Internal transcribed spacer rRNA gene sequencing analysis of fungal diversity in Kansas City indoor environments
William R.
Rittenour
a,
Christina E.
Ciaccio
b,
Charles S.
Barnes
b,
Michael L.
Kashon
ac,
Angela R.
Lemons
a,
Donald H.
Beezhold
a and
Brett J.
Green
*a
aAllergy and Clinical Immunology Branch, Health Effects Laboratory Division, National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention, Morgantown, WV 26505, USA. E-mail: dox6@cdc.gov
bThe Department of Allergy, Asthma, and Immunology, Children's Mercy Hospital, Kansas City, MO 64108, USA
cBiostatistics and Epidemiology Branch, Health Effects Laboratory Division, National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention, Morgantown, WV 26505, USA
First published on 20th November 2013
Abstract
Compared to traditional methods of fungal exposure assessment, molecular methods have provided new insight into the richness of fungal communities present in both indoor and outdoor environments. In this study, we describe the diversity of fungi in the homes of asthmatic children located in Kansas City. Fungal diversity was determined by sequencing the internal transcribed spacer (ITS) regions of ribosomal RNA derived from fungi collected in air and dust samples from 31 homes participating in the Kansas City Safe and Healthy Homes Program (KCSHHP). Sequencing results were then compared to data obtained using viable and non-viable fungal exposure assessment methods. ITS clone libraries were predominantly derived from the phylum Ascomycota in both air (68%) and dust (92%) samples and followed by the Basidiomycota and Zygomycota. The majority of Ascomycota clones belonged to four orders including the Pleosporales, Eurotiales, Capnodiales, and Dothideales. ITS sequencing revealed the presence of a number of rarely documented fungal species placed in the Pleosporales. Several species placed in the Basidiomycota were detected in ITS clone libraries but not by viable or non-viable methods. The prevalence of organizational taxonomic units (OTUs) was significantly higher in air than in dust samples (p < 0.0001); however, no differences between OTUs in air samples collected in the subjects' room and basement were observed. These sequencing results demonstrate a much broader diversity of Ascomycota and Basidiomycota communities in KCSHHP indoor environments than previously estimated using traditional methods of assessment.
Environmental impactInternal transcribed spacer (ITS) rRNA gene sequencing has been used as an approach to evaluate the diversity of fungi in indoor and outdoor environments. This methodological approach overcomes a number of limitations associated with traditional methods of fungal exposure assessment. In this study that was located in the Midwest of the United States, ITS rRNA gene sequencing revealed that species within the phylum Ascomycota were prominent in air and dust samples collected from the homes of children with mild or persistent asthma. Species placed in the Basidiomycota and Zygomycota were additionally identified but in lower proportions compared to the Ascomycota. This study provides new insights into the diversity of fungi associated with Kansas City indoor environments. |
1 Introduction
The Kingdom Fungi comprise a diverse group of eukaryotic macro- and microorganisms that predominantly function as saprophytes in the environment. Production of meiotic and mitotic spores and the fragmentation of hyphae result in fungal structures that are aerosolized and disseminated into air currents.1 In the indoor environment, fungi can proliferate and contaminate building materials that lead to elevated ambient concentrations of fungal bioaerosols. Occupants of damp fungal contaminated buildings are at increased risk of upper respiratory morbidity including cough, wheeze, and asthma in sensitized subjects.2–4 To date, fungi that belong to the phylum Ascomycota have predominantly been evaluated in animal and human exposure studies.5 Other prevalent phyla such as the Basidiomycota and Zygomycota have remained overlooked by many investigators.6 Confounding factors associated with traditional fungal exposure assessment methods have limited our understanding of the complete spectrum of fungal bioaerosols in indoor and occupational environments. Identifying and quantifying the complete diversity of fungal bioaerosols using standardized methodologies is critical in determining the fungi that are a burden to public and occupational health.Several fungal detection methods are commonly employed in exposure assessment studies, the most common being viable culture. In this method, samples are collected (air or dust), inoculated onto general nutrient media, and fungi are identified based on the macroscopic and microscopic morphological characteristics.7 Viable culture may favor species that out compete slower-growing species. Several different types of media and physiological conditions (e.g. temperature) may also be required to assess fungal diversity. Another common method of assessment is collection onto an adhesive surface followed by microscopic identification based on the morphological characteristics of the spores (size, shape, etc.). Termed non-viable methods, this approach overcomes several of the limitations introduced into viable analyses but can be confounded by subjectivity and observer bias of the examiner especially as diverse groups of fungi may produce spores and hyphal fragments that are morphologically indiscernible.1,8
Within the last decade, DNA sequencing technologies have been used to study prokaryotic and eukaryotic microbial diversity in soil, water, and air samples. These studies are possible due to the rapid expansion of curated DNA sequences in major databases such as NCBI. The most popular target for evaluating fungal taxonomic relationships has been the internal transcribed spacer (ITS) region; an area of non-functional RNA placed between ribosomal RNAs.9 The ITS region has been recognized as the official fungal barcode and is commonly used as a molecular marker in fungal ecology studies due to the large number of copies per cell and deposited sequences in international databases.10,11 Various molecular methodologies such as sanger and 454 sequencing technologies have provided new insight into previously undetected fungal species in indoor and outdoor air samples.12–19 Diverse collections of fungal taxa once overlooked, such as the phylum Basidiomycota, have been identified using these molecular approaches.13,16,18,20 However, some of these data sets have been confounded by limitations associated with DNA extraction and amplification biases.16,21
The ability to identify the richness of fungal bioaerosol communities that would otherwise be overlooked using traditional methods of assessment could aid in the identification of biomarker fungal species that may be associated with adverse health effects in indoor and occupational environments. Using ITS sequencing methods recently optimized to avoid DNA extraction limitations, we characterized the diversity of fungal bioaerosols in the homes of asthmatic children participating in the Kansas City Safe and Healthy Homes Program (KCSHHP).22 The secondary objective of this study was to compare these results with those obtained using traditional methods of fungal exposure assessment.
2 Experimental methods
2.1 Study population and home characteristics
Homes evaluated in this study consisted of a subset of homes participating in the KCSHHP.22 Homes in this program had at least one asthmatic child who resided in the home for a minimum of 4 days per week. The severity of the asthma of the participating children ranged from mild to severe. Environmental health professionals assessed homes in the program either before or after a series of targeted home improvement efforts that were designed to improve the indoor air quality of the home. Financial support was provided by the Department of Housing and Urban Development as a Healthy Homes Initiative Demonstration Grant awarded to Children's Mercy Hospital to provide for health assessments, home improvement, and analytical services.2.2 Air and dust sampling from homes
Three airborne samples were collected at two separate locations in every home on every sampling occasion. The three collected samples consisted of one collection for non-viable fungal analysis, one collection for viable fungal analysis, and one collection for ITS sequence analysis. In each home, the site of one sampling set was the bedroom of the asthmatic subject and the other sampling site was in the basement utility area of the home. Air sampling for nonviable spore estimation was accomplished using either a Buck Bioaire sampler (A.P. Buck, Inc, Orlando, FL, USA) or an Allergenco MK-III (Environmental Monitoring Systems, Inc., Charleston, SC, USA). Air was collected for 10 minutes at an inlet velocity of 15 liters per minute onto glass slides coated with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of silicon grease and Lubraseal (Thomas Scientific, Swedesboro, NJ, USA) dissolved in hexane. Airborne concentrations of observable spores were estimated by visual enumeration following ASTM Standard D739123 by examiners certified in spore identification by the National Allergy Bureau of the American Academy for Allergy, Asthma and Immunology. Airborne collections for viable spore estimation were taken using a BioStage Single-stage Viable Cascade Impactor (SKC, Eighty Four, PA, USA) sampling at 15 liters per minute for 10 minutes onto inhibitory mold agar (R01506; Remel, Lenexa, KS, USA). Air collections for viable airborne fungi were counted on the developing plate after 5 days at room temperature (RT), identified microscopically and reported as estimates of total fungal colony forming units (CFUs) per cubic meter of air for 25 taxa commonly identified in airborne samples as previously described.24 Air collections for ITS sequence analysis were performed using the National Institute for Occupational Safety and Health (NIOSH) Bioaerosol Cyclone Personal Sampler BC251. The samplers were operated with a SKC 224-PCXR9 personal air sampling pump at 2 liters per minute from between 10 and 90 minutes depending on the time allowed in the residence. The final stage filter for the BC251 sampler was a Millipore AAWP037P0 0.8 μm mixed cellulose ester membrane (EMD Millipore Corporation, Billerica, MA, USA).
:1 mixture of silicon grease and Lubraseal (Thomas Scientific, Swedesboro, NJ, USA) dissolved in hexane. Airborne concentrations of observable spores were estimated by visual enumeration following ASTM Standard D739123 by examiners certified in spore identification by the National Allergy Bureau of the American Academy for Allergy, Asthma and Immunology. Airborne collections for viable spore estimation were taken using a BioStage Single-stage Viable Cascade Impactor (SKC, Eighty Four, PA, USA) sampling at 15 liters per minute for 10 minutes onto inhibitory mold agar (R01506; Remel, Lenexa, KS, USA). Air collections for viable airborne fungi were counted on the developing plate after 5 days at room temperature (RT), identified microscopically and reported as estimates of total fungal colony forming units (CFUs) per cubic meter of air for 25 taxa commonly identified in airborne samples as previously described.24 Air collections for ITS sequence analysis were performed using the National Institute for Occupational Safety and Health (NIOSH) Bioaerosol Cyclone Personal Sampler BC251. The samplers were operated with a SKC 224-PCXR9 personal air sampling pump at 2 liters per minute from between 10 and 90 minutes depending on the time allowed in the residence. The final stage filter for the BC251 sampler was a Millipore AAWP037P0 0.8 μm mixed cellulose ester membrane (EMD Millipore Corporation, Billerica, MA, USA).
One house dust composite collection was also taken from each home either from the home vacuum (as long as the homeowner used it regularly within the home) or from 9 square feet of carpet, throw rug, or floor. Dust samples were collected in the homes by thoroughly vacuuming 9, one square foot areas at locations chosen from the following: child's bedroom, living room, family room, TV room, hallway, playroom, or den. Dust samples were returned to the laboratory and stored at −20 °C until processed. Samples were sieved through a 300-mesh screen (Thermo Fisher Scientific, Waltham, MA. USA) and 10 mg of sieved dust was shipped to the NIOSH for ITS sequence analysis.
2.3 Genomic DNA extraction
Air and dust samples were processed separately for DNA extraction using the High Pure PCR Template kit (Roche, Basel, Switzerland) as previously described.21 For dust samples, 10 mg of dust was added to a 2 mL bead-beater tube containing 300 mg glass beads (212–300 μm; Sigma, St. Louis, USA). The kit lysis buffer was added to each tube (350 μL) and then placed in a bead beater (BioSpec Products, Bartlesville, OK, USA) for 15 seconds at high speed. The tubes were centrifuged at 17000×g and the supernatant was added to 200 μL of the kit's binding buffer. The sample was washed and eluted according to the manufacturer's instructions. In each extraction, the DNA was eluted in 100 μL. The eluate was then reapplied to the filter for a final DNA volume of 100 μL. For air samples, each stage from the NIOSH BC251 air sampler was combined prior to DNA extraction. The after filter was sectioned into 6-pieces with a scalpel using aseptic methods. These pieces were placed into a 2 mL bead-beater tube containing 300 mg glass beads as described above. The tubes were placed in liquid nitrogen for 1 minute and processed in a bead beater for 30 seconds. The kit's lysis buffer (650 μL) was then sequentially added to the first and second stage tubes and vortexed in order to collect the fungal spores and fragments from the samples. The lysis buffer was added to the 2 mL bead-beater tube containing the macerated filter material. These tubes were processed with a bead beater for 30 seconds and then centrifuged for 1 minute at 17000×g. The supernatant was collected, mixed with 200 μL of the kit's binding buffer, and then washed and eluted as recommended by the manufacturer.
2.4 ITS amplification, cloning, and sanger sequencing
Fungal rRNA was targeted for PCR amplification as previously described.16,21 Briefly, fungal rDNA sequences were amplified using the primer pair Fun18Sf/ITS4 and Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA) according to the methods previously described.16 Five 50 μL replicate PCR reactions were run for each sample using 5 μL of DNA template. These replicates were then combined and the rDNA amplicons were purified using a PCR purification kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Purified product (5 μL) was then run on a 1% agarose gel containing 0.4 μg mL−1 ethidium bromide and examined for amplicons using an ultraviolet gel doc (Alpha Innotech, Santa Clara, CA, USA). Amplicons were then cloned into the pDRIVE vector using a PCR cloning kit (Qiagen, Valencia, CA, USA). Clone libraries were generated by transforming cloned plasmids into chemically competent Escherichia coli cells as previously described.21 Positive colonies (as determined colorimetrically by the inactivation of the lacZ gene) were selected and cultured for 16 hours at 37 °C in liquid Luria–Bertani media containing 100 μg mL−1 ampicillin. Resultant cells were centrifuged at 300×g and the pellet resuspended in 200 μL of 15% glycerol, and sent for Sanger sequencing of the fungal rRNA insert (Genewiz, Inc., South Plainsfield, NJ, USA). Inserts were sequenced in both directions, allowing for sequence analysis of both the ITS1 and the ITS2 regions.Sequencing results were downloaded as .ab1 chromatogram files from Genewiz Inc. Vector sequence data were trimmed and forward and reverse sequences were assembled using Geneious Software (Biomatters Ltd, Auckland, New Zealand). Sequence data were then clustered into operational taxonomic units (OTUs) with MOTHUR software25 using a 97% similarity cutoff. Sequences representative of each OTU were then used in a BLAST search against NCBI's database.21
2.5 Statistical analysis
Statistical analysis of OTUs quantified in air and dust samples was performed using SAS version 9.2 for Windows (SAS, Cary, NC). The data were log transformed prior to analysis and ProcMix was used to run a one way analysis of variance (ANOVA) with ‘experiment’ included as a random factor to measure statistical significance. Differences between experimental groups that resulted in a p value of less than 0.05 were considered significant. Significant differences between the proportions of fungal species collected in homes using viable, non-viable, and ITS sequencing methods were additionally determined using the Fishers exact test. One sided p values <0.05 indicated statistically significant differences between the respective methods.2.6 Nucleotide sequence accession numbers
The sequences from representative fungal OTUs were deposited in the GenBank database under the following accession numbers: KF800092–KF800697.3 Results
3.1 ITS clone library analysis
DNA was amplified with the universal fungal primer pair Fun18Sf/ITS4 from 30 dust samples and from 27 KCSHHP air samples. From these samples, 4626 ITS clones were collected and sequenced. The DNA sequences were clustered into 610 individual OTUs and 456 unique fungal OTUs were identified. Within the fungal OTU dataset, 73% were ≥97% identical to reference ITS sequences in the NCBI database. Additional OTUs were placed in the Amoebozoa (n = 2), Algae (n = 3), Plantae (n = 54), 51 OTUs were predicted chimeric, and 44 yielded no close hits to ITS sequences in the major databases.The mean number of fungal-specific OTUs quantified in air samples (x = 28) was significantly greater than the number of OTUs quantified in dust samples (x = 15; p < 0.0001). Analysis of air samples additionally demonstrated that there were no statistically significant differences between the mean number of OTUs quantified in air samples derived from the subject's room compared to the basement. Furthermore, there were no observable differences between the spectrum of fungal phyla identified in Level 1 and Level 2 homes (data not shown).
3.2 Prevalence of fungi in KCSHHP air and dust samples
A total of 3170 clones were generated and sequenced from the air samples and 2499 were determined to be of fungal origin. In dust samples, 1456 clones were sequenced and 1208 were determined to be of fungal origin (Table 1). The phylum Ascomycota was identified to be the most prominent fungal phylum in both air (68%) and dust (92%) clone libraries (Fig. 1). The Basidiomycota was the next most prevalent phylum and clones were more prevalent in air samples (30%) than dust samples (7%; Fig. 1). Members of the Zygomycota were additionally quantified in the analysis but only accounted for 2.2% and 1.7% of all fungal clones in air and dust samples, respectively (Fig. 1).| Air (n = 27a) | Dust (n = 30b) | ||||||
|---|---|---|---|---|---|---|---|
| Genus/species | Clone # | Clone % | Genus/species | Clone # | Clone % | ||
| a Total number of fungal clones identified in air samples n = 2499. b Total number of fungal clones in dust samples n = 1208. | |||||||
| Aspergillus spp. | 309 | 12.36 | Leptosphaerulina chartarum | 175 | 14.48 | ||
| A. aculeatus | 4 | 0.16 | Phoma medicaginis | 168 | 13.91 | ||
| A. candidus | 1 | 0.04 | Aspergillus spp. | 155 | 12.83 | ||
| A. fumigatus | 18 | 0.72 | A. penicilloides | 41 | 3.39 | ||
| A. niger | 2 | 0.08 | A. spp. BF8 | 2 | 0.17 | ||
| A. ochraceus | 3 | 0.12 | A. unguis | 2 | 0.17 | ||
| A. penicilloides | 21 | 0.84 | A. ustus | 1 | 0.08 | ||
| A. sclerotiorum | 2 | 0.08 | A. versicolor | 50 | 4.14 | ||
| A. spp. BF8 | 7 | 0.28 | A. vitricola | 59 | 4.88 | ||
| A. spp NRRL 4642 | 1 | 0.04 | Cladosporium spp. | 69 | 5.71 | ||
| A. terreus | 1 | 0.04 | Epicoccum spp. | 62 | 5.13 | ||
| A. tubingensis | 5 | 0.20 | Penicillium spp. | 49 | 4.06 | ||
| A. unguis | 1 | 0.04 | Cochliobolus spp. | 42 | 3.48 | ||
| A. ustus | 1 | 0.04 | Alternaria spp. | 38 | 3.15 | ||
| A. versicolor | 137 | 5.48 | Kabatiella microsticta | 38 | 3.15 | ||
| A. vitricola | 105 | 4.20 | Curvularia spp. | 34 | 2.81 | ||
| Epicoccum spp. | 299 | 11.96 | Eurotium spp. | 20 | 1.66 | ||
| Cladosporium spp. | 156 | 6.24 | Cryptococcus spp. | 16 | 1.32 | ||
| Leptosphaerulina chartarum | 134 | 5.36 | Stagonospora spp. | 16 | 1.32 | ||
| Ustilago spp. | 112 | 4.48 | Ustilago spp. | 13 | 1.08 | ||
| Didymella exitialis | 94 | 3.76 | |||||
| Kabatiella microsticta | 84 | 3.36 | |||||
| Irpex lacteus | 87 | 3.48 | |||||
| Penicillium spp. | 72 | 2.88 | |||||
| Phoma medicaginis | 73 | 2.92 | |||||
| Malassezia restricta | 65 | 2.60 | |||||
| Eurotium spp. | 63 | 2.52 | |||||
| Mucorales | 56 | 2.24 | |||||
| Davidiella macrospora | 57 | 2.28 | |||||
| Phanerochaete spp. | 51 | 2.04 | |||||
| Alternaria spp. | 47 | 1.88 | |||||
| Wallemia sebi | 44 | 1.76 | |||||
| Coniothyrium fuckelii | 44 | 1.76 | |||||
| Gloeoporus dichrous | 30 | 1.22 | |||||
| Cryptococcus spp. | 36 | 1.44 | |||||
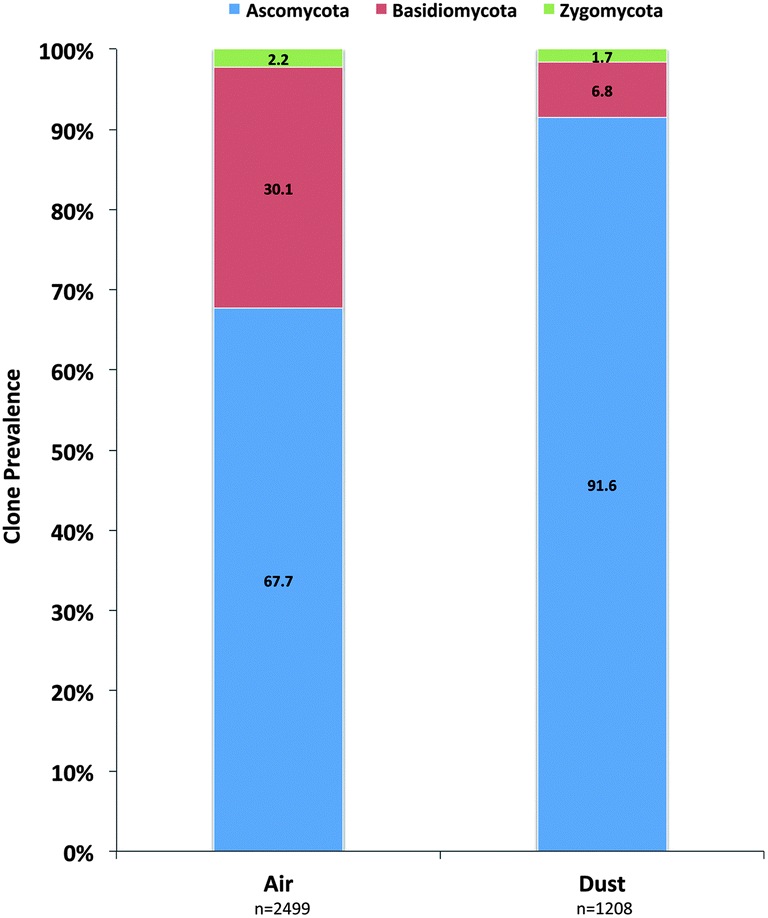 | ||
| Fig. 1 Prevalence of individual fungal phyla determined in the ITS sequence analysis of KCSHHP air and dust samples. | ||
The fungal order, Pleosporales, was the most prominent Ascomycota order and accounted for 46% and 54% of fungal clones in air and dust samples, respectively (Fig. 2A). The most prevalent Pleosporalean OTUs recovered in the clone analysis included Epicoccum nigrum, Leptosphaerulina chartarum, Didymella exitialis, and Phoma medicaginis (Fig. 2B). The Eurotiales accounted for 28% and 21% of clones in air and dust samples and the two most prevalent species that represented this order included Aspergillus versicolor and A. vitricola (Fig. 2C). Cladosporium cladosporiodes was the most common fungal species detected in the Capnodiales (Fig. 2D). Other orders including the Dothideales and Hypocreales were prevalent in air and dust samples and accounted for ≤5% of clones (Fig. 2A).
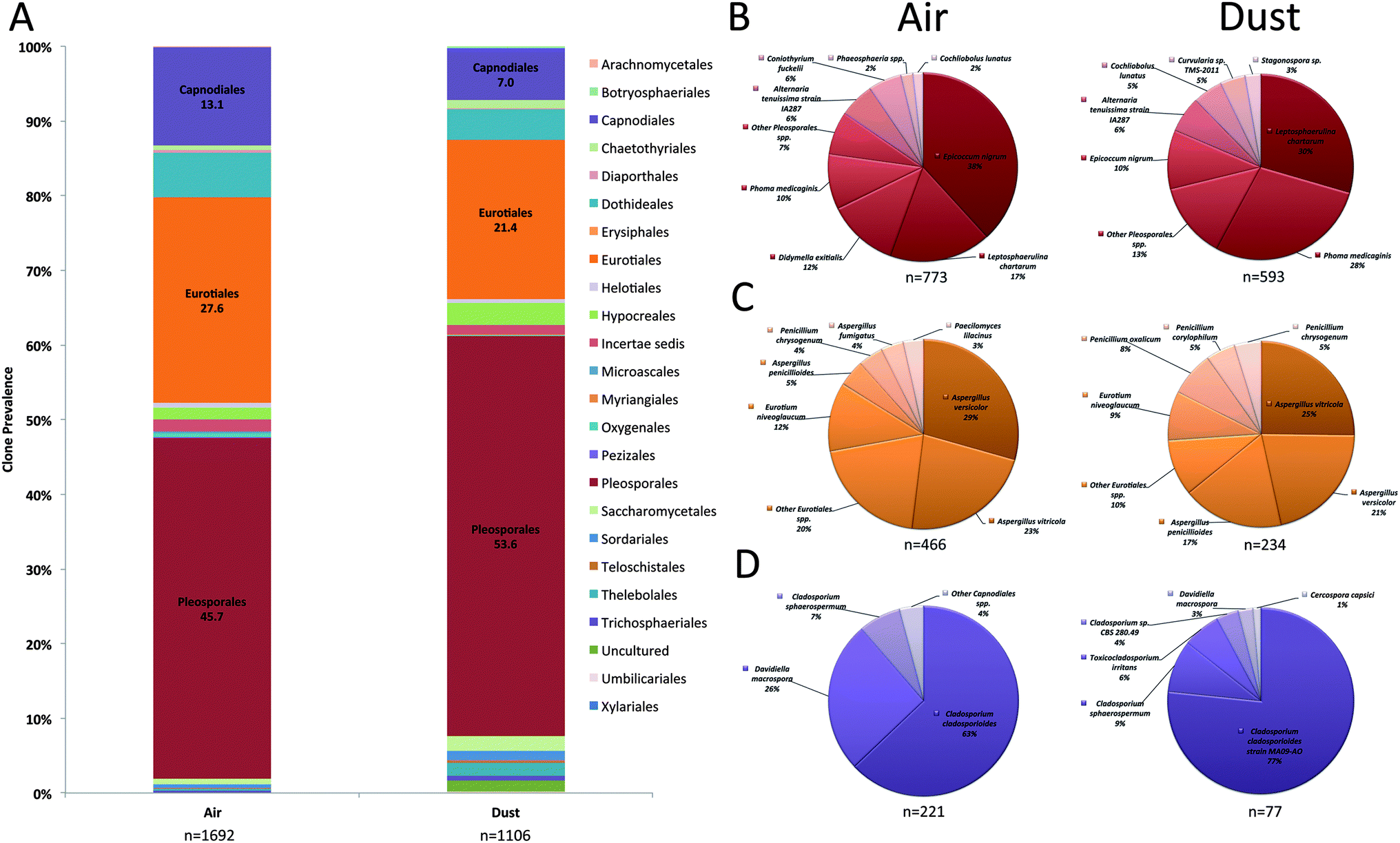 | ||
| Fig. 2 Prevalence of individual Ascomycota orders in KCSHHP air and dust samples (A). Distribution of the most prevalent species determined in the ITS sequence analysis of air (left pie chart) and dust (right pie chart) samples for the fungal orders Pleosporales (B), Eurotiales (C), and Capnodiales (D). n, denotes the number of clones identified. | ||
Prevalent Basidiomycota orders included the Ustilaginales, Tremellales, and Polyporales in both air and dust samples (Fig. 3A). Although accounting for a small proportion in dust samples, the orders Wallemiales, Malasseziales, Sporidiobolales, Pucciniales, Auriculariales, and Agaricales were additionally prevalent in air samples (Fig. 3A). The most prevalent Basidiomycota species included Ustilago drakensbergiana, Cryptococcus spp., and Irpex lacteus (Fig. 3B–D). In contrast, only six Zygomycota genera were identified in the analysis and Mucor and Blakeslea were the most prevalent identified in air and dust samples (Fig. 4).
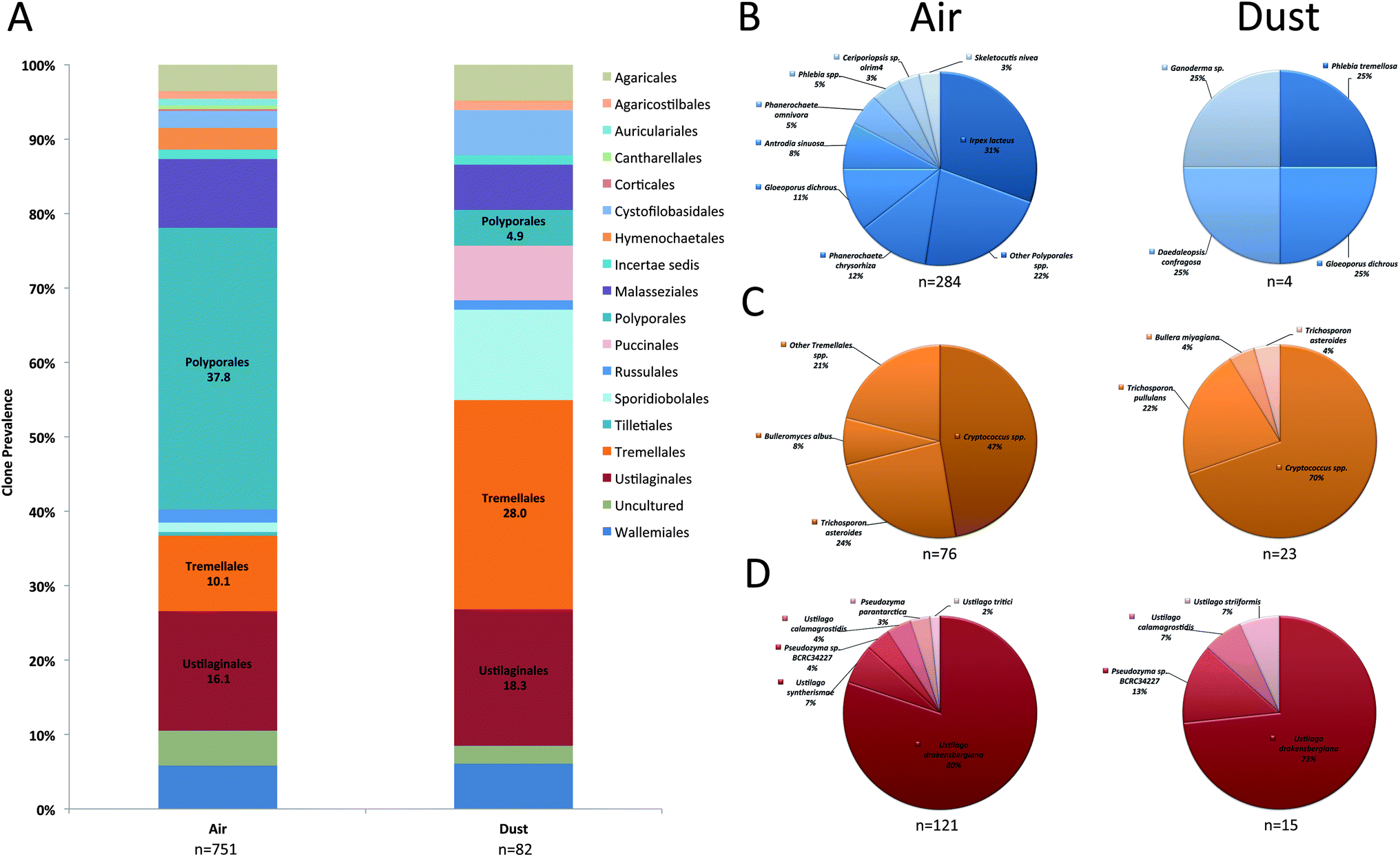 | ||
| Fig. 3 Prevalence of individual Basidiomycota orders in KCSHHP air and dust samples (A). Distribution of most prevalent species in the ITS sequence analysis of air (left pie chart) and dust (right pie chart) samples for the fungal orders Ustilaginales (B), Tremellales (C), and Polyporales (D). n, denotes the number of clones identified. | ||
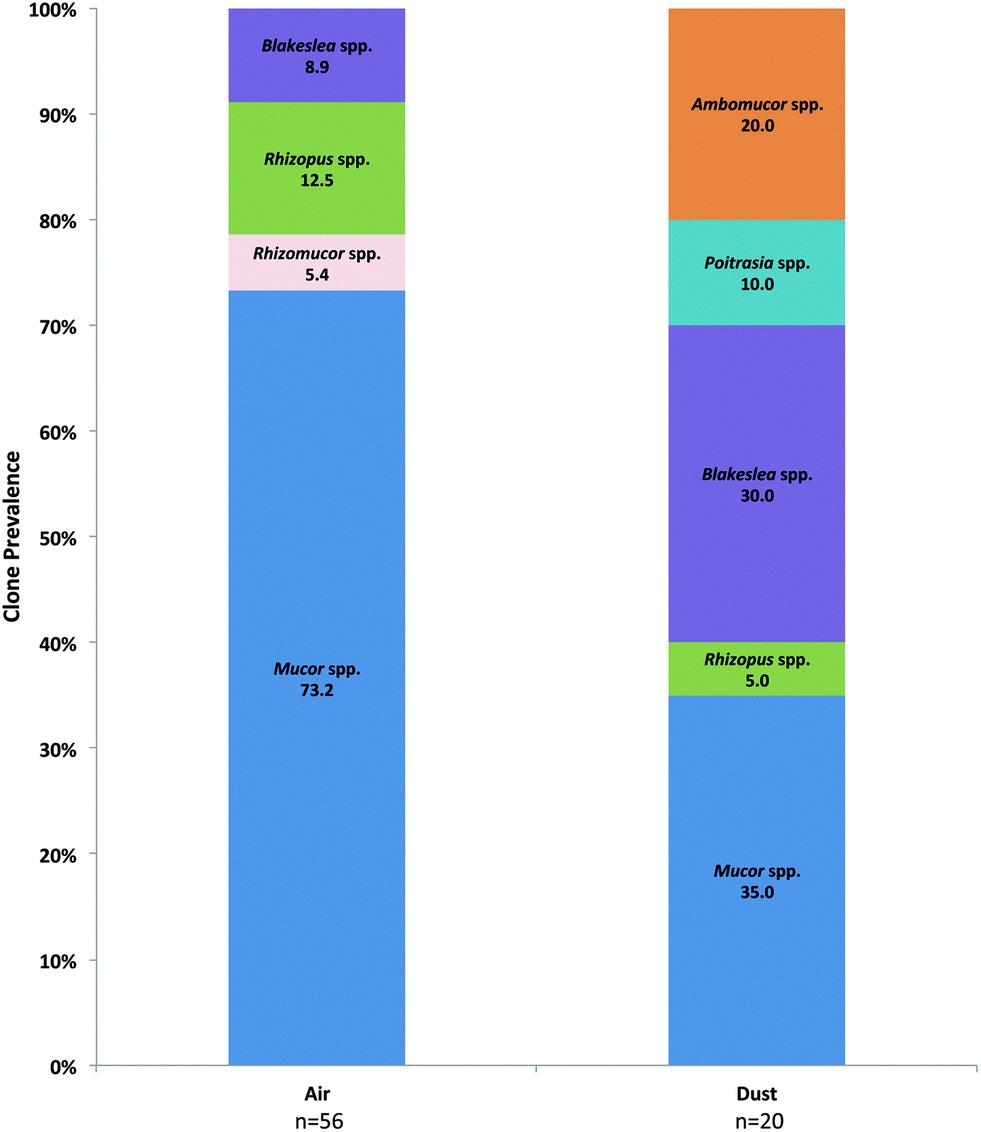 | ||
| Fig. 4 Prevalence of individual Zygomycota genera determined in the ITS sequence analysis of KCSHHP air and dust samples. | ||
The most prevalent individual fungal OTUs identified in air samples belonged to the order Eurotiales and the most common species included A. versicolor and A. vitricola (Table 1). Epicoccum spp., Cladosporium spp., L. chartarum, Ustilago spp., D. exitialis, K. microsticta, and I. lacteus were also prevalent and represented 38% of all fungal clones sequenced (Table 1). Fungal bioaerosols commonly reported in the literature including Penicillium spp., Mucorales and Alternaria spp. only accounted for 2.9, 2.2, and 1.9% of all fungal clones sequenced in the air samples. In contrast, the most prevalent individual fungal genera in dust samples comprised Pleosporalean fungi including L. chartarum and P. medicaginis, representing 14.5% and 14%, respectively of all fungal clones isolated from dust samples (Table 2). Compared to the diversity of fungi captured in air samples, dust samples contained a similar proportion of Aspergillus species (12.8%). The most common Aspergillus species identified in dust samples included A. penicillioides (3.4%), A. versicolor (4.1%), and A. vitricola (4.8%).
| Viable (n = 31) | Non-viable (n = 31) | OTU-air (n = 27) | OTU-DUST (n = 30) | ||||
|---|---|---|---|---|---|---|---|
| Species | % Homes | Species | % Homes | Species | % Homes | Species | % Homes |
| a # OTU air versus OTU dust, p < 0.05; ‡ OTU air versus viable, p < 0.05; * OTU air versus non-viable, p < 0.05; † OTU dust versus viable, p < 0.05; ⊕ OTU dust versus non-viable, p < 0.05. | |||||||
| Alternaria spp. | 51.6 | Alternaria spp. | 87.1 | Alternaria tenuissima | 57.7* | Alternaria tenuissima | 50.0⊕ |
| Aspergillus spp. | 61.3 | Aspergillus/Penicillium | 87.1 | Aspergillus versicolor | 57.7 | Aspergillus versicolor | 26.7 |
| Aspergillus vitricola | 57.7 | Aspergillus vitricola | 20.0⊕ | ||||
| Eurotium niveoglaucum | 46.2 | Eurotium niveoglaucum | 36.7 | ||||
| Ascospores | 0.0 | Ascospores | 80.6 | Ascospores | NA | Ascospores | NA |
| Basidiospores | 3.2 | Basidiospores | 87.1 | Basidiospores | NA | Basidiospores | NA |
| Cladosporium spp. | 87.1 | Cladosporium spp. | 93.5 | Cladosporium spp. | 92.3 | Cladosporium spp. | 73.3⊕ |
| Coniothyrium spp. | 0.0 | Coniothyrium spp. | 0.0 | Coniothyrium spp. | 23.1‡* | Coniothyrium spp. | 6.7 |
| Didymella exitialis | 0.0 | Didymella exitialis | 0.0 | Didymella exitialis | 61.5‡* | Didymella exitialis | 23.3#†⊕ |
| Epicoccum spp. | 0.0 | Epicoccum spp. | 67.7 | Epicoccum spp. | 69.2‡ | Epicoccum spp. | 50.0† |
| Pithomyces spp. | 9.7 | Pithomyces spp. | 87.1 | Pithomyces chartarum | 65.4‡ | Leptosphaerulina chartarum | 70.0† |
| Kabatiella/Aureobasidium | 0.0 | Kabatiella/Aureobasidium | 0.0 | Kabatiella/Aureobasidium | 50.0‡* | Kabatiella/Aureobasidium | 43.3†⊕ |
| Bipolaris/Drechslera | 9.7 | Bipolaris/Drechslera | 38.7 | Bipolaris/Drechslera | 23.1‡ | Bipolaris/Drechslera | 40.0† |
| Curvularia spp. | 12.9 | Curvularia spp. | 67.7 | Curvularia spp. | 34.6* | Curvularia spp. | 50.0† |
| Penicillium spp. | 80.6 | Aspergillus/Penicillium | 87.1 | Penicillium spp. | 57.7* | Penicillium spp. | 43.3†⊕ |
| Phoma spp. | 0.0 | Phoma spp. | 0.0 | Phoma spp. | 57.7‡* | Phoma spp. | 70.0†⊕ |
| Rhizopus spp. | 9.7 | Rhizopus spp. | 3.2 | Rhizopus spp. | 23.1* | Rhizopus spp. | 3.3# |
| Cryptococcus spp. | 0.0 | Cryptococcus spp. | 0.0 | Cryptococcus spp. | 57.7‡* | Cryptococcus spp. | 43.3†⊕ |
| Wallemia spp. | 0.0 | Wallemia spp. | 0.0 | Wallemia sebi | 46.2‡* | Wallemia sebi | 3.3# |
| Smuts/Myxomycetes | 0.0 | Smuts/Myxomycetes | 83.9 | Ustilago spp. | 38.5‡* | Ustilago spp. | 53.3†⊕ |
| Nigrospora spp. | 0.0 | Nigrospora spp. | 6.5 | Nigrospora spp. | 7.7 | Nigrospora spp. | 20.0† |
| Stagnospora spp. | 0.0 | Stagnospora spp. | 0.0 | Stagnospora spp. | 11.5 | Stagnospora spp. | 30.0†⊕ |
| Rhodotorula spp. | 0.0 | Rhodotorula spp. | 0.0 | Rhodotorula spp. | 7.7 | Rhodotorula spp. | 16.7†⊕ |
| Unknown | 71.0 | Unknown | 0.0 | Unknown | 53.8* | Unknown | 33.3†⊕ |
3.3 Comparison of fungal detection methods
Overall, the proportion of fungi collected in homes using each approach was not statistically different for Aspergillus and Cladosporium species (Table 2). When comparing viable culture to air clone libraries, statistically significant differences were observed for Coniothyrium spp., D. exitialis, Epicoccum spp., L. chartarum, Kabatiella spp., Bipolaris/Drechslera spp., Phoma spp., Cryptococcus spp., Wallemia sebi, and Ustilago spp. primarily as these organisms were not observed in viable culture (Table 2). Similar statistically significant differences were observed for ITS dust sample clone libraries although additional species including Curvularia spp., Penicillium spp., Nigrospora spp., Stagonospora spp., and Rhodotorula spp. were identified in greater proportions compared to viable culture analysis (Table 2).Compared to non-viable methodologies, greater proportions of Coniothyrium spp., D. exitialis, Kabatiella spp., Phoma spp., Rhizopus spp., Cryptococcus spp., and W. sebi were recovered in ITS air sample clone libraries but were lower for Curvularia spp., Penicillium spp., L. chartarum, and Ustilago species (Table 2). Significant differences between ITS dust sample clone libraries and viable culture were comparable to air sampling data, although additional differences were observed for Curvularia spp., Penicillium spp., Nigrospora spp., Stagnospora spp., Rhodotorula spp., and unknown species (Table 2). Interestingly, greater proportions of Alternaria spp., Cladosporium spp., Penicillium spp., and Ustilago spp. were observed in non-viable methods but not in ITS dust sample clone libraries. In addition, the proportions of D. exitialis, Rhizopus spp., and W. sebi were greater in ITS air sample clone libraries compared to ITS dust sample clone libraries. Despite the differences reported for the prevalence of specific fungal species across the homes sampled, it must be highlighted that the overall frequency of several of these species in the library was relatively low (<4%; Table 1).
4 Discussion
During the last decade, several monographs and review articles have concluded that sufficient epidemiological data was available to demonstrate an increased risk of adverse respiratory health effects in subjects that occupy damp or fungal contaminated buildings.2–4 However, the fungal bioaerosol burden that is associated with respiratory disease has remained uncharacterized due to the lack of standardized methods to assess fungal exposures. Recent advances in molecular quantification methods, such as qPCR, have provided new objective approaches to identify fungal bioaerosols in the environment.26,27 However, many fungal biomarker species have been selected from culture-based studies (http://www.epa.gov/microbes/moldtech.htm). As a result, the species richness of fungal communities has not been fully assessed in the indoor environment. Recently, large-scale studies based on the sequencing of ITS regions of fungal rRNA genes using sanger sequencing and more recently next generation sequencing technologies have provided new insights into the diversity of fungi in both indoor and outdoor air samples.14–21 In this study, a more complete fungal diversity was determined for homes of asthmatic children participating in the KCSHHP using ITS sequencing and these data were compared to diversity results derived from traditional methods of assessment.Several fungi were exclusively detected with ITS sequencing; the most common included P. medicaginis, D. exitialis, K. microsticta, Coniothyrium spp., I. lacteus, U. drakensbergiana, Cryptococcus spp., and W. sebi. The Ascomycota fungal genera Coniothyrium, Didymella, and Phoma belong to the fungal order Pleosporales, a broadly cross reactive and important allergenic fungal order for Kansas City residents.28,29 Allergen cross reactivity has been reported between Alternaria alternata and Phoma betae30 and D. exitialis has been associated with late summer asthma in subjects sensitized to A. alternata.31Coniothyrium spp. has also been reported as a possible aeroallergen source in Puerto Rico.32 Other Ascomycota including two closely related yeast-like genera that belong to the fungal order Dothideales, Aureobasidium and Kabatiella, were identified in the ITS sequencing analysis. Compared to Kabatiella spp. that are more difficult to identify using traditional methods of assessment, Aureobasidium is a prevalent genus in moist indoor environments and has been previously associated with asthma exacerbations.33 Basidiomycota that are often overlooked in exposure assessment studies were also identified and included the genera Cryptococcus and Wallemia. Although no pathogenic Cryptococcus spp. such as C. neoformans were detected in air or dust samples, nonpathogenic Cryptococcus species (e.g. C. albidus) were identified and have been previously shown to elicit skin prick test reactions in atopic patients.34 Recent studies using next generation sequencing technologies have also shown that Cryptococcus (only found with ITS sequencing) is a common genus identified in outdoor air,18 indoor air,17 and floor dust.19 This genus also appears to be associated with asthma development in homes with increased moisture.19 Other Basidiomycota such as W. sebi and I. lacteus were also identified but not by traditional methods of assessment. W. sebi is ubiquitous and frequently isolated in agricultural environments and clinical studies have previously shown that a significant number of patients with farmer's lung had W. sebi specific antibodies.35 To date, the contribution of other Basidiomycota such as I. lacteus to adverse health effects has either not been evaluated or requires further study.6 Although the spectrum of fungi identified using similar sequencing methods used in the present analysis vary from those previously reported in other environments,21 these findings further highlight the potential public health burden of overlooked Ascomycota and Basidiomycota species in Kansas City environments.
The prevalence of amerospore producing genera in air samples was often significantly greater in ITS sequencing compared to non-viable methods of assessment. These results highlight the subjectivity associated with microscopically differentiating morphologically indiscernible amerospores derived from a complex mixture of fungal bioaerosols. Commonly placed in a broad Aspergillus/Penicillium group in non-viable analyses, other fungal genera that were identified in ITS sequencing that also produce amerospores included Aspergillus spp., Penicillium spp., Phoma spp., Rhizopus spp., and W. sebi. Other smaller amerospore producing yeasts were also detected and included Aureobasidium, Cryptococcus, and Rhodotorula species. However, the comparative analysis of viable, non-viable and ITS sequence results should be interpreted with caution, as there were several limitations associated with the different air sampling methods employed in this study. Previous studies have shown that the collection efficiencies for smaller particles may vary between air sampling methods.36 The collection and identification of amerospore genera using ITS methods could be associated with the collection efficiency of the NIOSH Bioaerosol Cyclone Personal Sampler BC251.37
In addition to differentiating amerospores, ITS sequencing also provided an approach to readily differentiate individual fungal species compared to viable and non-viable methods that are subjective and require a trained mycologist. In the current study, 16 Aspergillus species were detected using ITS sequencing, including the teleomorph, Eurotium niveoglaucum. Differentiating between Aspergillus species is critically important as a number of individual species have been recently associated with opportunistic disease, asthma, or contain species-specific allergens.14,38,39
Aspergillus was among the most prevalent fungal genera detected in air and dust samples. Although this result is not surprising considering the prevalence of Aspergillus reported in studies utilizing traditional methods of assessment,7,40 previous sequencing studies have demonstrated Aspergillus species to comprise only a small proportion of the total number of ITS clone libraries.15,16 The method of DNA extraction used in ITS sequencing studies may significantly influence the composition of the resulting clone libraries. Recently, our laboratory has shown that DNA extraction from Aspergillus conidia can be highly variable depending on the method.21 In the current study, a Roche High Pure PCR Template kit was used to isolate DNA from fungal conidia and hyphae. This method was previously shown to efficiently extract DNA from a variety of amerospore producing species compared to other commercially available DNA extraction kits.21 Although this represents a potential confounding variable of the analysis, other sample processing biases may also be introduced to sequencing studies (either Sanger or high-throughput sequencing) and these variables must be empirically determined before sequence data is interpreted.12
The distribution of Ascomycota in air and dust samples was comparable. Members of the order Pleosporales were more prevalent in dust samples (54%) compared to air samples (46%), but represented a large portion in both libraries. In addition to Coniothyrium spp. and D. exitialis, other members of the Pleosporales such as Epicoccum spp. were commonly detected in non-viable and ITS sequencing analysis of air samples but not using viable methods of assessment. Compared to the amerospore producing species, Epicoccum produces large multicellular dictyospores that are morphologically discernible but can be outgrown by r strategist genera such as Aspergillus or Penicillium species. Although conidia viability was not evaluated in the present study, it may also be possible that many of the Epicoccum conidia were not viable and were unable to colonize nutrient agar plates. Non-viable Epicoccum conidia have been reported to release allergen41 and may represent an additional burden of Pleosporalean allergen in these KCSHHP indoor environments. Compared to other fungal orders, members of the Pleosporales contain several species that have important functional roles in exacerbating asthma, most notably, A. alternata. The allergen Alt a 1 has been shown to be a major contributing factor to allergic sensitization42 and it is conserved among several species within the Pleosporales but no other fungi.29 Interestingly, other members of the Pleosporales that contain Alt a 1 orthologues often express the allergen in higher quantities than A. alternata.29 In addition to L. chartarum, a prevalent Pleosporalean fungus identified in previous Kansas City assessment studies,40 several other members of the Pleosporales identified in the present analysis may also produce an Alt a 1 orthologue. We hypothesize that as an order, the Pleosporales may contribute significantly to the respiratory morbidity in subjects that live in these KCSHHP indoor environments.43
In contrast, the Basidiomycota represented a larger portion of sequences from air samples compared to dust samples. Although it was not possible to determine if the increased Basidiomycota burden in air samples was derived from outdoor sources, the largest group of Basidiomycota identified belonged to the order Polyporales (38% of Basidiomycota clones). This finding supports recent fungal ITS sequencing data derived from sanger sequencing20 and more recently 454 sequencing studies.17,18 The order Polyporales was comprised mainly of the genera Irpex, Phlebia, Ganoderma, Phanerochaete, and Gloeoporus. Species within this order typically produce bracket-like fruiting bodies on dead woody material and were likely derived from anthropogenic sources or introduced through openings in the building envelope. Given the specific nutrient requirements of members within this order and the morphologically indistinguishable spores that are produced, these fungi have remained overlooked in exposure assessment studies. Although the clinical relevance of Basidiomycota exposures has been previously demonstrated in a number of tropical environments, little is known about the contribution of the fungi within the Polyporales.44
Compared to air samples, dust samples contained a much lower proportion of Basidiomycota than Ascomycota. The orders Tremellales, Ustilaginales, and Sporidiobolales comprised the largest percentages. These orders were predominantly represented by the fungal genera Cryptococcus, Ustilago, and Rhodotorula, respectively. Each of these fungal genera has been previously associated with respiratory morbidity34,45–47 and may represent additional sources of exposure for subjects sensitized to fungal bioaerosols in these KCSHHP indoor environments. Other Basidiomycota that cause skin infections were additionally identified in the analysis and included the genera, Malassezia and Trichosporon. Interestingly, Malassezia species have been identified in a number of previous studies that have utilized this analytical approach (Sanger or high-throughput sequencing).13,14,16,18,20,21
Sequencing the fungal ITS regions can provide valuable insight into the richness of fungal communities in air and dust samples. To date, several limitations have often been overlooked that may confound the utility of this approach in fungal exposure assessment studies. Previous studies have shown that DNA extraction methods and PCR amplification steps introduce biases that may skew the data and confound quantitative conclusions that can be drawn from the quantity of Sanger sequences.10,21 These biases may also apply to high-throughput sequencing methods but this remains to be evaluated.12 Fungal ITS sequencing studies are also dependent on high-quality database entries but approximately 27% of the annotated sequences in the International Nucleotide Sequence Database (INSD) are not sufficiently annotated to species level.48 Although these limitations must be taken in consideration, future fungal ITS sequence studies may aid in the further development of standardized molecular methods, such as the Environmental Relative Moldiness Index (ERMI). ERMI, a qPCR-based methodology developed by the United States Environmental Protection Agency, enables the quantification of biomarker fungi in indoor environments.27 The method is used in commercial and academic sectors and has been utilized in epidemiological studies to demonstrate associations between asthma development and individual fungal species such as Aspergillus ochraceus, A. unguis, and Penicillium variabile.49 However, recent ITS sequencing studies have demonstrated that only a small proportion of ERMI biomarker fungi are actually present in contaminated homes.14 These findings have been confirmed as several of the most common fungal genera identified in the present ITS sequencing study are not currently included in the ERMI panel (e.g. Leptosphaerulina, Phoma, Didymella, Ustilago, and Irpex etc.).26 These preliminary ITS sequencing studies further demonstrate the variation of fungal diversity in different geographic environments.7,13,17–20 Additional ITS sequencing studies are required in other regions of the United States to evaluate the spatial and geographic distribution of fungal bioaerosols with the aim of producing regionally relevant biomarker panels for current molecular detection methods.
5 Conclusions
In this study, fungal ITS sequencing was compared to traditional methods of assessment to describe the diversity of fungi in the homes of asthmatic children participating in the KCSHHP. Several previously overlooked Ascomycota and Basidiomycota fungi were detected using ITS sequencing. Using this approach, it has been possible to explore the diversity of amerospore producing species that are often misidentified using traditional methods of assessment. Future studies aim to utilize fungal ITS sequencing to describe the temporal and spatial differences in indoor fungal composition, as both season and geography may strongly influence the diversity of fungi detected. Furthermore, the role of these overlooked fungi to respiratory morbidity currently remains unclear in the KCSHHP pediatric cohort and is the focus of future clinical studies.Acknowledgements
The findings and the conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health. The authors declare no conflict of interest. This study was supported in part by an interagency agreement with NIEHS IAA# AES 12007-00100000.Notes and references
- B. J. Green, D. Schmechel and R. C. Summerbell, in Fundamentals of mold growth in indoor environments and strategies for healthy living, ed. O. Adnan and R. A. Samson, Wageningen Academic Publishers, Amsterdam, The Netherlands, 2011, pp. 211–245 Search PubMed.
- M. J. Mendell, A. G. Mirer, K. Cheung, M. Tong and J. Douwes, Environ. Health Perspect., 2011, 119, 748–756 CrossRef CAS PubMed.
- Institute of Medicine (U.S.), Committee on Damp Indoor Spaces and Health, Damp indoor spaces and health, National Academies Press, Washington, DC, 2004 Search PubMed.
- WHO Guidelines for Indoor Air Quality: Dampness and Mould, Geneva, 2009 Search PubMed.
- W. Eduard, Crit. Rev. Toxicol., 2009, 39, 799–864 CrossRef CAS PubMed.
- F. E. Rivera-Mariani and B. Bolanos-Rosero, Aerobiologia, 2012, 28, 83–97 CrossRef.
- B. G. Shelton, K. H. Kirkland, W. D. Flanders and G. K. Morris, Appl. Environ. Microbiol., 2002, 68, 1743–1753 CrossRef CAS PubMed.
- B. J. Green, E. R. Tovey, J. K. Sercombe, F. M. Blachere, D. H. Beezhold and D. Schmechel, Med. Mycol., 2006, 44, S245–S255 CrossRef CAS PubMed.
- C. A. Petti, Clin. Infect. Dis., 2007, 44, 1108–1114 CrossRef CAS PubMed.
- E. Bellemain, T. Carlsen, C. Brochmann, E. Coissac, P. Taberlet and H. Kauserud, BMC Microbiol., 2010, 10, 189 CrossRef PubMed.
- U. Koljalg, R. H. Nilsson, K. Abarenkov, L. Tedersoo, A. F. Taylor, M. Bahram, S. T. Bates, T. D. Bruns, J. Bengtsson-Palme, T. M. Callaghan, B. Douglas, T. Drenkhan, U. Eberhardt, M. Duenas, T. Grebenc, G. W. Griffith, M. Hartmann, P. M. Kirk, P. Kohout, E. Larsson, B. D. Lindahl, R. Lucking, M. P. Martin, P. B. Matheny, N. H. Nguyen, T. Niskanen, J. Oja, K. G. Peay, U. Peintner, M. Peterson, K. Poldmaa, L. Saag, I. Saar, A. Schussler, J. A. Scott, C. Senes, M. E. Smith, A. Suija, D. L. Taylor, M. T. Telleria, M. Weiss and K. H. Larsson, Mol. Ecol., 2013, 22, 5271–5277 CrossRef CAS PubMed.
- A. S. Amend, K. A. Seifert and T. D. Bruns, Mol. Ecol., 2010, 19, 5555–5565 CrossRef CAS PubMed.
- A. S. Amend, K. A. Seifert, R. Samson and T. D. Bruns, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13748–13753 CrossRef CAS PubMed.
- M. W. Nonnenmann, G. Coronado, B. Thompson, W. C. Griffith, J. D. Hanson, S. Vesper and E. M. Faustman, J. Environ. Monit., 2012, 14, 2038–2043 RSC.
- M. Pitkaranta, T. Meklin, A. Hyvarinen, A. Nevalainen, L. Paulin, P. Auvinen, U. Lignell and H. Rintala, BMC Microbiol., 2011, 11, 235 CrossRef CAS PubMed.
- M. Pitkaranta, T. Meklin, A. Hyvarinen, L. Paulin, P. Auvinen, A. Nevalainen and H. Rintala, Appl. Environ. Microbiol., 2008, 74, 233–244 CrossRef CAS PubMed.
- R. I. Adams, M. Miletto, J. W. Taylor and T. D. Bruns, ISME J., 2013, 7, 1262–1273 CrossRef CAS PubMed.
- N. Yamamoto, K. Bibby, J. Qian, D. Hospodsky, H. Rismani-Yazdi, W. W. Nazaroff and J. Peccia, ISME J., 2012, 6, 1801–1811 CrossRef CAS PubMed.
- K. C. Dannemiller, M. J. Mendell, J. M. Macher, K. Kumagai, A. Bradman, N. Holland, K. Harley, B. Eskenazi and J. Peccia, Indoor Air, 2013, DOI:10.1111/ina.12072.
- J. Frohlich-Nowoisky, D. A. Pickersgill, V. R. Despres and U. Poschl, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12814–12819 CrossRef PubMed.
- W. R. Rittenour, J. H. Park, J. M. Cox-Ganser, D. H. Beezhold and B. J. Green, J. Environ. Monit., 2012, 14, 766–774 RSC.
- L. Johnson, C. Ciaccio, C. S. Barnes, K. Kennedy, E. Forrest, L. C. Gard, F. Pacheco, P. Dowling and J. M. Portnoy, Allergy Asthma Proc., 2009, 30, 377–385 CrossRef PubMed.
- ASTM International, ASTM D7391 - 09 Standard Test Method for Categorization and Quantification of Airborne Fungal Structures in an Inertial Impaction Sample by Optical Microscopy, West Conshohocken, PA, 2009, vol. D7391–D7399 Search PubMed.
- C. S. Barnes, K. Kennedy, L. Gard, E. Forrest, L. Johnson, F. Pacheco, F. Hu, M. Amado and J. M. Portnoy, Allergy Asthma Proc., 2008, 29, 197–204 CrossRef PubMed.
- P. D. Schloss, S. L. Westcott, T. Ryabin, J. R. Hall, M. Hartmann, E. B. Hollister, R. A. Lesniewski, B. B. Oakley, D. H. Parks, C. J. Robinson, J. W. Sahl, B. Stres, G. G. Thallinger, D. J. Van Horn and C. F. Weber, Appl. Environ. Microbiol., 2009, 75, 7537–7541 CrossRef CAS PubMed.
- S. Vesper, C. Barnes, C. E. Ciaccio, A. Johanns, K. Kennedy, J. S. Murphy, A. Nunez-Alvarez, M. T. Sandel, D. Cox, G. Dewalt and P. J. Ashley, J. Asthma, 2013, 50, 155–161 CrossRef PubMed.
- S. Vesper, C. McKinstry, R. Haugland, L. Wymer, K. Bradham, P. Ashley, D. Cox, G. Dewalt and W. Friedman, J. Occup. Environ. Med., 2007, 49, 829–833 CrossRef PubMed.
- C. Barnes, J. Tuck, S. Simon, F. Pacheco, F. Hu and J. Portnoy, Ann. Allergy, Asthma, Immunol., 2001, 86, 517–523 CrossRef CAS.
- M. Saenz-de-Santamaria, I. Postigo, A. Gutierrez-Rodriguez, G. Cardona, J. A. Guisantes, J. Asturias and J. Martinez, Mycoses, 2006, 49, 91–95 CrossRef CAS PubMed.
- A. Szantho, P. Osvath, Z. Horvath, E. K. Novak and E. Kujalek, J. Invest. Allergol. Clin. Immunol., 1992, 2, 84–90 CAS.
- M. G. Harries, J. Lacey, R. D. Tee, G. R. Cayley and A. J. Taylor, Lancet, 1985, 1, 1063–1066 CrossRef CAS.
- L. A. Roure and J. M. Ramirez, Caribb. J. Sci., 1970, 10, 141–158 Search PubMed.
- M. Niedoszytko, M. Chelminska, E. Jassem and E. Czestochowska, Ann. Allergy, Asthma, Immunol., 2007, 98, 153–156 CrossRef.
- A. Koivikko, K. Kalimo, E. Nieminen, J. Savolainen, M. Viljanen and M. Viander, Allergy, 1988, 43, 192–200 CrossRef CAS PubMed.
- G. Reboux, R. Piarroux, F. Mauny, A. Madroszyk, L. Millon, K. Bardonnet and J. C. Dalphin, Am J Respir Crit Care Med, 2001, 163, 1534–1539 CrossRef CAS PubMed.
- V. Aizenberg, T. Reponen, S. A. Grinshpun and K. Willeke, AIHAJ, 2000, 61, 855–864 CAS.
- W. G. Lindsley, D. Schmechel and B. T. Chen, J. Environ. Monit., 2006, 8, 1136–1142 RSC.
- D. Benndorf, A. Muller, K. Bock, O. Manuwald, O. Herbarth and M. von Bergen, Allergy, 2008, 63, 454–460 CrossRef CAS PubMed.
- V. P. Kurup, Med. Mycol., 2005, 43(suppl 1), S189–S196 CrossRef CAS PubMed.
- J. Meng, C. S. Barnes and L. J. Rosenwasser, Clin. Exp. Allergy, 2012, 42, 1448–1458 CrossRef CAS PubMed.
- B. J. Green, T. Z. Mitakakis and E. R. Tovey, J. Allergy Clin. Immunol., 2003, 111, 285–289 CrossRef PubMed.
- J. A. Asturias, I. Ibarrola, A. Ferrer, C. Andreu, E. Lopez-Pascual, J. Quiralte, F. Florido and A. Martinez, J. Allergy Clin. Immunol., 2005, 115, 1210–1217 CrossRef CAS PubMed.
- M. Oliveira, L. Delgado, H. Ribeiro and I. Abreu, J. Environ. Monit., 2010, 12, 1187–1194 RSC.
- S. K. Gupta, B. M. Pereira and A. B. Singh, J. Invest. Allergol. Clin. Immunol., 2000, 10, 83–89 CAS.
- H. C. Siersted and S. Gravesen, Allergy, 1993, 48, 298–299 CrossRef CAS PubMed.
- F. W. Wittich and E. C. Stakman, J. Allergy, 1937, 8, 189–193 CrossRef.
- K. Yoshida, M. Suga, H. Yamasaki, K. Nakamura, T. Sato, M. Kakishima, J. A. Dosman and M. Ando, Thorax, 1996, 51, 650–651 CrossRef CAS PubMed.
- R. H. Nilsson, M. Ryberg, E. Kristiansson, K. Abarenkov, K. H. Larsson and U. Koljalg, PLoS One, 2006, 1, e59 Search PubMed.
- T. Reponen, J. Lockey, D. I. Bernstein, S. J. Vesper, L. Levin, G. K. K. Hershey, S. Zheng, P. Ryan, S. A. Grinshpun, M. Villareal and G. LeMasters, J. Allergy Clin. Immunol., 2012, 130, 639–644 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |