Mimicking mineral neogenesis for the clean synthesis of metal–organic materials from mineral feedstocks: coordination polymers, MOFs and metal oxide separation†
Feng
Qi
a,
Robin S.
Stein
b and
Tomislav
Friščić
*a
aDepartment of Chemistry, McGill University and FRQNT Centre for Green Chemistry and Catalysis, 801 Sherbrooke St. W., Montreal, H3A 0B8 Canada. E-mail: tomislav.friscic@mcgill.ca; Fax: +1-514-398-3757; Tel: +1-514-398-3959
bBruker Ltd, 555 Steeles Avenue East, Milton, ON L9T 1Y6, Canada
First published on 30th August 2013
Abstract
We present a systematic study of a mild approach for the activation of metal oxides, involving reactivity and self-assembly in the solid state, which enables their solvent-free chemical separation and direct solvent-free and low-energy conversion into coordination polymers and open metal–organic frameworks (MOFs). The approach is inspired by geological biomineralization processes known as mineral weathering, in which long-term exposure of oxide or sulfide minerals to molecules of biological origin leads to their conversion into simple coordination polymers. This proof-of-principle study shows how mineral neogenesis can be mimicked in the laboratory to provide coordination polymers directly from metal oxides without a significant input of either thermal or mechanical energy, or solvents. We show that such “aging” is accelerated by increased humidity, a mild temperature increase and/or brief mechanical activation, enabling the transformation of a variety of high-melting (800 °C–2800 °C) transition (MnII, CoII, NiII, CuII, and Zn) and main group (Mg and PbII) oxides at or near room temperature. Accelerated aging reactions are readily scaled to at least 10 grams and can be templated for the synthesis of two-dimensional and three-dimensional anionic frameworks of Zn, Ni(II) and Co(II). Finally, we demonstrate how this biomineralization-inspired approach provides an unprecedented opportunity for solvent-free chemical segregation of base metals, such as Cu, Zn and Pb, in their oxide form under mild conditions.
Introduction
The development of environmentally-friendly and sustainable synthesis is one of the central tasks of modern science and technology.1 As the number of potential applications of coordination polymers and metal–organic frameworks (MOFs) grows to include hydrogen or methane storage, carbon sequestration, catalysis and even light-harvesting,2 it becomes of critical importance to address how such materials can be manufactured in a clean, sustainable and scalable manner from slightly soluble natural feedstocks (e.g. mineral concentrates consisting of metal oxides or sulfides), rather than from conventionally used derivatives that are also toxic or corrosive (metal nitrates, chlorides, and acetates).3 While the clean synthesis of coordination polymers and MOFs4 from soluble salts has been explored using sonochemistry,5 microwave synthesis,6 electrochemistry,7 mechanochemical synthesis8 and near-critical water conditions,9 we are interested in developing mild processes for directly transforming metal minerals or mineral concentrates into metal–organic materials. Metal oxides are recognized by researchers10 and industry3 as the ideal feedstock for inexpensive and clean synthesis. Also, the development of mild methods for metal oxide and sulfide transformation and separation offers benefits to mineral processing and refining industries by improving the energy- and solvent-intensive pyro- and hydrometallurgical protocols.11However, metal oxides are rarely used as starting materials due to their low solubility, high melting points and generally inert nature resulting from high lattice energies (typically 4–6 MJ mol−1). Transformations of metal oxides into metal–organic materials are energy-intensive, e.g. conversion of ZnO or CoO into porous frameworks via melt reactions can last for several days at 120 °C–160 °C.12 Consequently, activation of metal oxides near room temperature and without bulk solvents is a challenge that must be met by non-conventional approaches. Although mechanochemistry8 has recently enabled the conversion of main group or d10-oxides (ZnO, MgO, CdO, and Bi2O3)13–18 into MOFs and pharmaceuticals, it has been less efficient with transition metal oxides. Instead, mechanosyntheses of coordination compounds of Co, Ni or Cu use acetates, carbonates or halides.19,20
We are exploring a novel and potentially general methodology for converting transition metal oxides into metal–organic derivatives, which minimizes energy use and completely avoids organic solvents. Microorganisms, such as lichens, can transform inert minerals into metal–organic derivatives by excreting small molecules known as lichen acids.21 Such geological biomineralization, known as mineral weathering (neogenesis), is essential for the formation of secondary metal–organic minerals (also called “organic” minerals).22 For example, oxalic acid (H2ox) generated by lichens or found as ammonium oxalate in guano can slowly convert copper ores into the copper(II) oxalate mineral Moolooite.23,24 Other examples of mineral weathering by microorganisms include readily extractable deposits of metal oxalate biominerals humboldtine, lindbergite and glushinskite, based on iron(II), manganese(II) and magnesium, respectively.25In vitro experiments have also demonstrated the biomineralization of lead oxalate by Aspergillus niger or the lichen Diploschistes muscorum. Most oxalate biominerals are one-dimensional (1-D) coordination polymers (Scheme 1a).
 | ||
| Scheme 1 | ||
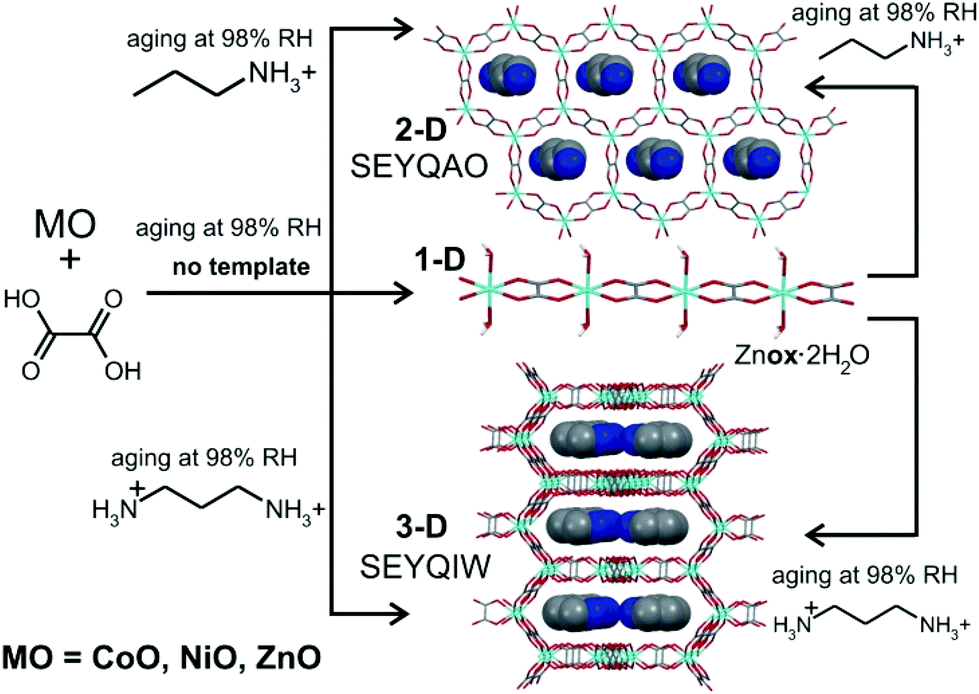
We are exploring whether such weathering of metal oxides can be reproduced in the synthetic chemical laboratory and adapted into an environmentally-friendly pathway to metal–organic materials. In this study we establish: (1) the conditions that enable the conversion of laboratory-scale samples of metal oxides into metal–organic materials; (2) the applicability of this methodology to a variety of metals and scale-up to multi-gram amounts; and (3) the use of templating to drive such a biomineralization-inspired reactivity for the synthesis of open MOFs.
Our work is influenced by that of Byrn and co-workers, who noted the complexation of MgO upon aging with pharmaceuticals.26 Our first report on “accelerated aging” demonstrated conversion of ZnO into zeolitic MOFs by aging with imidazole ligands at high humidity with an acid catalyst.27 For this systematic study we selected metal oxalates as models for three principal reasons. Oxalates are of obvious geological and biomimetic significance as the dominant type of “organic” secondary minerals and are of continuous interest as functional (magnetic, proton conducting) materials.28 There is a large variety of framework topologies accessible through oxalate ligands, providing an opportunity to explore how reactions of geological weathering can be adapted for the synthesis of open MOFs.29 The most significant metal oxalate topologies are the neutral 1-D polymer Mox·2H2O in monoclinic α-dihydrates of divalent metal oxalates (Scheme 1a),30 and the open two-dimensional (2-D) honeycomb and three-dimensional (3-D) topologies31 of the anion M2(ox)32− (M = divalent metal ion) (Scheme 1b). Formation of 2-D or 3-D M2(ox)32− MOFs is controlled by templates and depends on the configuration of the octahedral M(ox)3 subunits.32 Formation of oxalate MOFs in solution was extensively studied,28,29 and particularly important for this study is the work by Cheetham33 and Rao34 on templated solvothermal synthesis using organoammonium cations.
Experimental section
All chemicals were of reagent grade and obtained from commercial sources. MgO, MnO, CoO, NiO, ZnO and PbO were of 99%+ purity, obtained from Sigma-Aldrich, CuO was of 96% purity, obtained from Riedel-de Haën, and H2ox·2H2O was of 99%+ purity, obtained from American Chemicals Ltd. ZnO and MgO have been calcined (400 °C) to remove potential hydroxide and carbonate impurities, and were kept in a dry desiccator over P4O10. Aging reactions were conducted either in a walk-in incubator held at 45 °C, or at room temperature without a particular means of temperature control. The samples aged at high humidity were kept at 98% RH atmosphere established in a Secador® cabinet equilibrated with saturated aqueous K2SO4. In a typical experiment, 1 mmol of a metal oxide was mixed with 1 mmol of H2ox·2H2O and gently ground in an agate mortar and pestle for 30 seconds. The solid mixture was placed in a 20 ml open vial and stored under controlled conditions. All reactions were investigated by powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA), Fourier-transform infrared attenuated total reflection (FTIR-ATR) spectroscopy and, in some cases, UV-vis reflectance, solid-state NMR (SSNMR) and differential scanning calorimetry (DSC).Thermogravimetric analysis was conducted using a TA Instruments Q500 Thermogravimetric System with a Pt pan under a dynamic atmosphere of N2 or air with 40 ml min−1 balance flow and 60 ml min−1 purge flow. The upper temperature limit ranged from 500 °C to 800 °C with a heating rate of 10 °C min−1. DSC was performed on a TA Instruments Q2000 Differential Scanning Calorimeter using a standard aluminum pan of 40 μL. Nitrogen flow was set at 50 ml min−1 and the upper temperature limit ranged from 105 °C to 150 °C with a constant heating rate of 10 °C min−1.
UV-vis reflectance was measured on an Ocean Optics Jaz-Combo spectrometer using an LS-1 Tungsten Halogen Light Source (360–2000 nm), a 0.4 mm fiber optic reflection R400-Angle-UV probe with an RPH-1 reflection probe holder and a WS-1 PTFE diffuse reflection standard, from 400 nm to 800 nm.
SSNMR spectra of [pa]2[Zn2(ox)3]·3H2O were recorded on a standard-bore Bruker Avance III spectrometer operating at 500.13 MHz using a Bruker 4 mm double-resonance probe spinning at 5 kHz. Spectra were referenced using the chemical shift of the carbonyl carbon of glycine at 174.1 ppm with respect to TMS. SSNMR spectra of [pn][Zn2(ox)3]·3H2O were recorded on a 400 MHz Varian VNMRS spectrometer operating at 100.52 MHz using a T3HX probe with a 7.5 mm zirconia rotor spinning at 4.5 kHz. The spectrum was acquired in 1000 scans using cross-polarization for 1 ms and 2 s recycle delay.
PXRD patterns were collected in the 2θ range 3°–60° on a Bruker D2 Phaser diffractometer using a CuKα (λ = 1.54 Å) source. Data were analyzed using Panalytical X'pert HighScore Plus.
Results and discussion
As an initial foray into investigating the aging reactivity of oxalic acid and metal oxides, we prepared a stoichiometric 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of oxalic acid dihydrate (H2ox·2H2O) and ZnO. The mixture was prepared by gently grinding the reactants in a mortar and pestle for 30 seconds. Care was taken to avoid harsh grinding that could cause uncontrolled mechanical activation and, therefore, difficulties in reproducibility. Powder X-ray diffraction (PXRD) after 24 hours aging at room temperature (18 °C–22 °C) and a high (98%) relative humidity (RH) revealed the formation of a new product (Fig. 1).
:1 mixture of oxalic acid dihydrate (H2ox·2H2O) and ZnO. The mixture was prepared by gently grinding the reactants in a mortar and pestle for 30 seconds. Care was taken to avoid harsh grinding that could cause uncontrolled mechanical activation and, therefore, difficulties in reproducibility. Powder X-ray diffraction (PXRD) after 24 hours aging at room temperature (18 °C–22 °C) and a high (98%) relative humidity (RH) revealed the formation of a new product (Fig. 1).
 | ||
| Fig. 1 (a) Two coordination polymer chains connected by hydrogen bonds in Znox·2H2O (CSD QQQBOD04). PXRD patterns for aging reactions of ZnO and H2ox·2H2O at 98% RH: (b) H2ox·2H2O; (c) simulated for Znox·2H2O; (d) ZnO; (e) 1 day at room temperature; (f) 5 days at room temperature; (g) 1 day at 45 °C; (h) 5 days at 45 °C; (i) 1 day at room temperature with pre-milling; (j) 5 days at room temperature with pre-milling; and (k) 10 days at room temperature with pre-milling. (l) Completed 10 gram aging syntheses of Znox·2H2O and Niox·2H2O. | ||
Comparison with PXRD patterns simulated for known zinc oxalate structures revealed that the product was the 1-D coordination polymer zinc oxalate dihydrate (Znox·2H2O, CSD QQQBOD04) in the α-type structure.35 In one week, the conversion to the 1-D coordination polymer was complete. The composition Znox·2H2O was confirmed by thermogravimetric analysis (TGA) in air, which provided the assessment of weight fractions for the included water (measured: 18.5%, calculated: 19.0%) and ZnO residue (measured: 42.9%, calculated: 43.0%). Aging reactivity was further improved by storing the sample under 98% RH conditions at a mild temperature of 45 °C. Under such conditions, complete formation of α-Znox·2H2O, as established by PXRD (ESI Fig. S1†), was achieved in 5 days. When scaled to 10 grams, the reaction was completed in seven days (Fig. 1l). Transformation was also detected by Fourier-transform infrared attenuated total reflection (FTIR-ATR) spectroscopy (Fig. 2), specifically by the disappearance of O–H stretching bands of H2ox·2H2O at 3380 cm−1 and 3470 cm−1 and the appearance of the O–H stretching bands of Znox·2H2O at 3350 cm−1, as well as the appearance of characteristic Znox·2H2O bands at 1604 cm−1, 1356 cm−1 and 1311 cm−1.
 | ||
| Fig. 2 FTIR-ATR spectra of the final products of aging reactions for: (a) ZnO, 5 days at 45 °C; (b) CoO, 7 days at 45 °C; (c) NiO, 7 days at 45 °C; (d) CuO, 16 days at 45 °C; (e) MgO, 9 days at 45 °C; (f) MnO, 16 days at room temperature; (g) MnO, 16 days at 45 °C; (h) PbO, 14 days at 45 °C; and (i) MnO, aging of a pre-milled sample for 1 day at 45 °C. The spectrum of H2ox·2H2O is given under (j). | ||
Structure templating
After establishing that oxalic acid reacts with ZnO to form the closely packed Znox·2H2O, a templated synthesis of open structures was attempted. Rao and Cheetham have established that solvothermal syntheses of metal oxalates can be directed towards the formation of open MOFs by organoammonium templates.36–38 The design to introduce structure templating into aging reactivity involved mixtures of ZnO, H2ox·2H2O and either 1,3-propanediammonium oxalate [pn][ox] or propylammonium oxalate [pa]2[ox] in the respective stoichiometric ratio 2:2:1. Such templated aging reactions were expected to yield known materials [pn][Zn2(ox)3]·3H2O37 and [pa]2[Zn2(ox)3]·3H2O,36 based on the 2-D and 3-D forms of the [Zn2(ox)3]2− anionic framework, respectively (Fig. 3a).
 | ||
| Fig. 3 (a) Planned synthesis of open anionic 2-D and 3-D zinc oxalate frameworks; (b) templates pn2+ and pa+; and (c) a hydrogen-bonded layer in the crystal of (pa)2·(ox), with propyl chains omitted for clarity. | ||
The synthesis of [pn][ox] and [pa]2[ox] provided a minor challenge. The presence of water readily induced the formation of hydrogenoxalate hydrate salts [pa][Hox]·H2O (CSD QIGCOY, QIGCOY01)36 and [pn][Hox]2·2H2O (CSD PEPQEG, PEPQEG01).38 Hydrogenoxalates are not suitable for planned templating experiments, due to the inadequate ratio of cationic organoammonium templates and oxalate building blocks. The synthesis of [pn][ox] and [pa]2[ox] was devised by reacting anhydrous oxalic acid (obtained by drying H2ox·2H2O overnight at 125 °C) with the amine in methanol. In ethanol, the product contained a significant fraction of the hydrated hydrogenoxalate. The anhydrous nature of the material from methanol was confirmed by TGA. The structure and composition of [pn]2[ox] were also confirmed by single crystal X-ray diffraction (crystallographic data are given in ESI,† section 1). The structure consists of sheets of pa+ and ox2− held by N+–H⋯O− hydrogen bonds (Fig. 3c).
After 5 days at room temperature and 98% RH, a PXRD analysis (Fig. 4a and b) of the mixture of ZnO, H2ox·2H2O and [pn][ox] in a 2:2:1 ratio revealed the presence of Znox·2H2O, but also X-ray reflections consistent with the expected honeycomb-topology [pn][Zn2(ox)3]·3H2O (Fig. 5). Aging for a total of 16 days led to the complete disappearance of reflections of ZnO reactant and the Znox·2H2O intermediate, and the diffraction pattern displayed an excellent fit to that expected for the pn2+ salt of the Zn2(ox)32− framework. Since Znox·2H2O was an intermediate in the assembly of [pn][Zn2(ox)3]·3H2O, a room-temperature aging reaction of [pn][ox] with Znox·2H2O was attempted. After five days, the reaction mixture fully converted into [pn][Zn2(ox)3]·3H2O. Quantitative conversion was evident from PXRD (Fig. 4c and d) and TGA.‡
 | ||
| Fig. 4 PXRD patterns for templated aging syntheses of zinc oxalate MOFs: (a) Znox·2H2O; (b) ZnO, H2ox·2H2O and [pn][ox] in the ratio of 2:2:1 after 5 days; (c) Znox·2H2O and [pn][ox] in the ratio of 2:1 after 5 days; (d) simulated for 2-D MOF [pn][Zn2(ox)3]·3H2O (SEYQAO); (e) ZnO, H2ox·2H2O and [pa]2[ox] in the ratio of 2:2:1 after 5 days; and (f) simulated for 3-D MOF [pa]2[Zn2(ox)3]·3H2O (SEYQIW). Reactions were done at room temperature, 98% RH. The characteristic reflection of Znox·2H2O is marked with ‘*’. | ||
 | ||
| Fig. 5 Aging reactivity of ZnO, CoO and NiO towards H2ox·2H2O with and without organoammonium templates. | ||
Aging of a mixture of (pa)2(ox), H2ox and ZnO at room temperature and 98% RH was explored next. After five days, PXRD (Fig. 4e and f) and TGA‡ indicated complete conversion to the expected 3-D framework [pa]2[Zn2(ox)3]·3H2O (CSD SEYQIW, Fig. 5).36
Cross-polarization magic angle spinning 13C solid-state NMR (SSNMR, see ESI,† section 5) was consistent with the product being [pa]2[Zn2(ox)3]·3H2O, displaying the signals of ox2− and pa+. The 1H–13C HETCOR and 1H–1H INADEQUATE-style experiments enabled tentative assignment of the 1H SSNMR spectra of [pa]2[Zn2(ox)3]·3H2O, but with multiple signals expected from partially disordered water molecules unresolved. The HETCOR spectrum was consistent with the reported structure in which the framework interacts mostly with the methyl and ammonium moieties of the pa+. The 13C SSNMR spectrum of [pn][Zn2(ox)3]·3H2O was also consistent with the published structure. Differences between Znox·2H2O, [pn][Zn2(ox)3]·3H2O and [pa]2[Zn2(ox)3]·3H2O were also evident using FTIR-ATR (ESI Fig. S13†).
Reactivity of other metal oxides: product hydration state
The reactivity of other metal oxides with oxalic acid was also explored in 98% RH, at room temperature and at 45 °C. PXRD analysis revealed the complete conversion of the metal oxide into a metal oxalate structure for all investigated oxides, specifically MgO (Fig. 6a and b), MnO (Fig. 6c–g), NiO and CoO (Fig. 6h–j), CuO (Fig. 6k and l) and PbO (Fig. 6n–p). Table 1 lists the times required for the reflections of the metal oxide to disappear from the PXRD pattern of the reaction mixture (Fig. 6, ESI Fig. S2–S7†), demonstrating that aging reactions are applicable to a variety of metal oxides. | ||
| Fig. 6 PXRD patterns for selected reactions at 98% RH: (a) MgO; (b) Mgox·2H2O (5 days, 45 °C); (c) MnO; (d) Mnox·2H2O (pre-milled MnO, 1 day, room temperature); (e) Mnox·2H2O (9 days, 45 °C); (f) simulated α-Mnox·2H2O (FOZHUX11); (g) simulated γ-Mnox·2H2O (FOZHUX10); (h) NiO; (i) Niox·2H2O (3 days, 45 °C); (j) Coox·2H2O (6 days, room temperature, data were collected using CuKα radiation and energy-discriminating LynxEye detector); (k) CuO; (l) Cuox·0.5H2O (16 days, 45 °C; (m) Cuox·0.5H2O (pre-milled CuO, 10 days, room temperature); (n) PbO; (o) Pbox (pre-milled PbO, 9 days, 45 °C); (p) simulated Pbox (JAHVUJ). (q) Visual comparison of samples of oxide reactants with corresponding oxalate products obtained by aging (98% RH, 45 °C). | ||
| Oxide (melting point/°C) | Reaction time (days) | ||
|---|---|---|---|
| RTb | 45 °C | RTb with pre-millingc | |
| a Reactions were conducted using 2 mmol of the oxide and 2 mmol of H2ox·2H2O. b Room temperature. c Milling was conducted for 5 minutes in a 10 ml stainless steel jar. d PXRD pattern displayed no reflections of H2ox·2H2O or the metal oxide. e Very weak reflections of H2ox·2H2O were observable in the PXRD pattern. f An identical result was obtained at 45 ° and 98% RH after 1 day. | |||
| MgO (2852) | >9 | 9d | 5d |
| MnO (1945) | >16 | 9d | 1d |
| NiO (1955) | >25 | 3d | >10 |
| CuO (1201) | >25 | 16 | 10e,f |
| ZnO (1975) | >5 | 3d | 5d |
| PbO (888) | >30 | 9d | >10 |
The reactions could also be observed by FTIR-ATR spectroscopy (ESI Fig. S8–S12†). The results in Table 1 demonstrate the ability to transform metal oxides under mild conditions of temperature, despite their very high melting points. This is relevant in comparison to mechanochemistry, as analogous milling transformations of CoO, NiO, MnO or PbO into metal–organic derivatives have not yet been reported.8 The composition of the products was elucidated by the similarity of the PXRD patterns to those simulated for published structures (Fig. 6, ESI Fig. S2–S7†).
Product composition, indicating complete conversion, was corroborated by TGA (ESI Fig. S15–S28†). Notably, NiO demonstrated a similar level of reactivity as ZnO, with complete transformation into Niox·2H2O39 (isostructural to monoclinic Znox·2H2O) taking place within three days at 98% RH and 45 °C. The structural resemblance of Niox·2H2O to the zinc-based polymer was also evident from the similarity of the FTIR-ATR spectra (Fig. 2). Similar to ZnO, the synthesis of Niox·2H2O was also readily scaled to 10 grams (Fig. 1l). The slowest reaction was with CuO, for which the oxide was no longer observable by PXRD after 16 days aging at 98% RH and 45 °C. The product was partially hydrated copper(II) oxalate23 with traces of H2ox·2H2O evident in the PXRD pattern, explained by the technical (96%) purity of CuO. The product was analyzed as Cuox·0.5H2O, consistent with previous investigations.40 Like the oxides listed in Table 1, CoO also readily (in six days) converted into a pink material isostructural to Niox·2H2O and Znox·2H2O.
The diffraction pattern of the product exhibited X-ray fluorescence which impaired the detection of trace CoO or H2ox·2H2O using CuKα radiation. TGA was consistent with the formula Coox·2H2O,41 and the FTIR-ATR spectrum was almost identical to those of isostructural Ni(II) and Zn oxalates (Fig. 2). Subsequent PXRD studies using CoKα and CuKα radiation after energy discrimination by the LynxEye® detector (Fig. 6j) confirmed the absence of reactants and formation of Coox·2H2O. Although all transformations in Table 1 are conducted under 98% relative humidity, the obtained products are not the highest known hydrates of corresponding metal oxalates. MnO yielded Mnox·2H2O although a higher hydrate42 is also known. Similarly, PbO yields the anhydrous Pbox (CSD JAHVUJ, JAHVUJ01, Fig. 6n–p)43 although a dihydrate is also known.44 Copper produced the well-known partially hydrated structure, despite the existence of a trihydrate.45 For Pb(II) and Cu(II) the low content of water in products was also evidenced by FTIR-ATR spectra lacking the broad water absorption band at 3400 cm−1 (Fig. 2, also ESI Fig. S10 and S12†). These observations differentiate the herein reported reactions from previously studied cases of solid–gas reactivity46 where the reacting vapor becomes a structural component of the metal–organic product.
Formation of open structures based on Co and Ni
Structure templating by organoammonium cations to form MOFs was also applicable to aging reactions of CoO and NiO. Aging of either NiO or CoO (Fig. 7a–j) with oxalic acid dihydrate and [pn][ox] and [pa]2[ox] yielded materials that were, as established by PXRD, isostructural to the hydrated 2-D and 3-D MOFs [pn][Zn2(ox)3] and [pa]2[Zn2(ox)3], respectively. Framework formation was also corroborated by FTIR-ATR spectroscopy which demonstrated a high degree of mutual similarity in the set of three (Zn-, Co- and Ni-based) materials obtained in the presence of [pn][ox] (ESI Fig. S13†), and also in the set of three analogous materials obtained with [pa]2[ox] template. TGA was consistent with compositions [pn][Co2(ox)3]·3H2O, [pn][Ni2(ox)3]·3H2O and [pn][Zn2(ox)3]·3H2O. For pa+-templated systems, the analyses indicated formulas [pa]2[Co2(ox)3]·4H2O, [pa]2[Ni2(ox)3]·4H2O and [pa]2[Zn2(ox)3]·3H2O. It is not clear whether additional water in these systems is a result of impurity or a real difference in stoichiometric composition with respect to zinc MOFs. Formation of MOFs was also investigated by solid-state UV/vis reflectance spectroscopy (Fig. 8). Reflectance measurements showed a notable difference between the absorption spectra of either Coox·2H2O or Niox·2H2O and products of templated reactions. In contrast, the reflectance spectra of pa+- and pn2+-templated materials were similar to each other. Such observations are consistent with the difference in the coordination environment of the metal in the 1-D hydrated metal oxalate polymer, where the cation is octahedrally coordinated by two oxalate and two water ligands, and MOFs in which the metal is octahedrally surrounded by three oxalates. | ||
| Fig. 7 Selected PXRD patterns for templated aging synthesis of Ni(II) (green) and Co(II) (red) oxalate MOFs: (a) Niox·2H2O; (b) Coox·2H2O; (c) NiO, H2ox·2H2O and[pn][ox] in the ratio of 2:2:1 after 5 days; (d) Niox·2H2O and [pn][ox] in the ratio of 2:1 after 5 days; (e) CoO, H2ox·2H2O and[pn][ox] in the ratio of 2:2:1 after 5 days; (f) simulated 2-D MOF [pn][Zn2(ox)3]·3H2O (SEYQAO); (g) NiO, H2ox·2H2O and[pa]2[ox] in the ratio of 2:2:1 after 5 days; (h) Niox·2H2O and [pa]2[ox] in the ratio of 2:1 after 5 days; (i) CoO, H2ox·2H2O and[pa]2[ox] in the ratio of 2:2:1 after 5 days; (j) simulated 3-D MOF [pa]2[Zn2(ox)3]·3H2O (SEYQIW). Reactions were done at room temperature, 98% RH. Patterns of cobalt samples were recorded using CuKα radiation and an energy-discriminating detector. Characteristic reflections of Niox·2H2O, Coox·2H2O are marked with *. | ||
 | ||
| Fig. 8 Solid-state reflectance UV-vis spectra of cobalt(II) (top) and nickel(II) (bottom) oxalate dihydrates obtained by aging reactions. | ||
Effect of temperature
For all oxides except MnO, switching from room temperature to 45 °C only increased the reaction rate without changing the product. However, PXRD measurements using CuKα radiation (ESI Fig. S6†) showed that MnO produced the α-polymorph of Mnox·2H2O by aging at room temperature and only the reportedly more stable γ-form at 45 °C. The α-form is structurally similar to the other metal oxalate dihydrates (CSD FOZHUX11). In the γ-form inorganic connectivity between octahedrally coordinated Mn2+ ions is established by μ3-bridging oxalate oxygen atoms (Fig. 9a). The difference in polymorphic composition was also observable using FTIR-ATR (Fig. 10). For example, the O–H stretching bands in the α-form are located in a narrow region between 3300 cm−1 and 3360 cm−1, while the γ-form exhibits two broad maxima at 3090 cm−1 and 3320 cm−1. An earlier solution-based study47 indicated that the α-polymorph is metastable with respect to the γ-form. Therefore, the temperature-dependent change in polymorphic composition of the aging product suggests the formation of a kinetic product at lower temperatures. The formation of the thermodynamically stable form at 45 °C can be explained by higher mobility of molecules at a higher temperature. A subsequent PXRD study of the aged samples using a CoKα X-ray source (Fig. 9b–g), to avoid the X-ray fluorescence of manganese-based samples, indicated the presence of the γ-form also in the room temperature product. As the CuKα-based PXRD and FTIR-ATR measurements were all performed without delay and are mutually consistent, we explain the γ-form detected in the PXRD patterns obtained using CoKα radiation as a result of a spontaneous room-temperature transformation during sample transport and storage. Indeed, the transformation of the α-polymorph to the γ-form in moist air was noted by Huizing et al.47 and traces of the γ-polymorph are visible in the samples of the α-form after 16 days at room temperature and 98% RH. The α → γ transformation is not thermally reversible, since heating of the α-form did not result in a structural transformation, shown by DSC. | ||
| Fig. 9 (a) Crystal structures of α- (top) and γ-Mnox·2H2O (bottom). PXRD patterns were collected using CoKα radiation: (b) MnO; (c) MnO after 5 min milling; (d) reaction of MnO and H2ox·2H2O, room temperature, 98% RH; (e) reaction of MnO and H2ox·2H2O, 45 °C, 98% RH; (f) reaction of pre-milled MnO and H2ox·2H2O, room temperature, 98% RH; (g) reaction of pre-milled MnO and H2ox·2H2O, 45 °C, 98% RH. The full set of PXRD patterns is given in the ESI.† | ||
 | ||
| Fig. 10 Selected FTIR-ATR spectra for the reaction of MnO and H2ox·2H2O. Characteristic bands are labeled for α- (red line) and γ-forms (blue line) of Mnox·2H2O. | ||
Activation by milling: enabling room temperature reactivity
Although the ability to conduct aging reactions in the absence of a solvent and at mildly elevated temperature represents a considerable improvement over solution-based processes, the energy benefit can be reduced for long reaction times. Consequently, we explored a means to further accelerate the reactions by mechanically activating the reaction mixture by brief ball milling. Initial milling (Retsch MM400 mill operating at 30 Hz) of the reaction mixture for 5 minutes enabled most aging reactions to be completed at room temperature, eliminating the need for thermal treatment.48 The exception was the reaction of CuO that displayed traces of H2ox·2H2O even after 10 days. Most notable was the effect of milling on reactions of MnO, which completely converted into Mnox·2H2O in one day (Fig. 9f, g and 10). The product was the metastable α-Mnox·2H2O with a minor amount of the γ-form. Reaction acceleration was also achieved by pre-milling only MnO, and we speculate the success of pre-milling is due to introducing defects into the oxide structure.49 If the milled mixture of MnO and H2ox·2H2O was left to age at 45 °C and 98% RH the product was a mixture of the α- and γ-forms, indicating that pre-milling of reactants facilitates the kinetic formation of the metastable form, whereas increased temperature favors the thermodynamically stable one.Selective transformation of metal oxides in a mixture
Different metal oxides underwent aging reactions with oxalic acid at different rates implies the possibility of solvent-free segregation of metal oxide mixtures. A potential benefit of such selectivity in aging reactions would be the transformation of one of the metal oxides into a low density metal oxalate, allowing for its separation from the mixture using gravity, rather than aggressive dissolution reagents. A competitive aging experiment was conducted with a 1:1 stoichiometric mixture of CuO, the slowest reacting metal oxide in our study, and ZnO, one of the most reactive oxides in our study. The mixture contained only one equivalent of H2ox·2H2O. After five days of aging at 45 °C and 98% RH the initially black reaction mixture (5 grams) turned gray. PXRD analysis indicated that the mixture underwent spontaneous and solvent-free chemical separation of zinc from copper by selective conversion of ZnO into Znox·2H2O (eqn (1)):| CuO(s) + ZnO(s) + H2ox·2H2O(s) → Znox·2H2O(s) + Cuox(s) | (1) |
PXRD revealed the formation of a crystalline material isostructural to Znox·2H2O and the complete disappearance of X-ray reflections of ZnO (Fig. 11a–g). In contrast, the X-ray reflections of CuO were clearly observable and reflections of Cu(II) oxalate did not appear in the pattern, consistent with oxalic acid selectively reacting with ZnO. TGA (ESI Fig. S29†) indicated the complete conversion of ZnO, while the less reliable analysis of water loss upon heating indicated the selective conversion of ZnO over CuO in a stoichiometric ratio of 9:1. The FTIR-ATR spectrum of the reaction mixture was consistent with the formation of Znox·2H2O, but could not confirm the absence of copper(II) oxalate (ESI Fig. S14†). The near absence of copper(II) oxalate in the reaction mixture was confirmed by UV/vis reflectance spectroscopy (Fig. 11h). The spectrum of the aged mixture was almost identical to that of pure Znox·2H2O and different from the spectrum expected for a 1:1 mixture of Cu(II) and Zn oxalates. Thus, UV/vis spectra are consistent with the absence of any significant amounts of copper(II) oxalate in the aged mixture. Presumably, the large difference in densities of Znox·2H2O (density = 2.2 g cm−3) and CuO (density = 6.3 g cm−3) could allow for mechanical separation of copper from zinc by gravity,50 which is not possible for a mixture of raw oxides. Indeed, gravity separation with a “heavy liquid” CH2I2 (density 3.3 g cm−3)50,51 separated the reaction mixture into top and bottom layers that were identified by PXRD as Znox·2H2O and CuO, respectively (Fig. 11c and d). Thus, combining the solvent-free chemical separation of zinc and copper oxides with gravity separation allows the separation of copper and zinc oxides without the need for dissolution50 in aggressive solvents, such as sulfuric acid, or high temperatures. Preliminary results on accelerated aging mixtures of PbO and ZnO indicate similar selectivity (ESI Fig. S30†).
 | ||
| Fig. 11 PXRD patterns for the aging of a 1:1:1 mixture of ZnO, CuO and H2ox·2H2O: (a) after 5 days at 45 °C, 98% RH; (b) bottom layer from gravity separation using CH2I2; (c) top layer obtained by gravity separation using CH2I2; (d) Znox·2H2O; (e) Cuox; (f) ZnO; and (g) CuO. (h) Reflectance UV-vis spectra (top to bottom): average of Cuox and Znox·2H2O; reaction mixture after 5 days at 45 °C, 98% RH; pure Znox·2H2O and pure Cuox. PXRD and UV-vis spectra indicate selective transformation of ZnO into Znox·2H2O, leaving behind CuO. To highlight the selective conversion, selected X-ray reflections of CuO and Znox·2H2O are designated with * and ●, respectively. | ||
Conclusions
This proof-of-principle study demonstrated how mimicking the conditions responsible for mineral weathering can lead to solvent-free, low-energy approaches to making coordination polymers and open frameworks from mineral-like feedstocks, as well as to novel mild methodologies for metal oxide separation, potentially applicable in mineral industry. The transformation of a variety of metal (Mg, Mn, Co, Ni, Cu, Zn, and Pb) oxides was accessible at room temperature in mixtures prepared by gentle mixing of powders, despite high lattice stabilization energies52 and melting points of the oxides (800 °C–2500 °C). Mechanical activation or mild temperature increase accelerated the reactions, enabled the complete transformation of all explored metal oxides, and provided control over the polymorphic composition of the product (exemplified by reactions of MnO). The difference in aging reactivity of metal oxides enabled the solvent-free separation of metals in their oxide form without strong acids or high temperatures. As transition metal oxides are often slightly soluble and highly inert, the ability to conduct their chemical segregation in a solvent-free manner and under mild conditions is particularly surprising.53 After chemical separation, the metals were also mechanically separated by gravity. We believe that solvent-free separation by aging has considerable potential for reducing energy and solvent use in mineral industry.50§
It was demonstrated how the reactions can be directed towards the formation of 2-D or 3-D open MOFs. In our hands, this yielded two known open frameworks based on zinc, and four previously not described frameworks based on Co(II) or Ni(II). Whereas a potential criticism can be directed towards reactivity requiring several days, we note that accelerated aging reactions can take place over times that are comparable to those for conventional solvothermal syntheses, whilst being conducted quantitatively from a metal oxide54 and near room temperature. As demonstrated here for nickel and zinc oxalates, and by a recently reported synthesis of porous zeolitic imidazolate frameworks, accelerated aging can be conducted on a multi-gram scale.
It is worth noting that aging transformations are not unknown in large-scale applications, as illustrated by the Dutch process for manufacturing lead white paint. Mechanistic details of the herein presented aging reactivity are not known. We believe that the mobility of the organic phase plays a significant role, consistent with reports55 on vapor or gas-induced transformations of organic and organometallic solids, such as those by the Atwood56 and Braga groups.57 We are currently pursuing the reactivity of three- and four-valent metals, such as Sc, Ti, and Cr.
Acknowledgements
We acknowledge McGill University, the FRQNT Nouveaux Chercheurs Fund, the Canada Foundation for Innovation Leaders Opportunity Fund, the NSERC Discovery Grant and the FRQNT Centre for Self-Assembled Chemical Structures. Prof. D. S. Bohle is acknowledged for help in obtaining single crystal X-ray diffraction data. Bruker UK is acknowledged for SSNMR measurements. Dr N. Henderson and Mr S. Pharasi, Bruker AXS, are acknowledged for help with X-ray fluorescence. Preliminary results that inspired the research were obtained during the principal investigator's appointment as a Herchel Smith Research Fellow at the University of Cambridge. The support from the Royal Society (Research Grant) and the Herchel Smith Fund during that period is acknowledged. Advice from Prof. A. K. Cheetham, University of Cambridge, is appreciated.Notes and references
- (a) J. C. Crittenden and H. S. White, J. Am. Chem. Soc., 2010, 132, 4503 CrossRef CAS PubMed; (b) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphret, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC; (c) W. J. W. Watson, Green Chem., 2012, 14, 251 RSC; (d) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- (a) S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, CrystEngComm, 2012, 14, 3001 RSC; (b) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed; (c) S. A. K. Robinson, M.-V. L. Mempin, A. J. Cairns and K. T. Holman, J. Am. Chem. Soc., 2011, 133, 1634 CrossRef PubMed; (d) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed; (e) J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312 CrossRef CAS PubMed; (f) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650 CrossRef CAS PubMed; (g) C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858 CrossRef CAS PubMed; (h) C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084 CrossRef CAS PubMed.
- (a) A. Czaja, E. Leung, N. Trukhan and U. Müller, Industrial MOF Synthesis, in Metal-Organic Frameworks: Applications from Catalysis to Gas Storage, ed. D. Farrusseng, Wiley-VCH, 1st edn, 2011 Search PubMed; (b) B. Yilmaz, N. Trukhan and U. Müller, Chin. J. Catal., 2012, 33, 3 CrossRef CAS.
- (a) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed; (b) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed; (c) S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249 CrossRef CAS PubMed; (d) U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastré, J. Mater. Chem., 2006, 16, 626 RSC; (e) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- W.-J. Son, J. Kim, J. Kim and W.-S. Ahn, Chem. Commun., 2008, 6336 RSC.
- (a) S. H. Jhung, J.-H. Lee, P. M. Forster, G. Férey, A. K. Cheetham and J.-S. Chang, Chem.–Eur. J., 2006, 12, 7899 CrossRef PubMed; (b) J.-S. Choi, W.-J. Son, J. Kim and W.-S. Ahn, Microporous Mesoporous Mater., 2008, 116, 727 CrossRef CAS; (c) C.-M. Lu, J. Liu, K. Xiao and A. T. Harris, Chem.–Eng. J., 2010, 156, 465 CrossRef CAS.
- A. M. Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn and J. Gascon, Cryst. Growth Des., 2012, 12, 3489 Search PubMed.
- (a) A. Pichon, A. Lazuen-Garay and S. L. James, CrystEngComm, 2006, 8, 211 RSC; (b) F. Kotaro, A. Lazuen-Garay, J. Hill, E. Sbircea, Z. Pan, M. Xu, D. C. Apperley, S. L. James and K. D. M. Harris, Chem. Commun., 2010, 46, 7572 RSC; (c) S. L. James, P. Collier, I. Parkin, G. Hyatt, D. Braga, L. Maini, W. Jones, C. Bölm, A. Krebs, J. Mack, D. Waddell, W. Shearouse, A. G. Orpen, C. J. Adams, T. Friščić, J. W. Steed and K. D. M. Harris, Chem. Soc. Rev., 2012, 41, 413 RSC; (d) T. Friščić, J. Mater. Chem., 2010, 20, 7599 RSC; (e) D. Braga, S. L. Giaffreda, F. Grepioni, A. Pettersen, L. Maini, M. Curzi and M. Polito, Dalton Trans., 2006, 1249 RSC; (f) H. Sakamoto, R. Matsuda and S. Kitagawa, Dalton Trans., 2012, 41, 3956 RSC.
- I. A. Ibarra, P. A. Bayliss, E. Pérez, S. Yang, A. J. Blake, H. Nowell, D. R. Allan, M. Poliakoff and M. Schröder, Green Chem., 2012, 14, 117 RSC.
- (a) T. Friščić and L. Fábián, CrystEngComm, 2009, 11, 743 RSC; (b) A.-X. Zhu, R.-B. Lin, X.-L. Qi, Y. Liu, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Microporous Mesoporous Mater., 2012, 157, 42 CrossRef CAS.
- W. Zhang, Z. Zhu and C. Y. Cheng, Hydrometallurgy, 2011, 108, 177 CrossRef CAS.
- (a) J.-B. Lin, R.-B. Lin, X.-N. Cheng, J.-P. Zhang and X.-M. Cheng, Chem. Commun., 2011, 47, 9185 RSC; (b) M. Lanchas, D. Vallejo-Sánchez, G. Beobide, O. Castillo, A. T. Aguayo, A. Luque and P. Román, Chem. Commun., 2012, 48, 9930 RSC.
- T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621 Search PubMed.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493 RSC.
- W. Yuan, T. Friščić, D. Apperley and S. L. James, Angew. Chem., Int. Ed., 2010, 49, 3916 CrossRef CAS PubMed.
- (a) E. H. H. Chow, F. C. Strobridge and T. Friščić, Chem. Commun., 2010, 46, 6368 RSC; (b) T. Friščić, I. Halasz, F. C. Strobridge, R. E. Dinnebier, R. S. Stein, L. Fábián and C. Curfs, CrystEngComm, 2011, 13, 3125 RSC.
- P. Wang, G. Li, Y. Chen, S. Chen, S. L. James and W. Yuan, CrystEngComm, 2012, 14, 1994 RSC.
- V. M. André, A. Hardeman, I. Halasz, R. S. Stein, G. J. Jackson, D. G. Reid, M. J. Duer, C. Curfs, M. T. Duarte and T. Friščić, Angew. Chem., Int. Ed., 2011, 50, 7858 CrossRef PubMed.
- C. J. Adams, M. A. Kurawa, M. Lusi and A. G. Orpen, CrystEngComm, 2008, 10, 1790 RSC.
- Mechanochemical transformation of CuO into copper(II) acetate can be achieved only by extended milling with traces of water: F. C. Strobridge, N. Judaš and T. Friščić, CrystEngComm, 2010, 12, 2409 RSC.
- (a) G. M. Gadd, Y. J. Rhee, K. Stephenson and Z. Wei, Environ. Microbiol. Rep., 2012, 4, 270 CrossRef CAS PubMed; (b) P. Adamo and P. Violante, Appl. Clay Sci., 2000, 16, 229 CrossRef CAS; (c) G. M. Gadd, Microbiology, 2010, 156, 609 CrossRef CAS PubMed; (d) J. A. Sayer, M. Kierans and G. M. Gadd, FEMS Microbiol. Lett., 1997, 154, 29 CrossRef CAS PubMed; (e) M. Fomina, S. Hillier, J. M. Charnock, K. Melville, I. J. Alexander and G. M. Gadd, Appl. Environ. Microbiol., 2005, 71, 371 CrossRef CAS PubMed; (f) W. W. Barker and J. F. Banfield, Chem. Geol., 1996, 132, 55 CrossRef CAS; (g) Z. Wei, S. Hillier and G. M. Gadd, Environ. Microbiol., 2012, 14, 1744 CrossRef CAS PubMed; (h) J. A. Sayer and G. M. Gadd, Mycol. Res., 1997, 101, 653 CrossRef CAS; (i) Z. Wei, M. Kierans and G. M. Gadd, Geomicrobiol. J., 2012, 29, 323 CrossRef.
- (a) O. W. Purvis, Lichenologist, 1984, 16, 197 CrossRef CAS; (b) J. Chen, H.-P. Blume and L. Beyer, Catena, 2000, 39, 121 CrossRef CAS; (c) G. Sarret, A. Maceau, D. Cuny, C. van Haluwyn, S. Déruelle, J.-L. Hazemann, Y. Soldo, L. Eybert-Bérard and J.-J. Methonnex, Environ. Sci. Technol., 1998, 32, 3325 CrossRef CAS.
- O. W. Purvis and C. Halls, Lichenologist, 1996, 28, 571 CrossRef.
- R. M. Clarke and I. R. Williams, Mineral. Mag., 1986, 50, 295 CAS.
- (a) T. Echigo and M. Kimata, Can. Mineral., 2010, 48, 1329 CrossRef CAS; (b) U. M. Cowgill, Mineral. Mag., 1989, 53, 505 CAS; (c) M. M. Smits, A. M. Herrmann, M. Duane, O. W. Duckworth, S. Bonneville, L. G. Benning and U. Lundström, Fung. Biol. Rev., 2009, 23, 122 CrossRef; (d) G. M. Gadd and J. A. Raven, Geomicrobiol. J., 2010, 27, 491 CrossRef CAS; (e) E. J. Baran and P. V. Monje, Met. Ions Life Sci., 2008, 4, 219 CAS.
- (a) S. R. Byrn, W. Xu and A. W. Newman, Adv. Drug Delivery. Rev., 2001, 48, 115 CrossRef CAS; (b) X. Chen, J. G. Stowell, K. R. Morris and S. R. Byrn, J. Pharm. Biomed. Anal., 2010, 51, 866 CrossRef CAS PubMed.
- (a) M. J. C. Cliffe, C. Mottillo, R. S. Stein, D.-K. Bučar and T. Friščić, Chem. Sci., 2012, 3, 2495 RSC; (b) C. Mottillo, Y. Lu, M.-H. Pham, T.-O. Do and T. Friščić, Green Chem., 2013, 15, 2121 RSC.
- (a) M. Sadakiyo, H. Ōkawa, A. Shigematsu, M. Ohba, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 5472 CrossRef CAS PubMed; (b) J.-C. Tan, J. D. Furman and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 14252 CrossRef CAS PubMed; (c) E. Coronado, C. Martí-Gastaldo, J. R. Galán-Mascarós and M. Cavallini, J. Am. Chem. Soc., 2010, 132, 5456 CrossRef CAS PubMed; (d) M. Clemente-León, E. Coronado, C. Martí-Gastaldo and F. M. Romero, Chem. Soc. Rev., 2011, 40, 473 RSC; (e) J. Ling, C. M. Wallace, J. E. S. Szymanowski and P. C. Burns, Angew. Chem., Int. Ed., 2010, 49, 7271 CrossRef CAS PubMed; (f) J. Ling, J. Qiu and P. C. Burns, Inorg. Chem., 2012, 51, 2403 CrossRef CAS PubMed; (g) E. Cariati, R. Macchi, D. Roberto, R. Ugo, S. Galli, N. Casati, P. Macchi, A. Sironi, L. Bogani, A. Caneschi and D. Gatteschi, J. Am. Chem. Soc., 2007, 129, 9410 CrossRef CAS PubMed; (h) P. A. Prasad, S. Neeraj, S. Natarjan and C. N. R. Rao, Chem. Commun., 2000, 1251 RSC; (i) E. Gavilan, N. Audebrand and E. Jeanneau, Solid State Sci., 2007, 9, 985 CrossRef CAS; (j) E. Jeanneau, N. Audebrand and D. Louër, Chem. Mater., 2002, 14, 1187 CrossRef CAS.
- (a) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS PubMed; (b) A. K. Cheetham and C. N. R. Rao, Mater. Res. Bull., 2005, 30, 93 CrossRef CAS; (c) Q. Pan, Q. Chen, W.-C. Song, T.-L. Hu and X.-H. Bu, CrystEngComm, 2010, 12, 4198 RSC; (d) G. De Munno, M. Julve, F. Nicolo, F. Lloret, J. Faus, R. Ruiz and E. Sinn, Angew. Chem., Int. Ed., 1993, 32, 613 CrossRef; (e) G. De Munno, R. Ruiz, F. Lloret, J. Faus, R. Sessoli and M. Julve, Inorg. Chem., 1995, 34, 408 CrossRef CAS; (f) U. Baisch and D. Braga, CrystEngComm, 2009, 11, 40 RSC; (g) T. Bataille, J. P. Auffrédic and D. Louër, Chem. Mater., 1999, 11, 1559 CrossRef CAS.
- (a) D. Yu. Naumov, N. V. Podberezskaya, E. V. Boldyreva and A. V. Virovets, J. Struct. Chem., 1996, 37, 480 CrossRef; (b) R. Deyrieux and A. Peneloux, Bull. Chim. Soc. Fr., 1969, 2675 CAS.
- S. Descurtin, R. Pellaux, G. Antorrena and F. Palacio, Coord. Chem. Rev., 1999, 190–192, 841 Search PubMed.
- (a) S. Decurtins, H. W. Schmalle, P. Schneuwly, J. Ensling and P. Gütlich, J. Am. Chem. Soc., 1994, 116, 9521 CrossRef CAS; (b) S. Decurtins, H. W. Schmalle, P. Schneuwly and H. R. Oswald, Inorg. Chem., 1993, 32, 1888 CrossRef CAS; (c) F. Luo, Y.-X. Che and J.-M. Zheng, J. Mol. Struct., 2007, 827, 206 CrossRef CAS; (d) O. R. Evans and W. Lin, Cryst. Growth Des., 2001, 1, 9 CrossRef CAS.
- (a) S. Ayyapan, A. K. Cheetham, S. Natarajan and C. N. R. Rao, Chem. Mater., 1998, 10, 3746 CrossRef; (b) S. Natarajan, R. Vaidhyanathan, C. N. R. Rao, S. Ayyapan and A. K. Cheetham, Chem. Mater., 1999, 11, 1633 CrossRef CAS.
- (a) M. Dan and C. N. R. Rao, Angew. Chem., Int. Ed., 2006, 45, 281 CrossRef CAS PubMed; (b) A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780 RSC.
- G. Giester, Z. Kristallogr., 1997, 212, 720 CAS.
- R. Vaidhyanathan, S. Natarajan and C. N. R. Rao, J. Chem. Soc., Dalton Trans., 2001, 699 RSC.
- R. Vaidhyanathan, S. Natarajan, A. K. Cheetham and C. N. R. Rao, Chem. Mater., 1999, 11, 3636 CrossRef CAS.
- (a) R. Vaidhyanathan, S. Natarajam and C. N. R. Rao, J. Mol. Struct., 2002, 608, 123 CrossRef CAS; (b) J. C. Barnes, R. W. Longhurst and T. J. R. Weakley, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1347 Search PubMed.
- R. Deyrieux, C. Berro and A. Peneloux, Bull. Soc. Chim. Fr., 1973, 25 CAS.
- (a) A. Michalowicz, J. J. Girered and J. Goulon, Inorg. Chem., 1979, 18, 3004 CrossRef CAS; (b) H. Fichtner-Schmittler, Cryst. Res. Technol., 1984, 19, 1225 CrossRef CAS.
- (a) J. Bacsa, D. Eve and K. R. Dunbar, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, m58 Search PubMed; (b) E. Wisgerhof and J. W. Geus, Mater. Res. Bull., 1983, 18, 993 CrossRef CAS.
- (a) W.-Y. Wu, Y. Song, Y.-Z. Li and X.-Z. You, Inorg. Chem. Commun., 2005, 8, 732 CrossRef CAS; (b) X. Fu, C. Wang and M. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1348 CAS.
- A. N. Christensen, D. E. Cox and M. S. Lehmann, Acta Chem. Scand., 1989, 43, 19 CrossRef.
- A. V. Virovets, D. Yu. Naumov, E. V. Boldyreva and N. V. Podberezskaya, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 1882 CrossRef.
- W.-Y. Wu and L.-X. Zhai, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m429 CAS.
- (a) S. Libri, M. Mahler, G. M. Espallargas, D. C. N. G. Singh, J. Soleimannejad, H. Adams, M. D. Burgard, N. P. Rath, M. Brunelli and L. Brammer, Angew. Chem., Int. Ed., 2008, 47, 1693 CrossRef CAS PubMed; (b) G. M. Espallargas, M. Hippler, A. J. Florence, P. Fernandes, J. van de Streek, M. Brunelli, W. I. F. David, K. Shankland and L. Brammer, J. Am. Chem. Soc., 2007, 129, 15606 CrossRef PubMed.
- A. Huizing, H. A. M. van Hal, W. Kwestroo, C. Langereis and P. C. van Loosdregt, Mater. Res. Bull., 1977, 12, 605 CrossRef CAS.
- Consequently, the energy cost for such a mechanically activated reaction is reduced to the short period of operating the mill. At the laboratory scale this amounts to a small total input of 15 kJ per reaction, as measured in our laboratory for a Retsch MM400 mill which operates at 100 W (comparable to a light bulb) for two milling stations.
- (a) V. Šepelák, S. Bégin-Colin and G. Le Caër, Dalton Trans., 2012, 41, 11927 RSC; (b) V. Šepelák, S. M. Becker, I. Bergmann, S. Indris, M. Scheuermann, A. Feldhoff, C. Kübel, M. Bruns, N. Stürzl, A. S. Ulrich, M. Ghafari, H. Hahn, C. P. Grey, K. D. Becker and P. Heitjans, J. Mater. Chem., 2012, 22, 3117 RSC.
- The heavy liquid in gravity separation experiments does not act as a solvent, but serves for physical separation of substances without dissolving them. Whereas the use of CH2I2 heavy liquid is still a standard practice in a number of geology, chemistry and mineralogy laboratories, we note that there is a greener, but more costly, alternative based on aqueous sodium or lithium polytungstate solutions.51 In large-scale applications, heavy liquid separation is replaced by froth flotation in aqueous systems. For an overview of industrial froth flotation methods, see: (a) S. Bulatovic, Handbook of Flotation Reagents, Chemistry, Theory and Practice. Vol. 1 Flotation of Sulfide Ores and Vol. 2 Flotation of gold, PGM and oxide minerals, Elsevier, 2007 Search PubMed.
- (a) M. R. Gregory and K. A. Johnston, New Zeal. J. Geol. Geophys., 1987, 30, 317 CrossRef CAS; (b) G. A. Andreev and M. Hartmanová, Phys. Status Solidi, 1989, 116, 457l CrossRef; (c) T. L. Threlfall, Analyst, 1995, 120, 2435 RSC.
- Lattice energies of herein explored oxides range from 3.5 MJ mol−1 to 4.2 MJ mol−1, see: CRC Handbook of Chemistry and Physics, ed. R. C. Weast and M. J. Astle, CRC Press Inc., Boca Raton, FL, 61st edn, 1981 Search PubMed.
- The herein presented solvent-free metal oxide separation is based on solid-state reactivity which has potential to be generalized, and should not be confused with magnetic procedures unique to certain iron oxides (maghemite and magnetite).
- Reported rapid (minutes to hours) syntheses of MOFs in solution exhibit either poor yields (below 60%) or require an additional stoichiometric base, see: (a) A. F. Gross, E. Sherman and J. J. Vajo, Dalton Trans., 2012, 41, 5458 RSC; (b) D. J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553 CrossRef CAS.
- (a) D. Cinčić, I. Brekalo and B. Kaitner, Chem. Commun., 2012, 48, 11683 RSC; (b) D. Cinčić, I. Brekalo and B. Kaitner, Cryst. Growth Des., 2012, 12, 44 CrossRef; (c) M. A. Ziganshin, I. G. Efimova, V. V. Gorbatchuk, S. A. Ziganshina, A. P. Chuklanov, A. A. Bukharaev and D. V. Soldatov, J. Pept. Sci., 2012, 18, 209 CrossRef CAS PubMed; (d) R. Anneda, D. V. Soldatov, I. L. Moudrakovski, M. Casu and J. A. Ripmeester, Chem. Mater., 2008, 20, 2908 CrossRef; (e) G. M. Espallargas, M. Hippler, A. J. Florence, P. Fernandes, J. van de Streek, M. Brunelli, W. I. F. David, K. Shankland and L. Brammer, J. Am. Chem. Soc., 2007, 129, 15606 CrossRef PubMed; (f) B. D. Chandler, G. D. Enright, K. A. Udachin, S. Pawsey, J. A. Ripmeester, D. T. Cramb and G. K. H. Shimizu, Nat. Mater., 2008, 229 CrossRef CAS PubMed.
- (a) J. Tian, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2011, 133, 1399 CrossRef CAS PubMed; (b) J. Tian, P. Thallapally, J. Liu, G. J. Exarhos and J. L. Atwood, Chem. Commun., 2011, 47, 701 RSC; (c) P. K. Thallapally, B. P. McGrail, S. J. Dalgarno, H. T. Schaef, J. Tian and J. L. Atwood, Nat. Mater., 2008, 7, 146 CrossRef CAS PubMed.
- (a) D. Braga, S. L. Giaffreda, F. Grepioni, M. R. Chierotti, R. Gobetto, G. Palladino and M. Polito, CrystEngComm, 2007, 9, 879 RSC; (b) L. Maini, D. Braga, P. P. Mazzeo and B. Ventura, Dalton Trans., 2012, 41, 531 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic information for (pa)2(ox) in CIF format. Additional PXRD, FTIR-ATR, SSNMR and thermogravimetric data. CCDC 933546. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3gc41370e |
| ‡ TGA of [pn][Zn2(ox)3]·3H2O prepared by templated aging gave an excellent fit for the expected loss of three equivalents of water before 200 °C (measured weight loss = 10.1%, expected weight loss = 10.3%), followed by the decomposition of the metal–organic residue into ZnO (measured residue weight = 31.3%, calculated ZnO residue for [pn][Zn2(ox)3]·3H2O = 31.0%). Such a thermal behavior is consistent with that previously reported for solvothermally grown [pn][Zn2(ox)3]·3H2O.36 An excellent fit was also observed for TGA of [pa]2[Zn2(ox)3]·3H2O (measured water content = 9.1%, calculated = 9.5%; measured ZnO residue = 29.4%, calculated = 28.6%). Also see ESI.† |
| § Potential metallurgical and mineral processing applications of concepts and results of the work described in this manuscript are covered by the provisional patent application US 61/826,172. |
| This journal is © The Royal Society of Chemistry 2014 |