Magnetically recoverable fluorescence chemosensor for the adsorption and selective detection of Hg2+ in water†
Qiang
Lv
a,
Gang
Li
*a,
Zhuhong
Cheng
a,
Hong
Lu
b and
Xiaoxia
Gao
c
aState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: liganghg@dlut.edu.cn; Fax: +86-411-8498-6113; Tel: +86-411-8498-6113
bKey Laboratory of Industrial Ecology and Environmental Engineering (China Ministry of Education), School of Environmental Science and Technology, Dalian University of Technology, Dalian 116024, P. R. China
cSchool of Materials Science and Engineering, Dalian University of Technology, Dalian, 116024, P. R. China
First published on 23rd October 2013
Abstract
In order to conveniently and effectively detect the heavy metal ion Hg2+ existing in water, a magnetic fluorescence chemosensor has been strategically prepared by immobilizing a Rhodamine B derivative RhB–tris(2-aminoethyl)amine on Fe3O4@SiO2–Au@PSiO2 composites via gold particles. The adsorption and detection for Hg2+ ions were investigated with fluorophotometry. This chemosensor shows high sensitivity and high selectivity for Hg2+ over other metal cations owing to the ring opening of the rhodamine fluorophore selectively induced by Hg2+. In addition, the presence of Fe3O4 in the sensor also facilitates the magnetic separation of the Fe3O4@SiO2–Au–Hg2+@PSiO2 from the solution.
Environmental impactThe synthesized magnetic fluorescence chemosensor Fe3O4@SiO2–Au–RhB–Tren@PSiO2 possesses high sensitivity and selectivity in the adsorption and detection of Hg2+ ions in water. Importantly, this chemosensor can be separated conveniently from the solution with a magnet and exhibits good reversibility and regenerative ability. Likewise, magnetic chemosensors can be prepared and applied for the removal and detection of other heavy metals. |
1. Introduction
As is known, heavy metal ion Hg2+ is considered to be one of the most hazardous toxins that can cause a high risk to human health and the environment.1 Ionic mercury can bioaccumulate through skin, respiratory, and gastrointestinal tissues, and this will seriously damage the central nervous and endocrine systems.2–5 Therefore, the detection of Hg2+ in the environment at low concentration using a simple and efficient method is high desirable for us. Fluorescence spectrometry, as a powerful optical technique for many analytical applications, has received much interest in detecting Hg2+.5–15 Although a number of fluorescence-based Hg2+ probes have been reported previously,16–19 cost-effective production, reversibility and regeneration of the materials, are still our desired goals. So, with the development of nanotechnology, immobilizing probes on nanomaterials to detect heavy metal ions has attracted much attention recently.20–22 Silica-based mesoporous materials, such as SBA-15, MCM-41 and HMS, were reported as supports of fluorescence probes.23–30During the past decades, there have been some reports on magnetic silica nanoparticles functionalized with organic groups for the detection and separation of ions from water, which has become an interesting subject.31–38 However, most of these nanoparticles did not contain enough pores to accommodate more organic groups, and the functionalization process by silane agents may destroy the nanoparticles. Recently, noble metal nanoparticles (for example Au or Ag nanoparticles) modified with organic groups, forming Au–S, Au–N bonds,39,40 have received great attention for colorimetric sensing.41 But notable challenges still exist for these systems, such as their poor stability, inconvenient separation or regeneration. In order to overcome these deficiencies, a new chemosensor Au–HMS-probe with a worm-like mesoporous framework for selective detection of Hg2+ was prepared recently in our group by immobilizing a Rhodamine B derivative on Au–HMS via Au–N groups instead of silane agents.18 The material affords “turn-on” fluorescence enhancement and shows an excellent selectivity for Hg2+.
However, the Au particles located on the outer surface of the material may leach and aggregate in reactions. Moreover, the amount of Au particles loaded on the surface of silica as linkers between the support and probes is difficult to control. To solve these questions and further render the obtained material easier to separate, herein, a magnetically recoverable fluorescent chemosensor coated with mesoporous silica shell for the adsorption and detection of Hg2+ in water was prepared by immobilizing a Rhodamine B derivative on the Fe3O4@SiO2–Au@PSiO2via preformed gold particles, which provide active sites for the subsequent functionalization and the amount can be conveniently tuned (Fig. S1†). The preparation process of this material is illustrated in detail in Scheme 1. The fluorescent nanosized composites can be conveniently recovered because of the magnetic Fe3O4 cores, whilst the porous shell can adsorb ions in water and minimize the aggregation or leaching of Au particles.
 | ||
| Scheme 1 The fabrication process of a magnetic fluorescent chemosensor with a multilayered structure. | ||
2. Experimental
2.1 Chemicals
Rhodamine B (RhB), tris(2-aminoethyl)amine (Tren), 3-aminopropyl-triethoxysilane (APTS) were purchased from J&K. Tetraethylorthosilicate (TEOS), ethanol, methanol, 2-propanol, dichloromethane, sodium acetate, hydrous ferric chloride (FeCl3·6H2O), chloroauric acid (HAuCl4·4H2O), anhydrous sodium sulfate (Na2SO4), mercuric nitrate (Hg(NO3)2), ammonium hydroxide aqueous solution (28 wt%), sodium hydroxide, ethylene glycol and trisodium citrate were purchased from Shanghai Chemical Corp. Polyvinylpyrrolidone (PVP) K15 (Mw ∼ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and K30 (Mw ∼ 50000) were purchased from Aladdin. All reagents and solvents were used without further purification unless otherwise noted.
000) and K30 (Mw ∼ 50000) were purchased from Aladdin. All reagents and solvents were used without further purification unless otherwise noted.
2.2 Synthesis of APTS-modified Fe3O4@SiO2 microspheres
Fe3O4 particles were synthesized using a high-temperature solvothermal reaction reported previously.42 Then, the Fe3O4 particles were coated with a layer of silica through a modified Stöber process.43 Typically, the magnetite (0.10 g) synthesized above was mixed with a mixture of ethanol (120 mL) and aqueous ammonia (28 wt%, 7 mL) by vigorous mechanical stirring. TEOS (0.5 mL) was injected into the solution under continuous stirring. After magnetic separation with a magnet and washing with ethanol and water for three times, Fe3O4@SiO2 particles were obtained. The material was further modified using APTS to functionalize the silica surface with amino groups, which could interact with gold nanoparticles by coordination bonds. The Fe3O4@SiO2 particles were transferred to a mixture of isopropanol (100 mL) and APTS (25 μL) and kept at 80 °C for 2 h. Finally, with the help of a magnet the surface modified particles were washed with isopropanol and deionized water several times and re-dispersed in deionized water for further use.2.3 Preparation of Fe3O4@SiO2–Au@PSiO2
Firstly, gold nanoparticles, about 15 nm, were prepared according to Frens's method.44 The APTS-modified Fe3O4@SiO2 dispersed in deionized water (100 mL) was mixed with an aqueous solution of Au nanoparticles (150 mL) under sonication for 10 min. The Fe3O4@SiO2–Au particles were magnetically collected, re-dispersed in deionized water (40 mL), and then mixed with a PVP K30 aqueous solution (25 mL, 0.02 g mL−1). The mixture was further sonicated for 20 min to allow adequate polymer adsorption onto the particle surface. These colloids were magnetically separated from the original solution, dispersed in a mixture of ethanol (140 mL), water (20 mL) and aqueous ammonia (8 mL). TEOS (0.6 mL) was added to the system for the growth of the outer silica shell. Finally, the particles were washed with ethanol and dispersed in water (100 mL).The reported procedure of surface-protected etching was used.45 A mixture of above colloidal solution and PVP K15 (1 g) was refluxed under magnetic stirring at 100 °C for 3 h. NaOH solution (25 mL, 0.16 g mL−1) was injected to etch for 10 min at room temperature. The resulting composites defined as Fe3O4@SiO2–Au@PSiO2 (P means porous) with a porous silica shell were magnetically separated from the basic solution, washed with deionized water several times, and finally vacuum dried at 60 °C.
2.4 Synthesis of fluorescent molecular probe RhB–Tren
Under nitrogen, a solution of Rhodamine B (1 g) and tris(2-aminoethyl)amine (5 mL) in methanol was kept at reflux until the colour of solution turned to yellow from pink. After cooling to room temperature, the solvent was evaporated in vacuo. CH2Cl2 (100 mL) and water (200 mL) were added into the residue after evaporation, and the organic layer was separated. The CH2Cl2 layer was washed several times with water followed by drying over anhydrous Na2SO4. After removing Na2SO4 by filtrating and evaporating the solvent in a vacuum, a yellow oil was obtained. The obtained molecular probe RhB–Tren was dissolved in acetone (100 mL) for further use.2.5 Synthesis of Fe3O4@SiO2–Au–RhB–Tren@PSiO2
Fe3O4@SiO2–Au@PSiO2 (0.2 g) was mixed with the acetone solution (100 mL) containing RhB–Tren under sonication, then refluxed under magnetic stirring at 50 °C for 3 h. The as-obtained product was collected with a magnet, washed repeatedly with acetone and deionized water and vacuum dried at 80 °C. Finally, the magnetic fluorescent chemosensor Fe3O4@SiO2–Au–RhB–Tren@PSiO2 was obtained.2.6 Characterization of the materials
Transmission electron microscopy (TEM) images were taken on a FEI Company Tecnai G2 20 Stwin instrument with an acceleration voltage of 200 kV. X-ray diffraction (XRD) patterns were recorded on a Rigaku D/Max 2400 diffractometer using Cu Kα radiation run at 40 kV and 100 mA. UV-vis spectra were measured on a Jasco UV-550 spectrophotometer. FT-IR spectra were recorded on a Bruker EQUINOX55 spectrometer using a KBr pellet technique. The magnetic properties were measured with a Magnetic Properties Measurement System (MPMS XL-7) from Quantum Design. Fluorescence spectra of the solution were obtained using a FTI-700 spectrometer. Both excitation and emission slit widths were 5 nm, and the excitation wavelength was 520 nm. The detection process was performed in homogeneous aqueous media of pH 7.1 (1 mM HEPES buffer).2.7 Fluorescent detection
A stock solution (1.25 × 10−3 M) of the aqueous mercury nitrate (Hg(NO3)2) and a suspension of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 (0.1 g L−1) were prepared. For each run, a 2 mL Fe3O4@SiO2–Au–RhB–Tren@PSiO2 suspension was added into a quartz cuvette with a 1 cm optical path length, followed by a mercury nitrate stock solution injected into the quartz cell gradually using a micro-syringe. The volume of the added stock solution was less than 100 μL in order to keep the total volume of test solution roughly the same. A HITACHI F-7000 spectrometer was used to monitor the evolution of fluorescence intensity upon the addition of Hg2+. The excitation and emission slit widths were 5 nm, and the excitation wavelength was 520 nm. The addition of a TPAOH aqueous solution (2.5 × 10−4 M) to the Fe3O4@SiO2–Au–RhB–Tren–Hg2+@PSiO2 suspension caused an immediate fluorescence intensity decrease. Subsequently, with the addition of Hg2+, the Fe3O4@SiO2–Au–RhB-Tren@PSiO2 suspension gave a fluorescence increase.3. Results and discussion
Carboxyl group functionalized magnetic nanoparticles were prepared via a high temperature reaction with ethylene glycol in the presence of trisodium citrate. The characteristic peaks appearing in Fig. 1a confirm the successful fabrication of Fe3O4.46 The bands at 1652 and 1396 cm−1 in Fig. 2a are attributed to the carboxylate group, indicating that this sample has excellent dispersibility in polar solvents such as water and ethanol, which favors the subsequent coating or modification. In addition, due to relatively large particle sizes, the Fe3O4 cores interact strongly with external magnetic fields and can be easily separated from solution after reactions.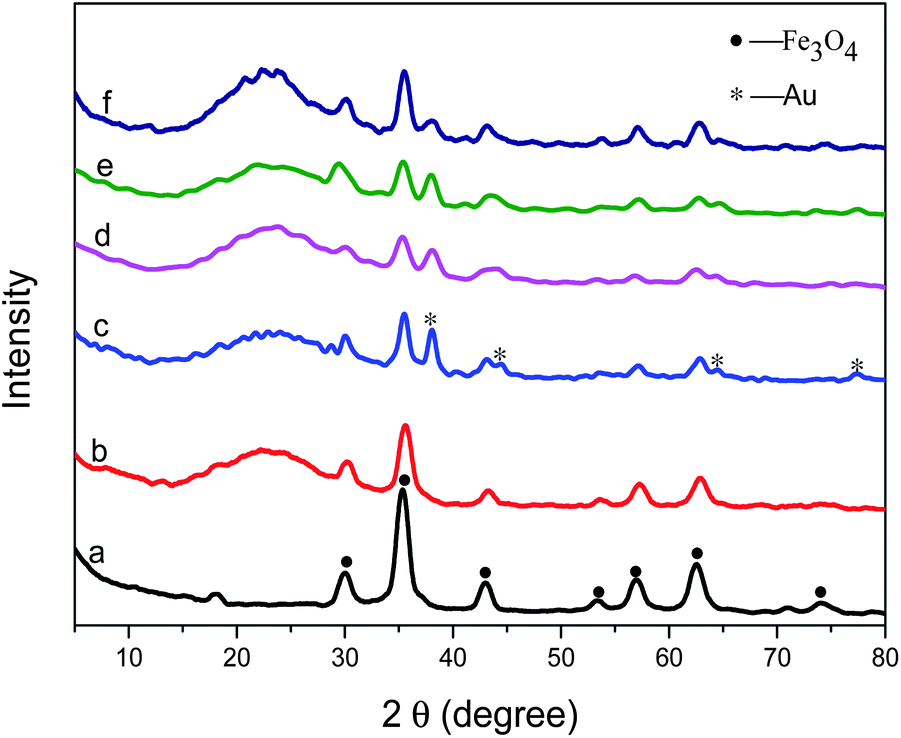 | ||
| Fig. 1 Wide-angle XRD patterns of (a) Fe3O4 particles, (b) Fe3O4@SiO2 microspheres, (c) Fe3O4@SiO2–NH2–Au microspheres, (d) Fe3O4@SiO2–Au@SiO2, (e) Fe3O4@SiO2–Au@PSiO2, (f) Fe3O4@SiO2–Au–RhB–Tren@PSiO2. | ||
 | ||
| Fig. 2 Fourier transform infrared (FT-IR) spectra of (a) Fe3O4, (b) Fe3O4@SiO2, (c) Fe3O4@SiO2–NH2, (d) Fe3O4@SiO2–Au@PSiO2, (e) Fe3O4@SiO2–Au–RhB–Tren@PSiO2. | ||
In order to protect the surface of Fe3O4, the particles were coated with a silica layer through a sol–gel process by the hydrolysis of tetraethylorthosilicate (TEOS) and condensation in an ethanol–water–ammonia mixture. As shown in Fig. 1b, Fe3O4 particles coated with SiO2 present the characteristic peaks of Fe3O4 as well as SiO2 (around 20°). Meanwhile, the characteristic bands of carboxyl group become weak clearly in Fig. 2b compared with that in Fig. 2a. TEM images (Fig. 3a and b) show that a layer of silica, about 150 nm, coats the outside of the Fe3O4 particles.
 | ||
| Fig. 3 TEM images of (a and b) Fe3O4@SiO2 particles, (c and d) Fe3O4@SiO2–NH2–Au microspheres, (e and f) Fe3O4@SiO2–Au@SiO2 and (g and h) Fe3O4@SiO2–Au@PSiO2 composites. | ||
After being modified using APTS, the microspheres present new peaks at 2920 and 2852 cm−1 in the FT-IR spectrum (Fig. 2c), which correspond to the –CH2– group of aminopropyl. The Fe3O4@SiO2 particles are successfully functionalized by APTS. The citrate-stabilized gold nanoparticles prepared using the method in Section 2.4 show a plasmon resonance band at 518 nm in the UV-vis spectrum (Fig. 4). The as-obtained gold nanoparticles can be loaded onto the surface of Fe3O4@SiO2–NH2 through the strong chemical affinity between Au and primary amines.47 TEM images (Fig. 3c and d) show that gold particles, about 15 nm, are uniformly deposited on the surface of Fe3O4@SiO2 microspheres. In addition, the amount of Au particles on the surface of the Fe3O4@SiO2 microspheres can be conveniently controlled by varying the concentrations of APTS during the modification.
 | ||
| Fig. 4 UV-vis spectrum of Au particles dispersed in an aqueous solution. | ||
With the forming of Fe3O4@SiO2–Au particles, characteristic diffraction peaks attributed to Au particles clearly appear, as shown in Fig. 1c. After that, Fe3O4@SiO2–Au composites were modified with PVP, and further encapsulated into another layer of silica through a sol–gel process. The broad peak of SiO2 in the wide-angle XRD pattern (Fig. 1d) became stronger. The TEM images (Fig. 3e and f) show that a layer of compact silica, about 50 nm, is coated on the outside of the gold particles. And then, with the protection of PVP, a sodium hydroxide solution was added to etch the outer silica layer to form a porous structure. The TEM images (Fig. 3g and h) clearly indicate a porous structure in the second silica layer after treatment with NaOH solution. At the same time, Au particles are preserved after the etching and Fe3O4@SiO2–Au@PSiO2 microspheres are obtained. The resultant Fe3O4@SiO2–Au@PSiO2 composites with porous shells show characteristic diffraction peaks indexed to Fe3O4, SiO2 and Au (Fig. 1e), which is in agreement with the observations from the TEM images. The outer porous silica layer allows the probe molecules or reactant to contact the inner Au particles, and at the same time it can prevent the leaching or aggregating of Au particles. That is, the outer porous silica layer can protect the inner Au particles and the probe on Au.
Finally, the Rhodamine B derivative RhB–Tren was synthesized and was further immobilized on Fe3O4@SiO2–Au@PSiO2via Au–N groups.48 The Fourier transform infrared (FT-IR) spectrum in Fig. 2e exhibited two absorption bands at 1510 and 1420 cm−1, which could be attributed to the stretching mode of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– and –C–N–, respectively. This confirmed the fluorescence probe molecules RhB–Tren were successfully attached on the surface of the Au particles. The as-obtained sample was denoted as Fe3O4@SiO2–Au–RhB–Tren@PSiO2 and further applied as a fluorescence chemosensor for the detection and adsorption of Hg2+ in water.
C– and –C–N–, respectively. This confirmed the fluorescence probe molecules RhB–Tren were successfully attached on the surface of the Au particles. The as-obtained sample was denoted as Fe3O4@SiO2–Au–RhB–Tren@PSiO2 and further applied as a fluorescence chemosensor for the detection and adsorption of Hg2+ in water.
In order to evaluate the magnetic response of the composites to an externally applied magnetic field, the mass magnetization was measured at 300 K by cycling the field between −15 and 15 kOe. The Fe3O4@SiO2–Au–RhB–Tren@PSiO2 exhibited super-paramagnetic properties similar to Fe3O4 (Fig. 5). Besides, Fe3O4@SiO2–Au–RhB–Tren@PSiO2 particles were easily dispersed in water owing to a suitable amount of hydrophilic hydroxyl groups on the surface of the porous silica. It was convenient to separate the composites from water by applying a magnet (inset of Fig. 5), although the saturation magnetization value of the composites was lower than that of Fe3O4.
 | ||
| Fig. 5 Hysteresis loops of (a) Fe3O4 particles and (b) Fe3O4@SiO2–Au–RhB–Tren@PSiO2 composites. Inset is photo of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 in deionized water responding to an external magnet. | ||
As Scheme 2 shows, in the absence of Hg2+, the aqueous suspension of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 is non-fluorescent. Addition of Hg2+ induces the spirolactam ring of the probe on the surface of the Au particles to open to give an enhanced fluorescence emission at 584 nm.13,49 Moreover, the fluorescent emission of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 can be quenched by treatment using tetrapropylammonium hydroxide (OH−) solution.
 | ||
| Scheme 2 Illustration of Hg2+-induced fluorescent effect and fluorescence quenching by OH−. | ||
Fluorescence spectra were recorded after the gradual addition of Hg2+ into the suspended solution of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 (Fig. 6). The Fe3O4@SiO2–Au–RhB–Tren@PSiO2 suspension showed very weak emission at 584 nm.50,51 Upon the increasing addition of Hg2+, the band at 584 nm appeared and became stronger, which could be ascribed to the delocalized xanthene moiety of the rhodamine group. Compared to RhB–Tren, Fe3O4@SiO2–Au–RhB–Tren@PSiO2, that possesses a polyhydroxy surface from the PSiO2, enhanced the contact of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 with Hg2+ in water and therefore improved the detection of Hg2+.
 | ||
| Fig. 6 Fluorescence spectra of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 in an aqueous HEPES buffer medium upon the increasing concentration of Hg2+ (0 to 7.5 × 10−5 M). | ||
The detection limit of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 for Hg2+ was also tested. Fig. 7 shows a plot of the emission intensity of Fe3O4@SiO2–Au–RhB–Tren@PSiO2versus Hg2+ concentration and a good linear relationship was obtained in the range of 0 to 6 × 10−6 M with a correlation coefficient of 0.998. The detection limit (DL) of Hg2+ using Fe3O4@SiO2–Au–RhB–Tren@PSiO2 was estimated from the linear part of the calibration plot, according to eqn (1), where k = 3, m and Sb stand for the slope of the calibration graph in the linear range and the standard deviation for the blank, respectively.35,52
 | (1) |
 | ||
| Fig. 7 Plot of fluorescence intensities for Fe3O4@SiO2–Au–RhB–Tren@PSiO2 (0.1 g L−1) as a function of Hg2+ concentration (0 to 6 × 10−6 M). | ||
The calculated standard deviation is 11.60% after eight successive measurements to assure the accuracy and precision of the detection method. The resultant DL value for Fe3O4@SiO2–Au–RhB–Tren@PSiO2 could be up to 2 × 10−8 M of Hg2+ in water.
To investigate the response time of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 with Hg2+, the fluorescence intensity of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 at 584 nm was detected at different times after the addition of Hg2+ (Fig. S2†). The intensity immediately started to increase and became stable after 70 seconds, which indicated the response time of this magnetic probe was about 70 seconds.
For biological and environmental applications as a fluorescence sensor, the sensing should be effective over a wide pH range. The effect of pH value on Fe3O4@SiO2–Au–RhB–Tren@PSiO2 in the absence and presence of Hg2+ was evaluated (Fig. S3†). At pH 5.5–10, the fluorescence emission intensity varied slightly in the absence of Hg2+. Upon addition of Hg2+, Fe3O4@SiO2–Au–RhB–Tren@PSiO2 responded stably from pH 5.5 to 7.8, which was the pH range at which the magnetic probe could be employed.
To further evaluate the performance of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 as an ion-selective fluorescence probe for Hg2+, the fluorescence emission changes of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 were investigated in the presence of other biologically and environmentally relevant metal ions such as Ni2+, Cd2+, Ca2+, Ba2+, Pb2+, Cu2+, Zn2+, Co2+, Mg2+, Ag+, Na+, Li+ and K+ (as shown in Fig. 8). As expected, the intensities of Fe3O4@SiO2–Au–RhB–Tren–Hg2+@PSiO2 almost were unaffected by the existence of Ni2+, Cd2+, Ca2+, Ba2+, Pb2+, Cu2+, Zn2+, Co2+, Mg2+, Ag+, Na+, Li+ and K+, indicating its selective detection for Hg2+ in the mixture of metal ions.
 | ||
| Fig. 8 Fluorescence intensity of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 (0.1 g L−1) with Hg2+ (2.25 × 10−6 M) in the presence of other metal ions such as Ni2+, Cd2+, Ca2+, Ba2+, Pb2+, Cu2+, Zn2+, Co2+, Mg2+, Ag+, Na+, K+ and Li+ ions (2.25 × 10−6 M). | ||
Next, we evaluated the effectiveness of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 in the adsorption and separation of Hg2+ from deionized water. As illustrated in Fig. 9, the HMS–Au-probe was selected to serve as a reference sample. Upon the addition of the HMS–Au-probe and Fe3O4@SiO2–Au–RhB–Tren@PSiO2, the Hg2+ aqueous solution turned pink and reddish brown respectively. When the Hg2+ solution containing Fe3O4@SiO2–Au–RhB–Tren@PSiO2 particles was placed in an externally applied magnetic field, Fe3O4@SiO2–Au–RhB–Tren@PSiO2 particles and Hg2+ ions could easily be separated. If adding HMS–Au-probe further to the above solution, the color of the solution did not exhibit pink again, which indicated that Fe3O4@SiO2–Au–RhB–Tren@PSiO2 was able to adsorb the most Hg2+ in water. The results indicated that Fe3O4@SiO2–Au–RhB–Tren@PSiO2 could work as both a chemosensor and an adsorbent for the Hg2+ ion. Thus, Fe3O4@SiO2–Au–RhB–Tren@PSiO2 could be a potentially useful and effective material for the selective separation and rapid removal of Hg2+ from water. The application of detecting Hg2+ in real samples consisting of tap water and rainwater were carried out by using an analogous procedure to that in purified water (Fig. S4†). The results indicated that no significant fluorescence intensity changes were observed in rainwater at the same Hg2+ concentration, while the fluorescence intensity slightly decreased in tap water. Thus, Fe3O4@SiO2–Au–RhB–Tren@PSiO2 has potential applications for the determination of Hg2+ in real water samples.
 | ||
| Fig. 9 The adsorption of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 for Hg2+ in water. HMS–Au-probe is used as a reference sample. | ||
Fig. 10 demonstrates the reversibility and regeneration of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 in the detection of Hg2+, which was of importance for sensing analytes in practical applications. The fluorescence quenching occurs upon the addition of basic TPAOH (tetrapropylammonium hydroxide) solution into the suspension of Fe3O4@SiO2–Au–RhB–Tren–Hg2+@PSiO2. Subsequently, Fe3O4@SiO2–Au–RhB–Tren–Hg2+@PSiO2 showed fluorescence again by adding Hg2+ and this process was reversible for at least four times, which suggested that Fe3O4@SiO2–Au–RhB–Tren@PSiO2 could be reproducible by a simple treatment with a basic solution.
 | ||
| Fig. 10 Fluorescence intensities of Fe3O4@SiO2–Au–RhB–Tren@PSiO2 in water with the alternative addition of Hg2+ and OH−. | ||
4. Conclusions
In conclusion, a magnetic fluorescence chemosensor Fe3O4@SiO2–Au–RhB–Tren@PSiO2 was fabricated and applied in the selective and sensitive detection of Hg2+ in water. The magnetite core in the composites makes it convenient to be recovered from solution for further use. The compact silica layer coated on the magnetite particles can protect the magnetite core and at the same time serve as the support for Au particles. The Au nanoparticles loaded on the surface of Fe3O4@SiO2 graft the RhB probe onto the composite. The outer porous silica layer allows the probe molecules to contact the inner Au particles, and protects the inner Au particles and the probe on Au. Hg2+ in water can pass through the outer pore and be detected selectively. Besides, the OH− groups existing on the surface of the silica layer promote the hydrophilicity of this fluorescence chemosensor, which facilitates the detection of metal ions in water. The chemosensor Fe3O4@SiO2–Au–RhB–Tren@PSiO2 presents high selectivity for Hg2+ over the other competing ions investigated, and successfully adsorbs and separates Hg2+ from water. Importantly, Fe3O4@SiO2–Au–RhB–Tren@PSiO2 exhibits reversibility and regenerative ability by treatment with tetrapropylammonium hydroxide solution. From our point of view, this strategy is suitable for the development of a category of tailor-made biocompatible systems built by immobilization of appropriate fluorogenic receptors on the surface of other novel nanomaterials to study the environmental function of toxic metal contaminants. Furthermore, many similar structured materials can also be prepared and applied in fields such as catalysis.Acknowledgements
The authors acknowledge the financial support from the Program for New Century Excellent Talents in University (NCET-04-0270), National Natural Science Foundation of China (no. 20406005) and Central University basic research fund (DUT10LK11).Notes and references
- S. S. Leonard, J. J. Bower and X. Shi, Mol. Cell. Biochem., 2004, 255, 3–10 CrossRef CAS.
- X. Chen, S.-W. Nam, M. J. Jou, Y. Kim, S.-J. Kim, S. Park and J. Yoon, Org. Lett., 2008, 10, 5235–5238 CrossRef CAS PubMed.
- D. W. Boening, Chemosphere, 2000, 40, 1335–1351 CrossRef CAS.
- J. Mutter, J. Naumann, R. Schneider, H. Walach and B. Haley, Neuroendocrinol. Lett., 2005, 26, 439–446 Search PubMed.
- E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed.
- V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386–7387 CrossRef CAS.
- Y. K. Yang, K. J. Yook and J. Tae, J. Am. Chem. Soc., 2005, 127, 16760–16761 CrossRef CAS PubMed.
- E. M. Nolan and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 5910–5919 CrossRef CAS PubMed.
- L. Zhou, Y. Lin, Z. Huang, J. Ren and X. Qu, Chem. Commun., 2012, 48, 1147–1149 RSC.
- Y. Tao, Y. Lin, Z. Huang, J. Ren and X. Qu, Talanta, 2012, 88, 290–294 CrossRef CAS PubMed.
- X. Peng, Y. Wang, X. Tang and W. Liu, Dyes Pigm., 2011, 91, 26–32 CrossRef CAS PubMed.
- J. H. Jung, J. H. Lee and S. Shinkai, Chem. Soc. Rev., 2011, 40, 4464–4474 RSC.
- M. H. Lee, S. J. Lee, J. H. Jung, H. Lim and J. S. Kim, Tetrahedron, 2007, 63, 12087–12092 CrossRef CAS PubMed.
- M. Park, S. Seo, I. S. Lee and J. H. Jung, Chem. Commun., 2010, 46, 4478–4480 RSC.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2013 DOI:10.1021/cr300508p.
- D. Wu, W. Huang, C. Duan, Z. Lin and Q. Meng, Inorg. Chem., 2007, 46, 1538–1540 CrossRef CAS PubMed.
- M. H. Lee, J.-S. Wu, J. W. Lee, J. H. Jung and J. S. Kim, Org. Lett., 2007, 9, 2501–2504 CrossRef CAS PubMed.
- N. Zhang, G. Li, Z. Cheng and X. Zuo, J. Hazard. Mater., 2012, 229, 404–410 Search PubMed.
- B. Zhu, J. Zhao, H. Yu, L. Yan, Q. Wei and B. Du, Chem. Eng. J., 2013, 219, 411–418 CrossRef CAS PubMed.
- S. JináLee and J. HwaáJung, Analyst, 2010, 135, 2802–2805 RSC.
- H. Son, J. H. Lee, Y.-R. Kim, I. S. Lee, S. Han, X. Liu, J. Jaworski and J. H. Jung, Analyst, 2012, 137, 3914–3916 RSC.
- H. S. Jung, M. Park, J. H. Han, J. H. Lee, C. Kang, J. H. Jung and J. S. Kim, Chem. Commun., 2012, 48, 5082–5084 RSC.
- Z. Jin, X. B. Zhang, D. X. Xie, Y. J. Gong, J. Zhang, X. Chen, G. L. Shen and R. Q. Yu, Anal. Chem., 2010, 82, 6343–6346 CrossRef CAS PubMed.
- L. Mercier and T. J. Pinnavaia, Environ. Sci. Technol., 1998, 32, 2749–2754 CrossRef CAS.
- M. Dong, Y. W. Wang and Y. Peng, Org. Lett., 2010, 12, 5310–5313 CrossRef CAS PubMed.
- R. Métivier, I. Leray, B. Lebeau and B. Valeur, J. Mater. Chem., 2005, 15, 2965–2973 RSC.
- C. Kresge, M. Leonowicz, W. Roth, J. Vartuli and J. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- N. Zhang, G. Li, J. Zhao and T. Liu, New J. Chem., 2013, 37, 458–463 RSC.
- W. Huang, P. Zhou, W. Yan, C. He, L. Xiong, F. Li and C. Duan, J. Environ. Monit., 2009, 11, 330–335 RSC.
- P. Zhou, Q. Meng, G. He, H. Wu, C. Duan and X. Quan, J. Environ. Monit., 2009, 11, 648–653 RSC.
- M. Yin, Z. Li, Z. Liu, X. Yang and J. Ren, ACS Appl. Mater. Interfaces, 2011, 4, 431–437 Search PubMed.
- Z. Wang, D. Wu, G. Wu, N. Yang and A. Wu, J. Hazard. Mater., 2012, 244, 621–627 Search PubMed.
- Y. Zhao, W. Zhang, Y. Lin and D. Du, Nanoscale, 2013, 5, 1121–1126 RSC.
- A. Abou-Hassan, R. Bazzi and V. Cabuil, Angew. Chem., Int. Ed., 2009, 48, 7180–7183 CrossRef CAS PubMed.
- Y. Wang, B. Li, L. Zhang, P. Li, L. Wang and J. Zhang, Langmuir, 2012, 28, 1657–1662 CrossRef CAS PubMed.
- P. Figueira, C. Lopes, A. Daniel-da-Silva, E. Pereira, A. Duarte and T. Trindade, Water Res., 2011, 45, 5773–5784 CrossRef CAS PubMed.
- J. Zhao, B. Zhu, H. Yu, L. Yan, Q. Wei and B. Du, J. Coll. Inter. Sci., 2013, 389, 46–52 CrossRef CAS PubMed.
- P. I. Girginova, A. L. Daniel-da-Silva, C. B. Lopes, P. Figueira, M. Otero, V. S. Amaral, E. Pereira and T. Trindade, J. Coll. Inter. Sci., 2010, 345, 234–240 CrossRef CAS PubMed.
- Z. Liu, J. Hu, S. Tong, Q. Cao and H. Yuan, Spectrochim. Acta, Part A, 2012, 97, 737–740 CrossRef CAS PubMed.
- J. Du, L. Jiang, Q. Shao, X. Liu, R. S. Marks, J. Ma and X. Chen, Small, 2012, 9, 1467–1481 CrossRef PubMed.
- V. Tharmaraj and K. Pitchumani, Nanoscale, 2011, 3, 1166–1170 RSC.
- J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., Int. Ed., 2009, 48, 5875–5879 CrossRef CAS PubMed.
- Q. Zhang, J. Ge, J. Goebl, Y. Hu, Y. Sun and Y. Yin, Adv. Mater., 2010, 22, 1905–1909 CrossRef CAS PubMed.
- G. Frens, Nature, 1973, 241, 20–22 CrossRef CAS.
- Z. Teng, G. Zheng, Y. Dou, W. Li, C. Y. Mou, X. Zhang, A. M. Asiri and D. Zhao, Angew. Chem., Int. Ed., 2012, 51, 2173–2177 CrossRef CAS PubMed.
- S. Yu and G. M. Chow, J. Mater. Chem., 2004, 14, 2781 RSC.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS PubMed.
- Y.-K. Yang, K.-J. Yook and J. Tae, J. Am. Chem. Soc., 2005, 127, 16760–16761 CrossRef CAS PubMed.
- W. Shi and H. Ma, Chem. Commun., 2008, 1856–1858 RSC.
- W. Shi, S. Sun, X. Li and H. Ma, Inorg. Chem., 2010, 49, 1206–1210 CrossRef CAS PubMed.
- Y. Wang, B. Li, L. Zhang, L. Liu, Q. Zuo and P. Li, New J. Chem., 2010, 34, 1946–1953 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3em00462g |
| This journal is © The Royal Society of Chemistry 2014 |