Continuous flow synthesis of toxic ethyl diazoacetate for utilization in an integrated microfluidic system†
Ram Awatar
Maurya‡
a,
Kyoung-Ik
Min‡
b and
Dong-Pyo
Kim
*b
aDivision of Medicinal Chemistry and Pharmacology, CSIR-Indian Institute of Chemical Technology, Hyderabad 500007, India
bNational Creative Research Center of Applied Microfluidic Chemistry (CAMC), Department of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Republic of Korea. E-mail: dpkim@postech.ac.kr; Fax: +82-54-279-5598; Tel: +82-54-279-2272
First published on 15th October 2013
Abstract
An integrated microfluidic system for multiple reactions and separations of hazardous ethyl diazoacetate is presented. The integrated techniques include: a droplet technique for liquid–liquid and/or gas–liquid separation and in situ generation of the toxic reagent, a dual channel membrane technique based on a cheap polymeric microseparator for liquid–liquid separation, and a capillary microreactor for carrying out cascade reactions in a sequential and continuous manner.
Developing safe and efficient routes for manipulating toxic, explosive and hazardous chemicals is an ever present challenge in chemistry.1 Diazomethane and ethyl diazoacetate are amongst the most common diazo-compounds, which remarkably, have attracted huge interest for fine chemical and pharmaceutical production.2 Despite their commercial availability, the storage, transportation, and reaction of diazo-chemicals have raised significant safety concerns due to their instability, high reactivity and explosive nature. Although in most cases the diazo reagents are freshly prepared prior to use, the potential for detonation of the chemical reagent inventory cannot be fully ignored. Recently microreactors have attracted much interest because they could resolve these safety concerns owing to their miniaturised reaction volume, continuous flow processing and fast heat and mass transfer ability.1,3
In our earlier communication, we reported the generation of diazomethane and the successive processes in PDMS dual channels.4 Despite controlling various parameters, only up to 63% of the total generated diazomethane could be separated and utilized for further reactions. Thus, it was disadvantageous that the safe manipulation of diazomethane was associated with a significant amount of hazardous waste. Additionally, the durability and productivity of the microfluidic device (∼1 mmol per day) were quite low. We therefore set out to develop a robust microfluidic set-up for advanced production of fine chemicals and/or pharmaceuticals using toxic, explosive and hazardous reagents (such as CH2N2, N2CHCOOEt, organic azides, etc.) without compromising on safety or waste hazard concerns.
The reactions of toxic, explosive and unstable reagents in a microreactor alone only partially address the associated safety concerns.5 Hence in situ generation and subsequent reaction of diazo-chemicals in microreactors appears to be the best way to overcome the safety and hazard concerns.4,6 However for multi-step reactions in flow it is crucial to separate or purify the intermediate from the main stream, as the unreacted precursor, excess reagents, solvent and other impurities may negatively affect or even prevent subsequent reaction of the intermediate with other chemicals downstream. The use of immobilized scavengers or packed bed columns to purify the intermediate is certainly inappropriate because of the necessity of periodically changing columns and deposition of by-products on polymer supports. A liquid–liquid and/or gas–liquid separation process using a simple microfluidic system appears more attractive and reasonable.7
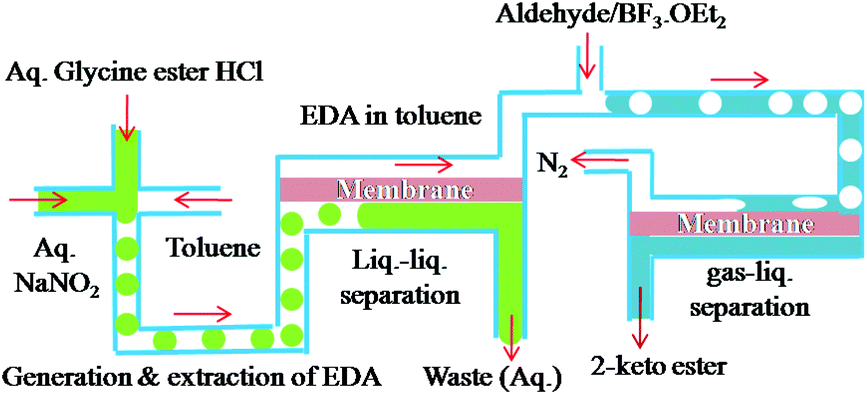
Presented in this communication is a microfluidic system that is integrated to facilitate multiple reactions and multiple separations involving hazardous materials. In the system, the toxic and explosive reactant is synthesized in situ and then reacted to produce a valuable intermediate for the synthesis of fine chemicals and pharmaceuticals. Complete separation of the product is made possible through the use of a droplet microreactor in the system, in which the reactants and the toxic product separate by themselves, i.e., self-separation without any input from outside such as heating or a polymer support, which in turn decreases decomposition of the product. The construction material for the subsequent separation and further reaction of the intermediate is such that the throughput of the final product is two orders of magnitude higher than that obtained earlier in the one-pot, non-integrated system.4 Ethyl diazoacetate (EDA) was taken as a model hazardous and explosive reagent to illustrate the concept. The integrated microfluidic system is shown in Fig. 1. Our integrated system approach not only allows safer and more efficient manipulation of hazardous chemicals but also enables much improved productivity.
 | ||
| Fig. 1 Schematic illustration of continuous generation, extraction, separation, and reaction of EDA in an integrated microfluidic system. | ||
The reaction of nitrous acid with glycine ethyl ester hydrochloride was selected as the reaction of choice for the synthesis of EDA since it produces a high yield of EDA under mild conditions.8 However, the reaction is very sensitive to temperature and is usually carried out by cooling the reaction mixture in a salt-ice bath. Additionally EDA, once synthesized, must be quickly extracted as it starts to decompose in the acidic reaction medium.8
Therefore, continuous reaction and extraction using droplet microreactors could be quite promising for the synthesis of EDA in a continuous flow system. Droplet microreactors have emerged as a powerful synthetic tool due to their quick heat dissipation and the enhanced convectional mixing that occurs inside droplets.9 Additionally they provide very a large surface area per unit volume for efficient extraction of EDA from the acidic reaction mixture which could help to minimize its decomposition. Thus, a simple microfluidic system composed of capillary and mixing units was assembled to generate droplets of EDA precursors in organic solvents that can extract EDA.
For safer and more efficient handling of the hazardous chemical, a chemically, thermally and mechanically robust microfluidic device was required. The PDMS dual channel system has a well known swelling problem in most non-polar organic solvents.10 We, therefore, decided to use a polyimide (PI) film microseparator because of its easy fabrication and excellent mechanical, thermal and chemical stability.11 A dual channel type of microseparator was fabricated using a commercially available polyimide film and PTFE membrane. Briefly, two laser ablated PI films and the PTFE membrane were mechanically bonded in a sandwich manner (for details of fabrication, see ESI†). The droplet microreactor was then connected to this PI dual channel separation device, which in turn was connected with a PFA capillary microreactor for the cascade reactions (Fig. 1).
To accomplish simultaneous in situ droplet synthesis, extraction and separation of EDA, solutions of glycine ethyl ester hydrochloride in acetate buffer (pH 3.5), aqueous NaNO2 and extracting solvent were introduced into the droplet microreactor. Initially we attempted to mix the aqueous solutions of glycine and NaNO2 and then disperse the mixture into toluene using two T-junctions (for details, see ESI†). However, there was a significant loss of EDA when two T-junctions were used, although cooling of the reaction mixture into ice bath did diminish degradation. We, therefore, decided to use an X-junction (see Fig. 1) for mixing the two aqueous solutions and instant dispersion of the mixture into the extracting solvent to minimize the decomposition of EDA in the acidic medium.
Aqueous solutions of glycine ester and NaNO2 were allowed to merge at the X-junction and disperse into the continuous toluene phase. The hydrophobic PFA capillaries (id = 800 μm) are quite suitable for forming aqueous droplets in toluene, which give superior yields of EDA compared to that obtained using other extracting solvents such as diethyl ether or dichloromethane (Table 1). The experiments were performed at ambient temperature and at various flow rates of the aqueous reactants and the organic extracting solvent. It was observed that a flow rate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (aq. glycine ester, aq. NaNO2 and toluene) led to well dispersed aqueous droplets in toluene using the PFA capillary. It is noteworthy that real time extraction of the synthesized EDA, from the aqueous droplets into the toluene medium, was efficiently conducted because of their tiny volume and large surface to volume ratio. Thus the possibility of decomposition of the synthesized EDA in the acidic reaction mixture was minimized. A residence time of 2 min was enough for the droplet reaction, extraction and separation of EDA, resulting in 99% yields of EDA. Lower residence times (<2 min) resulted in lower yields of EDA. It is noteworthy that contrary to batch reactions, nitrosation of glycine ester in a droplet reaction could be performed at room temperature without external cooling, revealing the energy saving characteristics of the microfluidic device. The subsequent continuous separation of the aqueous and organic mixture was realized in a microseparator where a PTFE membrane was sandwiched between the PI dual channels. The thin fluoropolymer membrane with a pore size of 0.45 μm was preferentially wetted by the organic solvent which allowed passage through the membrane holes, while the non-wetting aqueous phase did not penetrate the membrane. Complete separation of the aqueous (waste) and organic phases (EDA) was achieved at high flow rates (up to 300 μL min−1) without any problems, by regulating the back pressures of aqueous and organic outlets. It should be pointed out that the reaction and successive double separation steps were performed in a simple microfluidic set up that produced virtually no hazardous waste. The daily output of EDA was calculated to be 213.84 mmol (for the calculation in detail, see ESI†).
:1:1 (aq. glycine ester, aq. NaNO2 and toluene) led to well dispersed aqueous droplets in toluene using the PFA capillary. It is noteworthy that real time extraction of the synthesized EDA, from the aqueous droplets into the toluene medium, was efficiently conducted because of their tiny volume and large surface to volume ratio. Thus the possibility of decomposition of the synthesized EDA in the acidic reaction mixture was minimized. A residence time of 2 min was enough for the droplet reaction, extraction and separation of EDA, resulting in 99% yields of EDA. Lower residence times (<2 min) resulted in lower yields of EDA. It is noteworthy that contrary to batch reactions, nitrosation of glycine ester in a droplet reaction could be performed at room temperature without external cooling, revealing the energy saving characteristics of the microfluidic device. The subsequent continuous separation of the aqueous and organic mixture was realized in a microseparator where a PTFE membrane was sandwiched between the PI dual channels. The thin fluoropolymer membrane with a pore size of 0.45 μm was preferentially wetted by the organic solvent which allowed passage through the membrane holes, while the non-wetting aqueous phase did not penetrate the membrane. Complete separation of the aqueous (waste) and organic phases (EDA) was achieved at high flow rates (up to 300 μL min−1) without any problems, by regulating the back pressures of aqueous and organic outlets. It should be pointed out that the reaction and successive double separation steps were performed in a simple microfluidic set up that produced virtually no hazardous waste. The daily output of EDA was calculated to be 213.84 mmol (for the calculation in detail, see ESI†).
|
|
|||
|---|---|---|---|
| Entry | Res. timeb (min) | Extracting solvent | Yield of EDAc (%) |
|
a Composition of EDA precursors: 1.50 M EtOOCCH2NH2·HCl in acetate buffer (acetate conc. 2.0 M, pH 3.5), 1.51 M NaNO2 in water; experiments were carried out at room temperature; flow rate ratio of aqueous solutions and extracting solution was 1:1:1.
b Residence time for both droplet reaction and extraction in PFA capillary (id = 800 μm, length = 120 cm, internal volume = 600 μL), (50 + 50 + 50) μL min−1 for 4 min, (100 + 100 + 100) μL min−1 for 2 min of the residence time.
c Determined by GC–MS using anisole as an internal standard.
|
|||
| 1 | 4.0 | Toluene | 99 |
| 2 | 2.7 | Toluene | 98 |
| 3 | 2.0 | Toluene | 99 |
| 4 | 1.3 | Toluene | 87 |
| 5 | 2.0 | CH2Cl2 | 75 |
| 6 | 2.0 | Et2O | 80 |
Next we focused on the cascade reactions using EDA generated in situ. Coupling of EDA with aldehydes is an attractive, atom economical organic transformation for the synthesis of fine chemicals and pharmaceuticals. Although the reaction was originally known to be catalyzed by strong bases (e.g. butyllithium, lithiumdiisopropylamide, sodium hydride, potassium hydroxide), recently several mild organocatalytic approaches have been developed.12 In this work, we attempted 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) catalyzed coupling of benzaldehyde with EDA generated in situ in the microfluidic system (Table 2).
|
|
|||
|---|---|---|---|
| Entry | Res. time (min) | Temp (°C) | Yieldb (%) |
| a EDA was generated in situ as in Table 1. Flow rates of EDA and benzaldehyde containing DBU: (50 + 20) μL min−1 for 36 min, (75 + 30) μL min−1 for 24 min, (100 + 40) μL min−1 for 18 min of the residence time. b Isolated yield on 1 mmol scale. c Ultrasonication (power = 330 W, frequency = 40 kHz). | |||
| 1 | 36 | rt | 42 |
| 2 | 36 | 40 | 51 |
| 3 | 36 | 60 | 57 |
| 4 | 36 | 80 | 55 |
| 5c | 36 | 40 | 80 |
| 6c | 24 | 40 | 81 |
| 7c | 18 | 40 | 72 |

A solution of benzaldehyde in toluene (3.0 M) containing 20 mol% DBU was allowed to mix with the stream from the organic outlet of the PI dual channel separator (1.5 M EDA in toluene) to allow the aldol reaction to occur. Flow rates of both reactants were adjusted so that the EDA was a little in excess compared to the benzaldehyde in the reaction mixture (stoichiometry of EDA to aldehyde = 1.25:1).
The reaction mixture was then introduced into a PFA capillary (id = 800 μm, length = 5 m, internal volume = 2.5 mL) and the reaction was quenched in saturated aq. NaHCO3. It was observed that at room temperature the reaction of EDA with benzaldehyde was incomplete after the 36 min residence time. Better product yields were achieved at higher temperatures. However, we were pleased to have achieved a high yield of the desired product in 24 min of residence time using ultrasonic irradiation at 40 °C (Table 2). It is, therefore, obvious that ultrasonic irradiation of the reaction mixture greatly promoted the reaction rate, presumably due to enhanced chaotic mixing and the hot-spot effects caused by collapsed bubbles.13 It is notable that the productivity of the microfluidic set-up was significantly higher (104.98 mmol per day; see ESI† for calculations) when compared to our earlier PDMS dual channel system (∼1 mmol per day),4 with an increase in the throughput of two orders of magnitude.
Inspired by the successful cascade reaction of the EDA generated in situ with benzaldehyde, we expanded the scope of the reaction to a series of other aromatic, heteroaromatic and aliphatic aldehydes. Since aliphatic aldehydes are prone to undergo self aldol reactions in the presence of DBU, we did not pre-mix the DBU with the aldehyde solution. Instead, we kept the DBU and aldehyde in two separate syringes so that all three components, DBU, aldehyde and EDA from PI dual channel device, could be mixed together at the X-junction (for a detailed schematic illustration, see ESI†). Using the optimized protocol (40 °C, ultrasonication, 24 min of residence time), we achieved high yields of aldehyde-EDA adducts (Fig. 2). It is worth noting that our results for this DBU catalyzed aldol reaction of EDA with aldehydes were comparable to, or somewhat better than, the reported results obtained using anhydrous solvent and inert atmospheric conditions.12b The use of ultrasonic agitation in the microreactor not only decreased the reaction time but also increased the yield. The microfluidic system provides sufficiently pure and anhydrous EDA for DBU catalyzed aldol reactions. The automation, simplicity, high safety, and excellent control over the reaction parameters facilitates small scale laboratory research of EDA reactions.
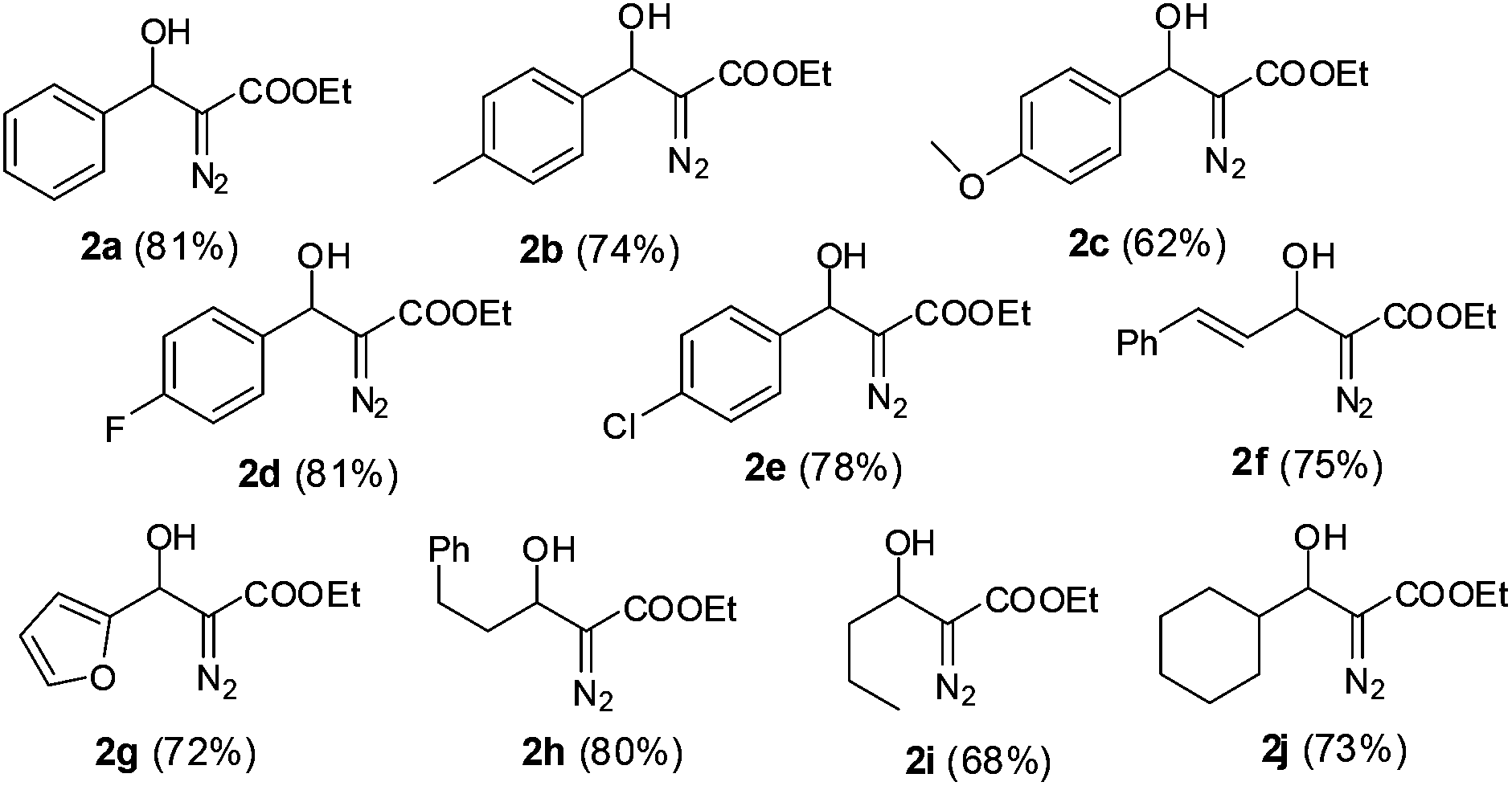 | ||
| Fig. 2 Cascade generation, separation and reaction of EDA with various aldehydes (yields in the parentheses are of isolated product on a 5 mmol scale). | ||
To show that the integrated microfluidic system is adaptable enough to handle reactions involving separation of gaseous products, we proceeded to study the synthesis of 2-keto esters from aldehydes. The reaction is catalysed by Lewis acids and accompanied by evolution of nitrogen.14 Handling of gas either as a reactant or a bi-product is quite challenging in microreactors as it disturbs the flow rates and makes control other reaction parameters difficult.
The reaction of EDA with hydrocinnamaldehyde catalyzed by BF3·OEt2 was taken as a model reaction for the synthesis of 2-keto esters. EDA was generated, extracted and separated as aforementioned and the outgoing organic phase from the first PI dual channel separator was connected to a T-junction where it met with a solution of hydrocinnamaldehyde (3.0 M) containing 5 mol% BF3·OEt2. The flow rates were adjusted in such a way that the stoichiometric content of EDA was in slight excess of hydrocinnamalaldehyde. Immediately after mixing the two solutions (EDA and aldehyde), the reaction mixture started generating nitrogen and the product stream was pushed rapidly into the outside collector. Although the yield of 2-keto ester 3h was good (see Fig. 4), the propulsion of the product stream could be a source of safety problems. Therefore, to remove the generated nitrogen from the reaction channel, another PI dual channel separator with a PTFE membrane was added to the system as shown in Fig. 3.
 | ||
| Fig. 3 Cascade generation, extraction, separation and reaction of EDA with adehydes to yield 2-keto esters with subsequent gas–liquid separation. | ||
 | ||
| Fig. 4 Automated generation, separation and reaction of EDA with aldehydes to yield 2-keto esters (yields in parentheses are of isolated product on 5 mmol scale). | ||
The PFA capillary microreactor (id = 800 μm, length = 5 m, internal volume = 2.5 mL) was then connected to a gas–liquid microseparator (PI dual channel). The PI dual channel separator completely separated the gas–liquid mixture, which made it possible to efficiently produce the desired 2-keto ester without compromising safety. Using the same microfluidic arrangement, we were able to synthesize a series of 2-keto esters using various aldehydes. All the reactions were carried out with the rates of 50 μL min−1 (EDA) and 20 μL min−1 (aldehyde). The yields were fairly good with aliphatic aldehydes but only moderate with aromatic aldehydes such as benzaldehyde (Fig. 4).
The integrated microfluidic system has been proven to be resilient and robust in handling hazardous reagents, even when subjected to continuous operation of the system over long periods of time. The durability can readily be attributed to the chemical inertness of the materials used (PFA, PTFE and PI). The integrated microfluidic system was repeatedly used for multiple optimizations and reactions over several months without any noticeable change in system performance.
Conclusions
In conclusion, we have demonstrated the continuous flow process for reactions and separations of hazardous reagents, using ethyl diazoacetate as a model reaction, in a safer and more efficient manner using an integrated microfluidic system. The integrated techniques include: a droplet technique for liquid–liquid and/or gas–liquid separation and in situ generation of the toxic reagent, a dual channel membrane technique based on a cheap polymeric microseparator, and a capillary microreactor for carrying out cascade reactions in a sequential and continuous manner. The novel, safe, microfluidic droplet approach allows real time extraction of EDA, a hazardous reagent chosen because of its importance in producing intermediates for fine chemicals and pharmaceuticals, from its acidic reaction mixture. The generation and separation of EDA are so efficient with 99% yields of EDA that the toxic hazardous waste produced is almost negligible. This integrated microfluidic approach not only allows safer and more efficient manipulation of hazardous and explosive EDA but also allows much improved production of fine chemicals. In addition, the integrated system can be adapted to accommodate different combinations of reactors and phase separators for the purpose of producing chemicals on demand.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2008-0061983 and NRF-2013-Global Ph.D. Fellowship Program).Notes and references
- (a) P. B. Palde and T. F. Jamison, Angew. Chem., Int. Ed., 2011, 50, 3525–3528 CrossRef CAS PubMed; (b) M. O'Brien, I. R. Baxendale and S. V. Ley, Org. Lett., 2010, 12, 1596–1598 CrossRef CAS PubMed; (c) M. Irfan, T. N. Glasnov and C. O. Kappe, Org. Lett., 2011, 13, 984–987 CrossRef CAS PubMed; (d) B. Gutmann, J.-P. Roduit, D. Roberge and C. O. Kappe, Angew. Chem., Int. Ed., 2010, 49, 7101–7105 CrossRef CAS PubMed; (e) R. Trotzki, M. Nüchter and B. Ondruschka, Green Chem., 2003, 5, 285–290 RSC; (f) D. R. J. Acke and C. V. Stevens, Green Chem., 2007, 9, 386–390 RSC; (g) A. R. Katritzky, S. Zhang and Y. Fang, Org. Lett., 2000, 2, 3789–3791 CrossRef CAS PubMed; (h) K. C. Basavaraju, S. Sharma, R. A. Maurya and D.-P. Kim, Angew. Chem., Int. Ed., 2013, 52, 6735 CrossRef CAS PubMed; (i) S. Sharma, R. A. Maurya, K.-I. Min, G.-Y. Jeong and D.-P. Kim, Angew. Chem., Int. Ed., 2013, 52, 7564 CrossRef CAS PubMed.
- For selected reviews, see: (a) Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 1015–1026 CrossRef; (b) G. Maas, Angew. Chem., Int. Ed., 2009, 48, 8186–8195 CrossRef CAS PubMed; (c) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–936 CrossRef CAS PubMed; (d) D. M. Hodgson, F. Y. T. M. Pierard and P. A. Stupple, Chem. Soc. Rev., 2001, 30, 50–61 RSC.
- For selected reviews and papers on microreactors, see: (a) R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed; (b) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC; (c) Y. Chen, Y. Zhao, M. Han, C. Ye, M. Danga and G. Chen, Green Chem., 2013, 15, 91–94 CAS; (d) C. P. Park, R. A. Maurya, J. H. Lee and D.-P. Kim, Lab Chip, 2011, 11, 1941–1945 RSC; (e) B. Pieber and C. O. Kappe, Green Chem., 2013, 15, 320–324 RSC; (f) A. Odedra, K. Geyer, T. Gustafsson, R. Gilmour and P. H. Seeberger, Chem. Commun., 2008, 3025–3027 RSC; (g) R. A. Maurya, P. H. Hoang and D.-P. Kim, Lab Chip, 2012, 12, 65–68 RSC.
- R. A. Maurya, C. P. Park, J. H. Lee and D.-P. Kim, Angew. Chem., Int. Ed., 2011, 50, 5952–5955 CrossRef CAS PubMed.
- (a) L. J. Martin, A. L. Marzinzik, S. V. Ley and I. R. Baxendale, Org. Lett., 2011, 13, 320–323 CrossRef CAS PubMed; (b) H. E. Bartrum, D. C. Blakemore, C. J. Moody and C. J. Hayes, J. Org. Chem., 2010, 75, 8674–8676 CrossRef CAS PubMed.
- (a) M. Struempel, B. Ondruschka, R. Daute and A. Stark, Green Chem., 2008, 10, 41–43 RSC; (b) H. E. Bartrum, D. C. Blakemore, C. J. Moody and C. J. Hayes, Chem.–Eur. J., 2011, 17, 9586–9589 CrossRef CAS PubMed; (c) D. X. Hu, M. O'Brien and S. V. Ley, Org. Lett., 2012, 14, 4246–4249 CrossRef CAS PubMed; (d) Z. Yu, Y. Lv, C. Yu and W. Su, Tetrahedron Lett., 2013, 54, 1261–1263 CrossRef CAS PubMed; (e) M. Chen and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 4247–4250 CrossRef CAS PubMed.
- (a) J. G. Kralj, H. R. Sahoo and K. F. Jensen, Lab Chip, 2007, 7, 256–263 RSC; (b) T. Noël, S. Kuhn, A. J. Musacchio, K. F. Jensen and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 5943–5946 CrossRef PubMed; (c) H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704–5708 CrossRef CAS PubMed; (d) R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 899–903 CrossRef CAS PubMed.
- (a) E. B. Womack and A. B. Nelson, Org. Synth., Coll., Vol. 3, 1955, 392 Search PubMed; (b) W. Wang, K. Shen, X. Hu, J. Wang, X. Liu and X. Feng, Synlett, 2009, 1655–1658 Search PubMed.
- (a) P. H. Hoang, C. T. Nguyen, J. Perumal and D.-P. Kim, Lab Chip, 2011, 11, 329–335 RSC; (b) P. H. Hoang, H. Park and D.-P. Kim, J. Am. Chem. Soc., 2011, 133, 14765–14770 CrossRef CAS PubMed.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS PubMed.
- K.-I. Min, T.-H. Lee, C. P. Park, Z.-Y. Wu, H. H. Girault, I. Ryu, T. Fukuyama, Y. Mukai and D.-P. Kim, Angew. Chem., Int. Ed., 2010, 49, 7063–7067 CrossRef CAS PubMed.
- (a) B. M. Trost, S. Malhotra and B. A. Fried, J. Am. Chem. Soc., 2009, 131, 1674–1675 CrossRef CAS PubMed; (b) N. Jiang and J. Wang, Tetrahedron Lett., 2002, 43, 1285–1287 CrossRef CAS; (c) W. Yao and J. Wang, Org. Lett., 2003, 5, 1527–1530 CrossRef CAS PubMed.
- (a) R. H. Liu, R. Lenigk, R. L. Druyor-Sanchez, J. Yang and P. Grodzinski, Anal. Chem., 2003, 75, 1911–1917 CrossRef CAS; (b) G. G. Yaralioglu, I. O. Wygant, T. C. Marentis and B. T. Khuri-Yakub, Anal. Chem., 2004, 76, 3694–3698 CAS; (c) R. L. Hartman, J. R. Naber, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Org. Process Res. Dev., 2010, 14, 1347–1357 CrossRef CAS; (d) D. F. Rivas, P. Cintas and H. J. G. E. Gardeniers, Chem. Commun., 2012, 48, 10935–10947 RSC; (e) B. S. Singh, H. R. Lobo, D. V. Pinjari, K. J. Jarag, A. B. Pandit and G. S. Shankarling, Ultrason. Sonochem., 2013, 20, 287–293 CrossRef CAS PubMed.
- For selected recent publications on Lewis acid catalyzed reaction of EDA with aldehyde, see: (a) M. R. Fructos, M. M. Díaz-Requejo and P. J. Pérez, Chem. Commun., 2009, 5153–5155 RSC; (b) H. Murata, H. Ishitani and M. Iwamoto, Tetrahedron Lett., 2008, 49, 4788–4791 CrossRef CAS PubMed; (c) W. Li, J. Wang, X. Hu, K. Shen, W. Wang, Y. Chu, L. Lin, X. Liu and X. Feng, J. Am. Chem. Soc., 2010, 132, 8532–8533 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc41226a |
| ‡ R. A. Maurya and K.-I. Min equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2014 |