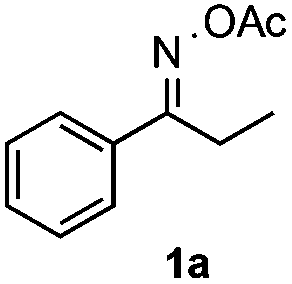
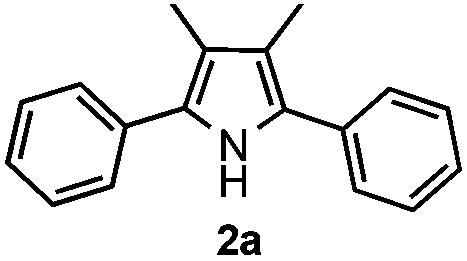
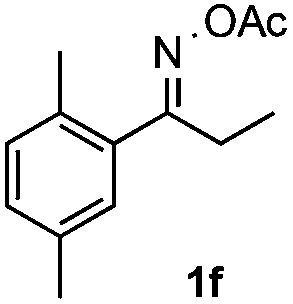
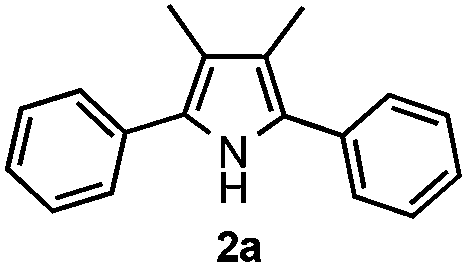
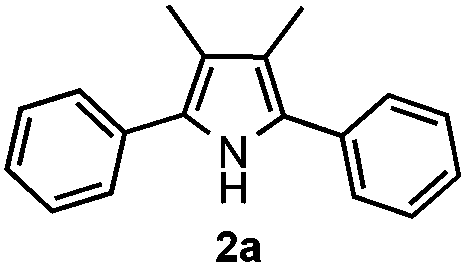
Propiophenone oxime acetate 1a was selected as a model substrate in the initial experiments to optimize the reaction conditions. The symmetrical pyrrole 2a was obtained in 37% yield when CuBr was used as the catalyst in DMSO at 120 °C (Table 1, entry 1). Then NaHSO3, which we found to be a good additive in our previous copper-catalyzed coupling of oxime acetates with aldehydes, was added to the reaction.15a The yield of pyrrole 2a was dramatically improved to 68% (Table 1, entry 2). Na2SO3 shows an inferior yield compared to NaHSO3. Next, a series of copper catalysts, such as CuI, CuCl, CuCl2, CuBr2 and Cu(OAc)2 were screened to determine if they improved the reaction efficiency (Table 1, entries 4–8). And various solvents, such as DMF and CH3CN, were screened as well (Table 1, entries 9–10). However, we found that CuBr and DMSO are still the most effective catalyst and solvent respectively. The reaction temperature was also varied; the yield of 2a was further increased to 72% when the reaction was conducted at 140 °C (Table 1, entry 11). Additionally, no reaction was observed in the absence of copper catalyst, confirming that Cu species did indeed act as the catalyst in the reaction (Table 1, entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Yieldb (%) |
| a Reaction conditions: propiophenone oxime acetate 1a (0.3 mmol), catalyst (5 mol%), NaHSO3 (1.2 equiv.) in solvent (3 mL) under Ar at 120 °C for 2 h. b Isolated yields. c The reaction was run at 140 °C. | ||||
| 1 | CuBr | — | DMSO | 37 |
| 2 | CuBr | NaHSO3 | DMSO | 68 |
| 3 | CuBr | Na2SO3 | DMSO | 30 |
| 4 | CuI | NaHSO3 | DMSO | 35 |
| 5 | CuCl | NaHSO3 | DMSO | 64 |
| 6 | CuCl2 | NaHSO3 | DMSO | 62 |
| 7 | CuBr2 | NaHSO3 | DMSO | 59 |
| 8 | Cu(OAc)2 | NaHSO3 | DMSO | 31 |
| 9 | CuBr | NaHSO3 | DMF | 24 |
| 10 | CuBr | NaHSO3 | CH3CN | 39 |
| 11 | CuBr | NaHSO3 | DMSO | 72 c |
| 12 | — | NaHSO3 | DMSO | 0 |
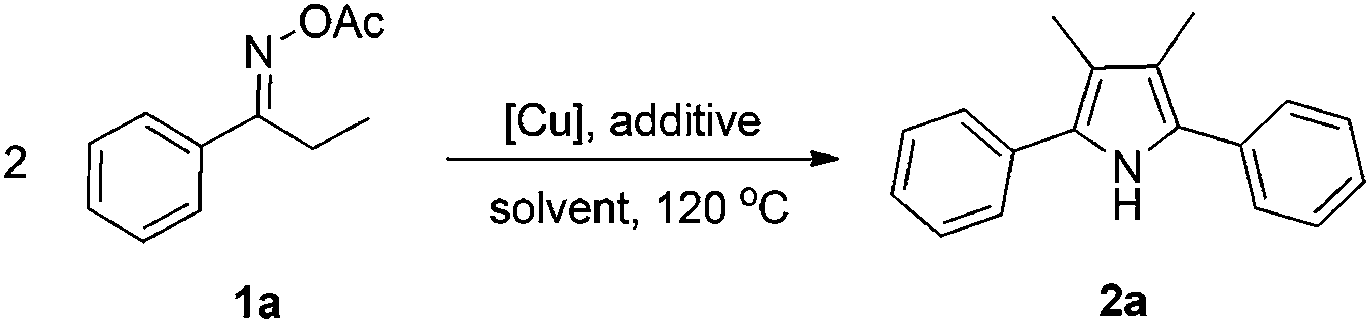
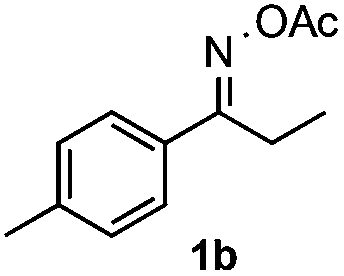
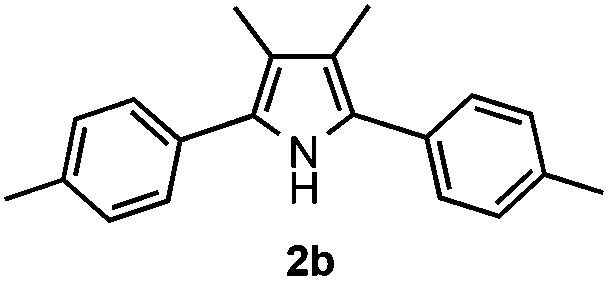
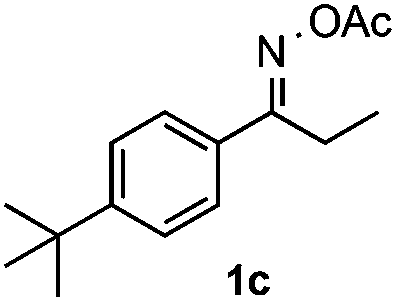
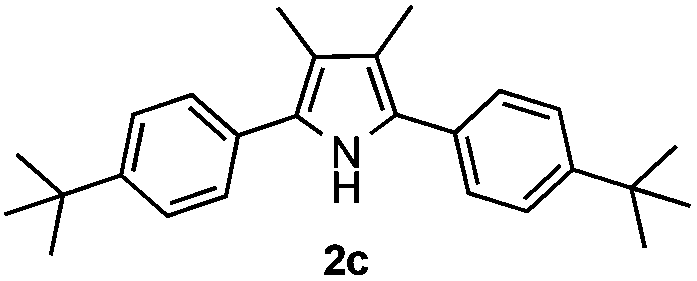
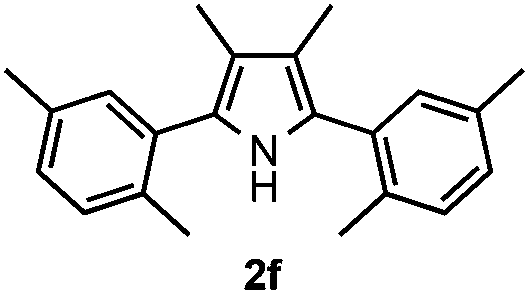
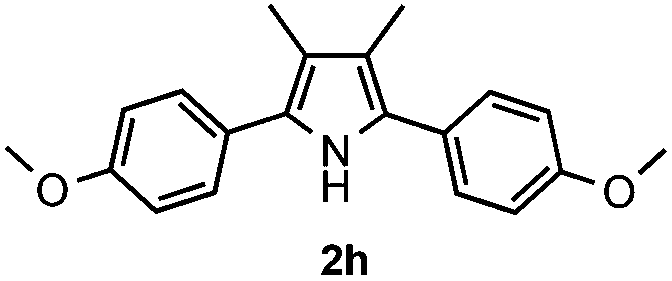
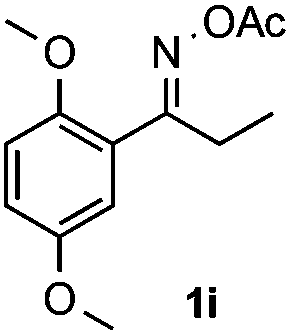
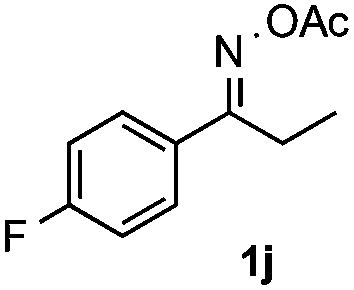
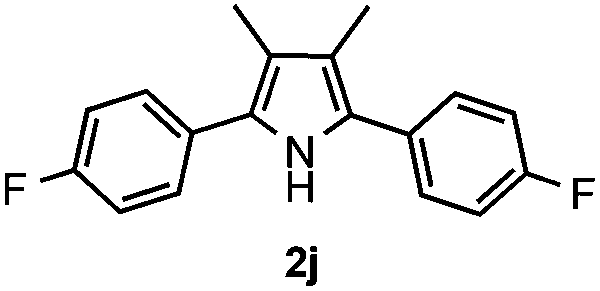
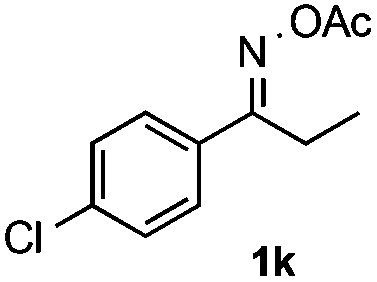
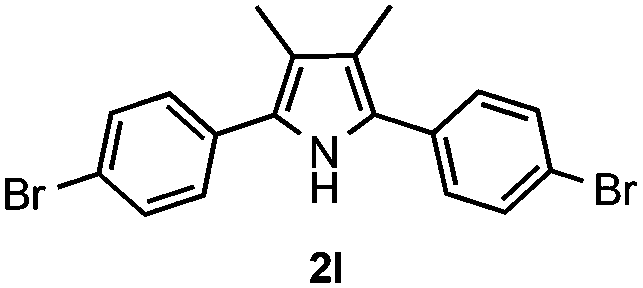
With the optimized reaction conditions established, the scope of the reaction was investigated (Table 2). The reaction showed good functional group tolerance and proved to be a general methodology for the preparation of symmetrical pyrroles. And the reaction was insensitive to the electronic effects of the substrates. Propiophenone oxime acetates with methyl, alkyl, methoxyl, fluoro and electron-withdrawing groups such as chloro and bromo groups on the aromatic rings all gave the corresponding symmetrical pyrroles 2b–2l in good yields. In this manner, the resulting Cl or Br substituted products 2k–2l, could be further expanded to a wider variety of functionalized symmetrical pyrroles by undergoing subsequent cross-coupling reactions. In addition, ortho-methyl and methoxyl substituted substrates reacted smoothly and resulted in the desired symmetrical pyrroles 2f–2g and 2i in 65–70% yields. These results indicated that steric hindrance on the aromatic rings of the substrates has little influence on the reaction (Table 2, entries 6–7, and 9).
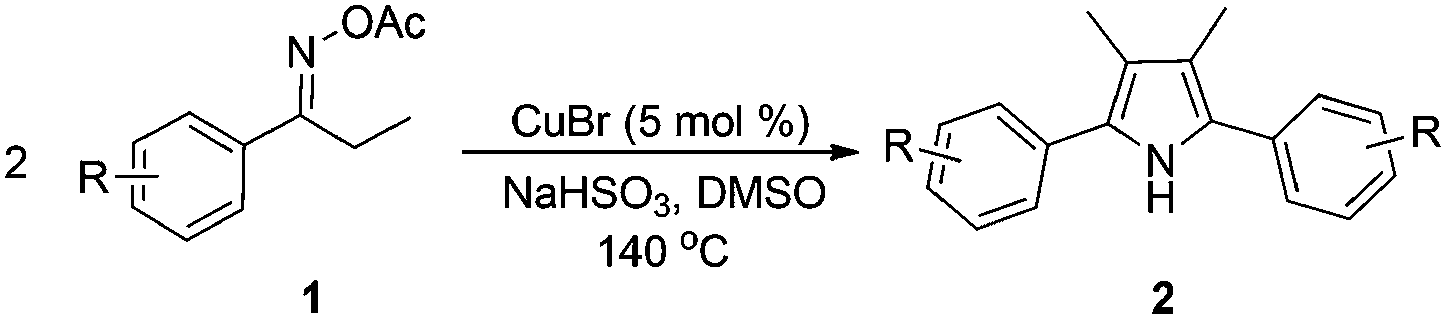



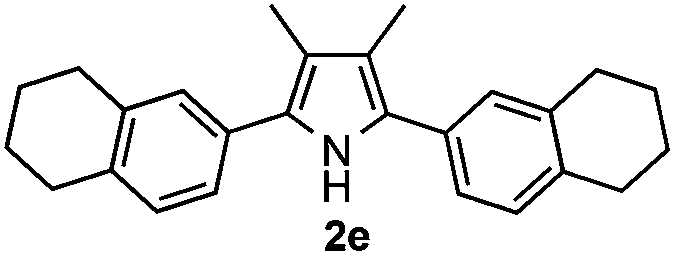






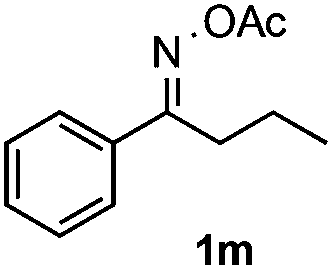
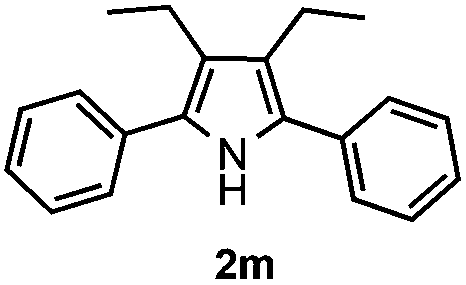
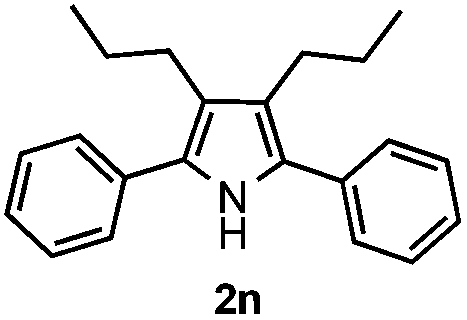
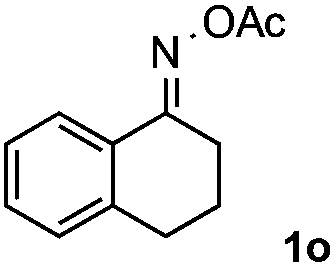
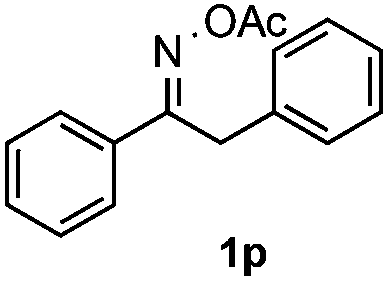
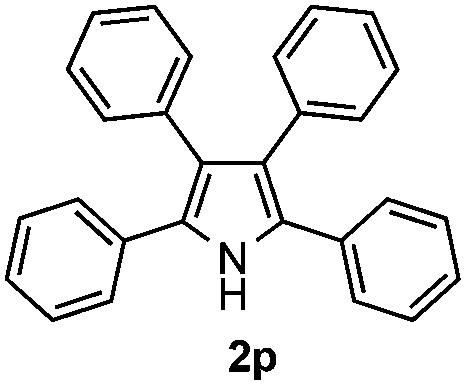
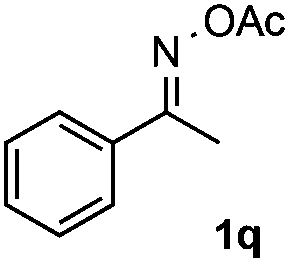
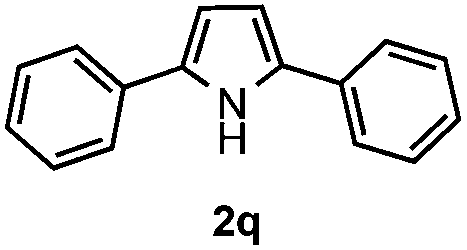
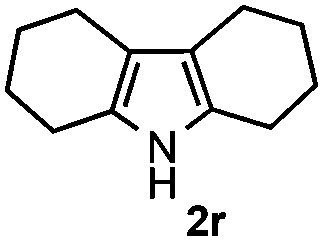
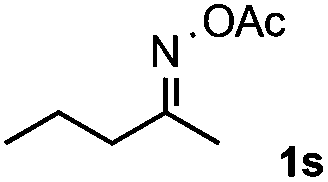
Furthermore, a series of phenyl alkyl ketoxime acetates were investigated to extend the reaction scope (Table 3). Ketoxime acetates derived from butyrophenone, valerophenone and α-tetralone reacted smoothly to give the desired symmetrical pyrroles 2m–2o in 56–68% yields (Table 3, entries 1–3). It should be noted that tetraphenyl pyrrole 2p, which has fluorophores for glassy blue-light-emitting diodes,2d was obtained in 52% yield when 1,2-diphenylethanone oxime acetate 1p was used as the substrate (Table 3, entry 4). However, acetophenone oxime acetate 1q gave only 10% yield of the corresponding 2,5-diphenyl pyrrole 2q under the aforementioned conditions (Table 3, entry 5). And no desired pyrrole products were obtained when the alkyl alkyl ketoxime acetates, such as cyclohexanone oxime acetate 1r and pentan-2-one oxime acetate 1s were employed as the substrates under the standard conditions (Table 3, entries 6–7). Finally, different propiophenone oxime carboxylates were investigated to determine their reactivity. It was found that the reactivity of the different carboxylates follows the sequence: acetate > propionate > pentafluorobenzoate > benzoate (Table 3, entries 8–10).






On the basis of the above results and previous studies, the tentative mechanisms for this homocoupling reaction are proposed in Scheme 1. Firstly, the reaction starts from a two-step single-electron-transfer reduction of ketoxime acetate 1 by CuI to generate imino-CuII complex B.14a,b,15 Tautomerization of the imino-CuII complex B gives the enamino-CuII intermediate C. Then, two pathways are possible for the following steps. In one case (path a), cleavage of the N–Cu bond of the enamino-CuII intermediate C forms a radical intermediate D.16 Radical coupling of the intermediate D affords diimine intermediate E.17 Intramolecular cyclization of intermediate E followed by elimination of NH3 produces the pyrrole 2. Alternatively, condensation of the enamino-CuII intermediate C with a second ketoxime acetate 1 gives the intermediate D′.15a,18 Elimination of intermediate D produces an enimine intermediate E′. Intramolecular radical cyclization of intermediate E′ assisted by CuII sequence with tautomerization of intermediate H′ produces the symmetrical pyrrole 2 (path b).8d,15e,16
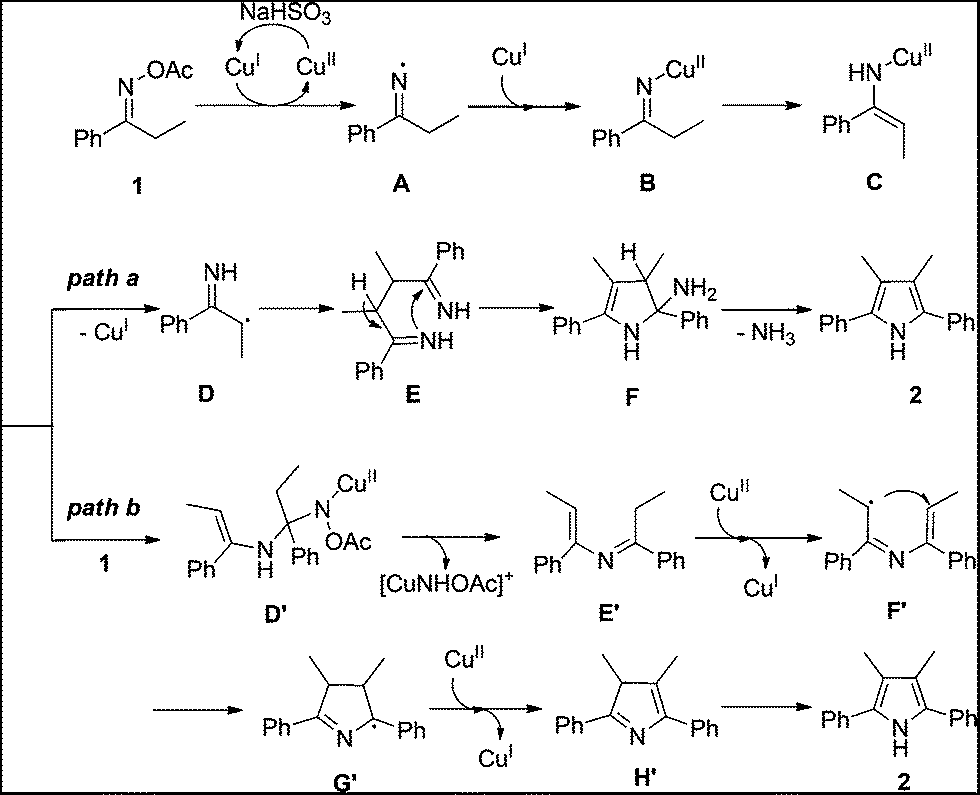 | ||
| Scheme 1 Proposed mechanism for the Cu-catalyzed homocoupling of ketoxime carboxylates. | ||
To gain insight into the mechanism of the reaction, 1,4-diphenylbutane-1,4-dione dioxime diacetate 1w was reacted under the standard conditions (Scheme 2). If the reaction proceeded via path b, we would have expected to observe the pyrrole 2q. However, no pyrrole 2q was observed in the reaction. This result indicated that path b is less likely.
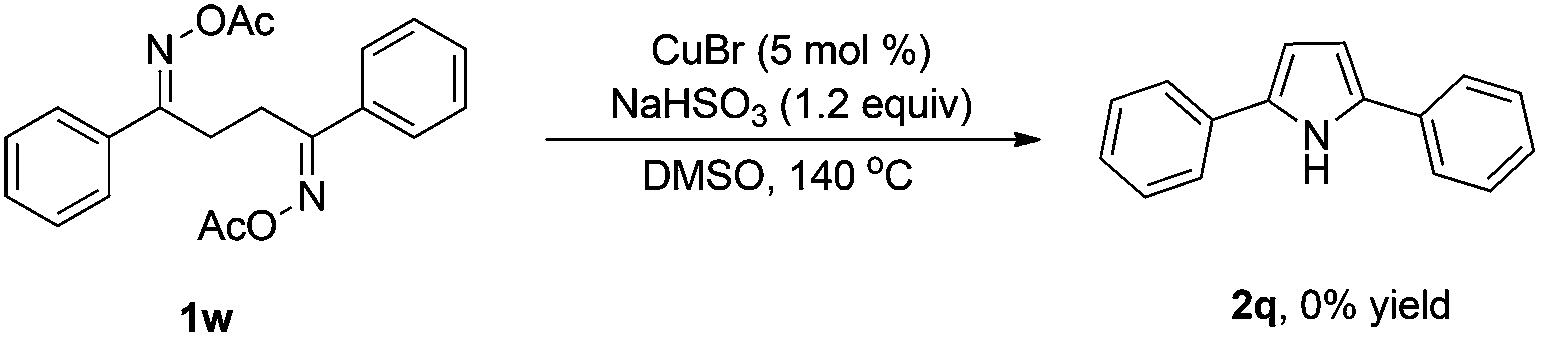 | ||
| Scheme 2 Intramolecular homocoupling of diketoxime diacetate 1w. | ||
In summary, we have developed a novel and efficient copper-catalyzed homocoupling of ketoxime carboxylates for the synthesis of symmetrical pyrroles. The reaction tolerates a wide range of functional groups and is a reliable method for the rapid elaboration of readily available ketoxime carboxylates into a variety of valuable symmetrical pyrroles. Further scope and mechanistic studies of the reaction are underway and will be reported in due course.
This work was supported by generous grants from the National Natural Science Foundation of China (NSFC-21272183, 21002077), and Fund of the Rising Stars of Shanxi Province (2012KJXX-26).
Notes and references
- (a) H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed; (b) M. Wang, H. Xu, T. Liu, Q. Feng, S. Yu, S. Wang and Z. Li, Eur. J. Med. Chem., 2011, 46, 1463 CrossRef CAS PubMed; (c) P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207 CrossRef PubMed.
- (a) M. Munde, M. Lee, S. Neidle, R. Arafa, D. W. Boykin, Y. Liu, C. Bailly and W. D. Wilson, J. Am. Chem. Soc., 2007, 129, 5688 CrossRef CAS PubMed; (b) B. Portevin, C. Tordjman, P. Pastoureau, J. Bonnet and G. D. Nanteuil, J. Med. Chem., 2000, 43, 4582 CrossRef CAS PubMed; (c) J. Lehuédé, B. Fauconneau, L. Barrier, M. Ourakow, A. Piriou and J.-M. Vierfond, Eur. J. Med. Chem., 1999, 34, 991 CrossRef; (d) W.-J. Kuo, Y.-H. Chen, R.-J. Jeng, L.-H. Chan, W.-P. Lin and Z.-M. Yang, Tetrahedron, 2007, 63, 7086 CrossRef CAS PubMed.
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed.
- (a) A. Hantzsch, Ber. Dtsch. Chem. Ges., 1890, 23, 1474 CrossRef; (b) C. Paal, Ber. Dtsch. Chem. Ges., 1885, 18, 367 CrossRef.
- (a) Z.-H. Guan, L. Li, Z.-H. Ren, J. Li and M.-N. Zhao, Green Chem., 2011, 13, 1664 RSC; (b) L. Li, M.-N. Zhao, Z.-H. Ren, J. Li and Z.-H. Guan, Synthesis, 2012, 532 CAS; (c) M.-N. Zhao, H. Liang, Z.-H. Ren and Z.-H. Guan, Adv. Synth. Catal., 2013, 355, 221 CrossRef CAS; (d) B. Pan, C. Wang, D. Wang, F. Wu and B. Wan, Chem. Commun., 2013, 49, 5073 RSC.
- (a) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2010, 39, 4402 RSC; (b) D. Hong, Y. Zhu, Y. Li, X. Lin, P. Lu and Y. Wang, Org. Lett., 2011, 13, 4668 CrossRef CAS PubMed; (c) X. Wang, X.-P. Xu, S.-Y. Wang, W. Zhou and S.-J. Ji, Org. Lett., 2013, 15, 4246 CrossRef CAS PubMed; (d) M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384 CrossRef CAS PubMed; (e) W. B. Liu, H. F. Jiang and L. B. Huang, Org. Lett., 2010, 12, 312 CrossRef CAS PubMed; (f) Y. Hu, C. Wang, D. Wang, F. Wu and B. Wan, Org. Lett., 2013, 15, 3146 CrossRef CAS PubMed.
- (a) S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140 CrossRef CAS PubMed; (b) D. Srimani, D. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 4012 CrossRef CAS PubMed; (c) K. Iida, T. Miura, J. Ando and S. Saito, Org. Lett., 2013, 15, 1436 CrossRef CAS PubMed.
- (a) B. Li, N. Wang, Y. Liang, S. Xu and B. Wang, Org. Lett., 2013, 15, 136 CrossRef CAS PubMed; (b) L. Wang and L. Ackermann, Org. Lett., 2013, 15, 176 CrossRef CAS PubMed; (c) S. Rakshit, F.-W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585 CrossRef CAS PubMed; (d) R. L. Yan, J. Luo, C. Y. Wang, C. W. Ma, G. S. Huang and Y. M. Liang, J. Org. Chem., 2010, 75, 5395 CrossRef CAS PubMed.
- (a) L. Meng, K. Wu, C. Liu and A. Lei, Chem. Commun., 2013, 49, 5853 RSC; (b) Z. Shi, M. Suri and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 4892 CrossRef CAS PubMed; (c) Z. Chen, B. Lu, Z. Ding, K. Gao and N. Yoshikai, Org. Lett., 2013, 15, 1966 CrossRef CAS PubMed.
- (a) F. Chen, T. Shen, Y. Cui and N. Jiao, Org. Lett., 2012, 14, 4926 CrossRef CAS PubMed; (b) Q. Li, A. Fan, Z. Lu, Y. Cui, W. Lin and Y. Jia, Org. Lett., 2010, 12, 4066 CrossRef CAS PubMed; (c) M. Periasamy, G. Srinivas and P. Bharathi, J. Org. Chem., 1999, 64, 4204 CrossRef CAS.
- (a) A. Y. Sukhorukov and S. L. Ioffe, Chem. Rev., 2011, 111, 5004 CrossRef CAS PubMed; (b) K. Narasaka and M. Kitamura, Eur. J. Org. Chem., 2005, 4505 CrossRef CAS.
- (a) C. Ramalingan and Y.-T. Park, J. Org. Chem., 2007, 72, 4536 CrossRef CAS PubMed; (b) M. Hashimoto, Y. Obora, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2008, 73, 2894 CrossRef CAS PubMed.
- (a) E. Choi, C. Lee, Y. Na and S. Chang, Org. Lett., 2002, 4, 2369 CrossRef CAS PubMed; (b) L. D. Luca, G. Giacomelli and A. Porcheddu, J. Org. Chem., 2002, 67, 6272 CrossRef PubMed.
- (a) H. Liang, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Chem.–Eur. J., 2013, 19, 9789 CrossRef CAS PubMed; (b) Z.-H. Guan, Z.-Y. Zhang, Z.-H. Ren, Y.-Y. Wang and X. Zhang, J. Org. Chem., 2011, 76, 339 CrossRef CAS PubMed; (c) W. Tang, A. Capacci, M. Sarvestani, X. Wei, N. K. Yee and C. H. Senanayake, J. Org. Chem., 2009, 74, 9528 CrossRef CAS PubMed.
- (a) Z.-H. Ren, Z.-Y. Zhang, B.-Q. Yang, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2011, 13, 5394 CrossRef CAS PubMed; (b) T. Gerfaud, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 572 CrossRef CAS PubMed; (c) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed; (d) S. Liu and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 6918 CrossRef CAS PubMed; (e) X. Tang, L. Huang, C. Qi, W. Wu and H. Jiang, Chem. Commun., 2013, 49, 9597 RSC.
- (a) S. E. Allen, R. R. Walvoors, R. Padilla-Salinas and M. C. Kozlwski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed; (b) J.-S. Tian and T.-P. Loh, Chem. Commun., 2011, 47, 5458 RSC; (c) J.-S. Tian and T.-P. Loh, Angew. Chem., Int. Ed., 2010, 49, 8417 CrossRef CAS PubMed.
- M.-N. Zhao, H. Liang, Z.-H. Ren and Z.-H. Guan, Synthesis, 2012, 1501 CAS.
- K. Takai, N. Katsura and Y. Kunisada, Chem. Commun., 2001, 1724 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data, 1H and 13C NMR spectra of products 2. See DOI: 10.1039/c3gc41800f |
| This journal is © The Royal Society of Chemistry 2014 |