Assessment of environmentally persistent free radicals in soils and sediments from three Superfund sites†
Albert Leo N.
dela Cruz
*a,
Robert L.
Cook
a,
Barry
Dellinger
a,
Slawomir M.
Lomnicki
a,
Kirby C.
Donnelly
b,
Matthew A.
Kelley
b and
David
Cosgriff
c
aLouisiana State University, Department of Chemistry, 338 Choppin Hall, Baton Rouge, LA 70803, USA. E-mail: adelac2@lsu.edu; Fax: +1-225-5780276; Tel: +1-225-5789094
bSchool of Rural Public Health, Texas A&M University, College Station, TX 77843, USA
cArrowhead Engineering, 1504 Kaniksu, Libby, MT 5992, USA
First published on 7th November 2013
Abstract
We previously reported the presence of environmentally persistent free radicals (EPFRs) in pentachlorophenol (PCP) contaminated soils at a closed wood treatment facility site in Georgia. The reported EPFRs were pentachlorophenoxyl radicals formed on soils under ambient conditions via electron transfer from PCP to electron acceptors in the soil. In this study, we present results for soil and sediment samples from additional Superfund sites in Montana and Washington. Paramagnetic centers associated with different chemical environments were characterized by distinct g-factors and line widths (ΔHp-p). EPFR concentrations in contaminated samples were ∼30×, ∼12×, and ∼2× higher than background samples at the Georgia, Montana, and Washington sites, respectively. EPR signals in the Montana contaminated soils were very similar to those previously observed for pentachlorophenol contaminated soils at the Georgia site, i.e., g = 2.00300 and ΔHp-p = 6.0 G, whereas signals in the Washington sediment samples were similar to those previously observed for other PAH contaminated soils, i.e., g = 2.00270 and ΔHp-p = 9.0 G. Total carbon content measurements exhibited direct correlation with EPFR concentration. The presence of radicals in sites contaminated a decade to a century ago suggests continuous formation of EPFRs from molecular contaminants in the soil and sediment.
Environmental impactOur group discovered a new class of emerging pollutant, the Environmentally Persistent Free Radicals (EPFRs). We originally reported that EPFRs are only formed at post-combustion conditions. However, it is more ubiquitous than we thought when we discovered their presence at environmentally relevant conditions in pentachlorophenol-contaminated soils from a Superfund site. The novelty of this current research work is to further characterize the chemical identity of our initial findings from contaminated soils through assessment of soils from other Superfund sites contaminated with other hazardous materials. This study will broaden the existing knowledge of understanding of the current methods and knowledge of mitigating environmental and human health impact on soils polluted with hazardous organic compounds. |
Introduction
Epidemiological studies have reported linkages of exposure to hazardous waste contaminated soils with cardiopulmonary dysfunctions, birth defects, certain types of cancers, and other diseases.1–4 Soil is a vast reservoir of complex chemical structures (such as macromolecular species and weakly associated molecular assemblies (WAMs), clay/mineral and soil organic matter (SOM) components, etc.) and may sorb or accumulate anthropogenic organic contaminants.5,6 Soil properties, like acidity, reactive metals, and chemical functional residues such as sulfates, phenolates, enolates, carboxylates, quinones, peroxides, have all been proposed as the causative agents of observed human health impacts.7–12 Additionally, risk factors may arise from the reactions of contaminants during remediation, e.g. formation of dioxins13–18 and chlorinated diphenoquinones.19,20There is evidence in the literature of radical formation from anthropogenic hazardous substances. Aromatic (chlorophenoxyl-type) radical cation formation was reported upon adsorption of chlorinated phenols on copper(II)-smectite with simultaneous reduction of Cu(II) to Cu(I).13,14 Furthermore, we recently reported the detection of pentachlorophenoxyl environmentally persistent free radical (EPFR) in the soil at a Superfund site contaminated with pentachlorophenol (PCP) for over 25 years,21 concluding that EPFRs were formed within the available mixture of organic, inorganic and biological components via electron transfer to a soil substrate. This led us to consider that EPFRs may be more common than previously suspected or envisioned, especially at sites contaminated with hazardous wastes. This manuscript addresses the potential for EPFRs formation from other Superfund sites with soils or sediments contaminated by hazardous compounds such as PCP, polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs), and polybrominated biphenyl ethers (PBDEs). The motivation and importance of this work from a toxicological point of view are as follows: (i) recent works has shown that EPFRs may initiate oxidative stress in lungs22,23 and (ii) SOM in lung epithelial cells reduces antioxidant levels in the lining fluid and is postulated to directly produce hydroxyl radicals.24 From an environmental point of view, the findings of the presented work are of great relevance as it questions the long held belief that sorption of an organic pollutant is a method of mitigating its environmental impact as an ecosystem perturbant. It should be noted that the above discussion implies that SOM result in the production of ROS rather than scavenging them and thus making the finds of the presented work in terms of elevated EPFR levels in contaminated soil and sediment samples more troubling and of importance.
Results and discussion
Soil component analysis
The compositional analyses of the Georgia samples showed significant difference between the contaminated and non-contaminated samples (cf.Table 1). However, the differences in the samples from both the Washington and Montana sites were within experimental error. Physical and chemical analyses of the soils indicated that on average the contaminated soil or sediment from each site contained more total carbon (TC) than their non-contaminated counterpart, suggesting that additional organic matter (pollutants) are present in the contaminated samples (cf.Table 1). However, for the Washington samples the total carbon differs insignificantly. The sample for Washington was a sediment sample thus it is different than the soil samples. The relatively close results of total carbon between its contaminated and non-contaminated sample can be explained by a large amount of the carbon associated with the contamination being washed away. This also implies that the organic matter component that the EPFRs are associated with is less water-soluble and supports our previous finding that EPFRs are associated with the clay/humin fraction of soils.| Superfund location | Classification | % Moisture loss on drying | % Total carbon | % Ash | % Volatiles |
|---|---|---|---|---|---|
| a Sediments were dried prior to shipment to LSU. | |||||
| Georgia | Non-contaminated | 0.72 (±0.04) | 0.27 (±0.003) | 94.6 (±0.15) | 4.45 (±0.12) |
| Contaminated | 6.75 (±0.01) | 10.4 (±0.32) | 73.0 (±0.64) | 9.89 (±0.32) | |
| Montana | Non-contaminated | 0.25 (±0.15) | 1.49 (±1.17) | 95.5 (±2.63) | 2.71 (±1.29) |
| Contaminated | 0.31 (±0.18) | 2.40 (±0.80) | 93.2 (±1.71) | 4.14 (±0.79) | |
| Washington | Non-contaminated | 1.03 (±0.25)a | 1.49 (±0.47) | 92.6 (±1.94) | 4.91 (±1.22) |
| Contaminated | 1.47 (±0.39)a | 1.69 (±0.51) | 91.6 (±2.13) | 5.28 (±1.29) | |
The moisture content varied for each sample location; in general, the contaminated soils and sediments contained higher moisture concentrations. This is a potentially important parameter, indicating the hydration properties of a soil and suggesting their capability to retain an organic chemical pollutant. The ash content is a good approximation of the mineral content (e.g. clays and metals). All three sites yielded a lower amount of ash from the combustion of the contaminated soils than their non-contaminated counterparts. This is due to the higher organic content. The volatile components also exhibited higher concentrations in the contaminated soil. Each sample was also analyzed for metal content and the results are summarized in Table 2, and showed the presence of redox-active transition metals in all samples.
| Superfund location | Transition metals and heavy metal, ppm (mg kg−1 of sample) | Copper (Cu) | Iron (Fe) | Manganese (Mn) | Nickel (Ni) | Lead (Pb) |
|---|---|---|---|---|---|---|
| a Values obtained from U.S. EPA Reports for Montana Superfund site. (“Investigation to Further Characterize the Upper Aquifer in the Former Source Areas, Libby Groundwater Site, Libby, Montana, Revision 0, URS Corporation, Project Number.22252225, February 9, 2012”; Date accessed: September 24, 2013). b Values obtained from U.S. EPA Reports for Washington Superfund site. (http://www.epa.gov/region10/pdf/sites/ldw/fs13/final_fs_sections_1-3_1.3112.pdf and http://yosemite.epa.gov/R10/CLEANUP.NSF/LDW/Lower+Duwamish+Waterway+Superfund+Site+Technical+Documents#FS; Date accessed: September 24, 2013). | ||||||
| Georgia | Non-Contaminated | 8.56 ± 0.56 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 199.98 ± 1589.50 199.98 ± 1589.50 |
46.27 ± 1.89 | 17.67 ± 0.92 | 10.27 ± 0.59 |
| Contaminated | 58.84 ± 0.89 | 16265.93 ± 54.78 |
288.61 ± 13.28 | 16.64 ± 1.02 | 64.29 ± 1.29 | |
| Montana | Contaminated | 14.21a (Mean) | 14767.61a (Mean) |
379.16a (Mean) | 8.87a (Mean) | 11.75a (Mean) |
| Washington | Contaminated | 106.00b (Mean) | 23875.00b (Mean) |
340.00b (Mean) | 28.00b (Mean) | 139.00b (Mean) |
Detection of EPFRs
Detailed examinations of the soil and sediment samples revealed the presence of paramagnetic centers associated with various chemical environments, which were characterized, by a distinct magnetic field center (g-factors) and line widths (ΔHp-p) (cf.Fig. 1). Two different types of paramagnetic species were discerned: (i) a single line signal (very intense for the contaminated soils with a g-factor of 2.00270–2.00340), and (ii) a much weaker six-line signal with hyperfine splitting of 89:90:94:94:97 and 89:90:92:94:97. The latter signal was not detected in the Washington sediment samples (cf.Fig. 1). The six line signal is typical for manganese(II) ion with an [Ar] 3d5 configuration and nuclear spin I = 5/2,25 thus indicating the presence of manganese in the Georgia and Montana soils which was validated by the elemental analyses reported on Table 2.
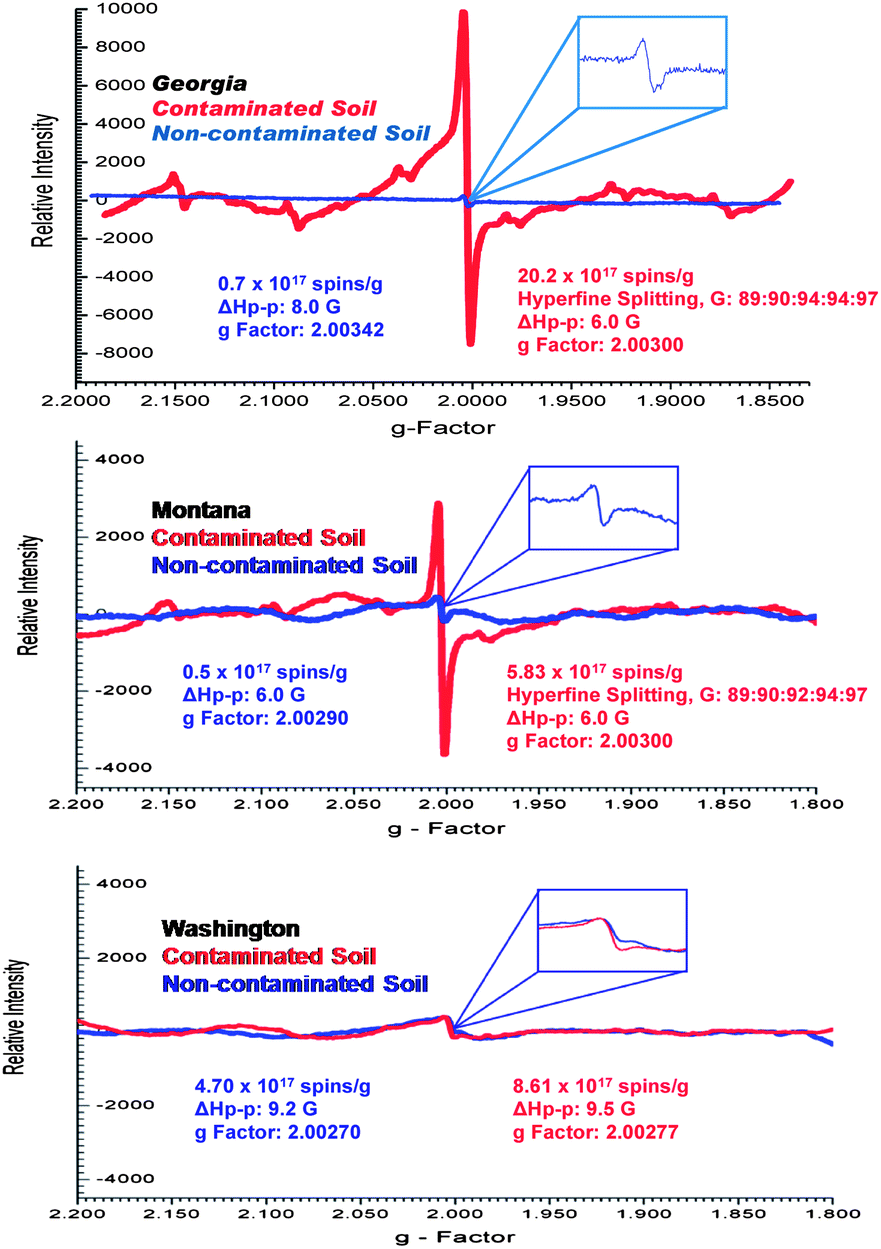 | ||
| Fig. 1 Example EPR spectra of the whole soils and sediments of contaminated (red) and non-contaminated (blue) locations collected from three different Superfund sites. | ||
For every contaminated sample, the most prominent EPR feature was the strong single line centered at ∼3430 Gauss, which was very weak in the non-contaminated samples. The spectral characteristics of this signal were as follows: g-factors of 2.00301–2.00307 and ΔHp-p of 5.8–6.2 Gauss for the Georgia samples, g-factors of 2.00275–2.00280 and ΔHp-p of 9.5–9.7 Gauss for the Washington samples, and g-factors of 2.00301–2.00304 and ΔHp-p of 6.1–6.3 Gauss for the Montana samples (cf.Fig. 1 and 2). In contrast, the non-contaminated/background soil samples exhibited a much weaker signal, a doublet signal for the Georgia soil (g-factors: ∼2.00340 and ΔHp-p: ∼8.0 Gauss), a singlet signal for the Montana soil (g-factors: ∼2.00290 and ΔHp-p: ∼6.0 Gauss), and a singlet signal for the Washington sediment (g-factors: ∼2.00270 and ΔHp-p: 9.0 Gauss). Such narrow lines with the g-factors between 2.003 and 2.008 are typical of the organic radicals.5,26–31 Based on the intensity of the observed radical signals relative to the mass of the sample, the radical concentration in the contaminated soils and sediments were ∼30×, ∼12×, and ∼2× higher than the non-contaminated counterpart at the Georgia, Montana, and Washington sites, respectively.
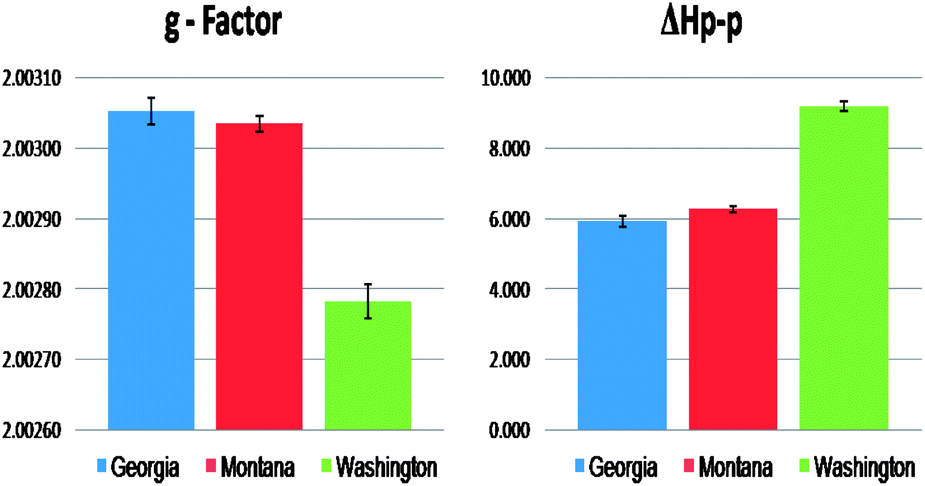 | ||
| Fig. 2 Comparison of the EPR signal parameters originating from the organic radicals for the three contaminated Superfund soil and sediment samples. | ||
The g-factor for the central singlet line for both the Georgia and Montana samples were ∼2.00300. Radical signals with g-factors of 2.003 and above are attributed to oxygen-centered radicals or carbon-centered radicals with a nearby heteroatom, such as oxygen or halogen that increases the spin–orbit coupling constant.5,26–31 Considering the similar source of the soil contamination for the Georgia and Montana sites, the increase in intensity of the radical signal for contaminated soils along with the similarities of the spectral parameters the single paramagnetic signal was attributed to pentachlorophenoxyl as a result of the PCP contamination history. The Washington sediment, which was contaminated primarily with PAHs, PCBs, and PBDEs, exhibited a weaker EPR signal with a g-factor of ∼2.00277, more typical to a carbon centered radical generated by PAH.32
The observed different g-factors between the soil and sediment resulted either from contaminant types or particle types. As can be seen from the EPR signal from the Washington sediments, the aromatic components of the pollutants, PAHs, PCBs, and PBDEs were selectively preserved in the sediment and thus resulted in an EPR signal with g-factors that is more carbon centered in nature. Additionally, the remaining native organic matter within the sediments is different than that found in soils, due to the potential washout of polar entities within the sediments' organic matter pool. Therefore resulting to a more carbon centered EPFRs in sediments. However, for the case of whole soils from Montana and Georgia sites, the major PCP contaminants were selectively preserved in the soil organic matter of the soil, and resulting in an EPR signal with higher g-factors that is more oxygen centered in nature. These effects of organic matter composition on the shifts of the g-factors concur with our previous study of pentachlorophenol EPFRs formation.31
The width (ΔHp-p) of the spectra also points to a different origin of the signal in the Georgia/Montana sites and the Washington site. The radical signals from Georgia and Montana samples were characterized by a narrow ΔHp-p of ∼6.0 G, whereas the signal in the Washington sample was broader, with a width of ΔHp-p = ∼9.0 G. The narrower spectrums for the Georgia and Montana samples are consistent with a single species. On the other hand, the broader Washington spectrum (ΔHp-p ∼ 9) is a convolution of multiple carbon-centered radicals from PAHs, PCBs, and PBDEs. The line broadening or narrowing of the EPR signal is a result of the sensitivity of the unpaired electrons towards its chemical environment. Spin exchange narrowing effect33 can contribute to the narrower EPR line width with soils dominantly contaminated with PCP. This effect is due to the higher heteroatom content that causes the delocalized unpaired electrons to undergo strong intermolecular interactions that slows down the relaxation processes involved.33 In contrast, the EPR signals for the Washington soil are broadened by association of the delocalized unpaired electrons with multiple functionalities present within the sample soil matrix, e.g. paramagnetic centers of aliphatic structures or smaller condensed aromatic structures. Association with the polyaromatic core causes disabling of electron delocalization which leads to increased electron spin–electron spin dipolar interactions resulting in additional line broadening of the EPR signals.8
Influence of pollutants on EPR spectra
The non-contaminated Georgia soil samples were analyzed and found to contain no PCP. In contrast, the Montana non-contaminated soil contained traces of PCP and the Washington non-contaminated sediment contained traces of PAHs, PCBs, and PBDEs. The concentration of PCP in the contaminated soil at the Georgia site was much greater than the Montana site, 200–5000 mg kg−1 of soil vs. 18–368 mg kg−1 of soil, respectively. Since the parameters of the radical signal in both the Georgia and Montana samples pointed to pentachlorophenol as a possible source of the radical species, it is rational that the radical concentration is dependent with the detected PCP content in the soils (cf.Fig. 3). However, when the two parameters (spins concentration and PCP concentration) from the data from Georgia and Montana samples are plotted, a non-linear plot is obtained that can be fitted with an exponential relations, (y = y0 + Aexp(−invTau*x)). The coefficients of determination of the non-linear correlation pertaining to the curves in Fig. 3 are 0.97 and 0.96 for Georgia and Montana contaminated soils, respectively. When the two data sets were combined (plot not shown), the coefficient of determination of the non-linear correlation was still high, viz. 0.85. The exponential fit values for Georgia samples are: y0 = 1.97 × 1018 (±2.12 × 1017), A = −1.72 × 1018 (±2.12 × 1017), and invTau = 9.31 × 10−5 (±3.61 × 10−5). The Montana samples, exponential fit values are: y0 = 1.38 × 1018 (±1.57 × 1017), A = −1.37 × 1018 (±1.16 × 1017), and invTau = 1.44 × 10−3 (±3.7 × 1037). Although more data points are desirable, this supports the common pollutant, PCP, in both soils was responsible for the observed EPR spectra. The observed non-linearity of the two parameters plotted in Fig. 3 is not surprising considering the complexity and heterogeneity of the whole soil and sediment system. However, the non-linear relationship can be explained as: the saturation of the active sites for EPFR formation in the environmental matrix is being approached, due to the very high concentration of the PCP contaminant, hence the plateauing of the lines in the plot. This saturation phenomenon is often observed in sorption studies of contaminants in soil matrix.
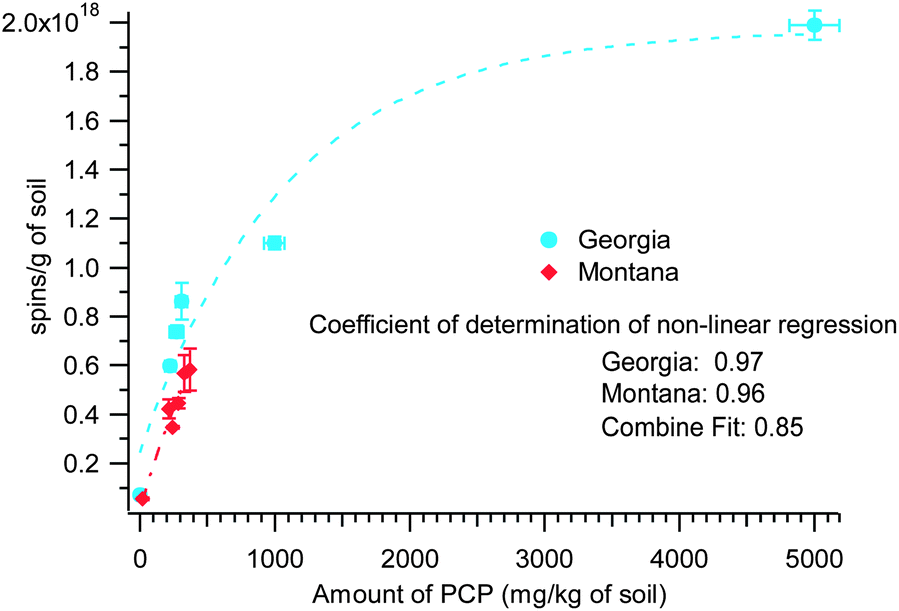 | ||
| Fig. 3 Non-linear regression of EPFR concentration and PCP concentration in contaminated Georgia and Montana soils. Coefficient of non-linear regression = 0.85 for the combined data. Fitted with exponential fit: y = y0 + Aexp(−invTau*x). | ||
For the Georgia site, in which we conducted our own sampling, a variation in EPFR concentration as a function of soil depth was observed (cf.Fig. 4). The middle depth soil (>10–20 cm) had the highest EPFR concentration, followed by the top layer soil (0–10 cm) and the bottom layer soil (>20–30 cm). We also correlated the variation of PCP concentration as a function of soil depth (data not shown) and again obtained strong correlations with R2 > 0.98. These results provided an overview of the PCP profile and the transport capability of PCP within the soil matrix. This further suggests and agrees with our previous report regarding the availability of PCP entrapped in the soil matrix to continuously form EPFRs.31
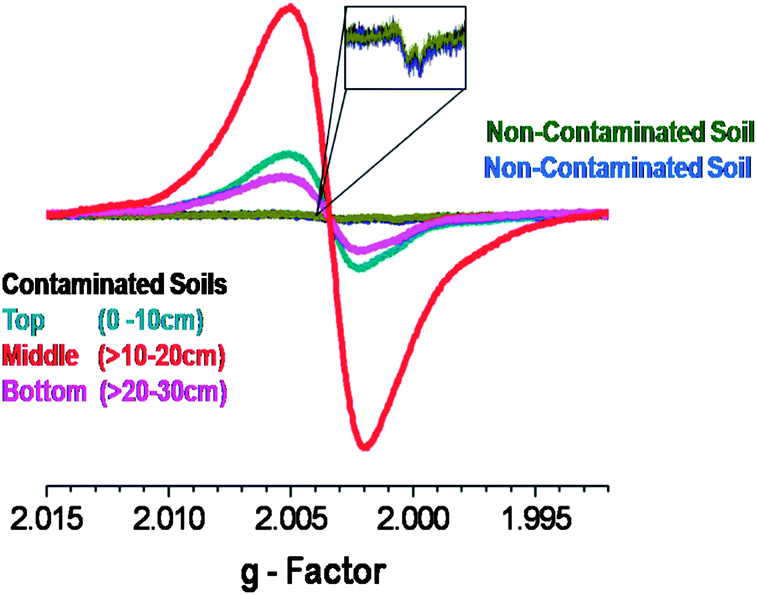 | ||
| Fig. 4 EPFR signal as a function of sampling depth for the Georgia Superfund site and comparison to EPFR concentration in non-contaminated soils. | ||
The concentration of major organic pollutants present in the soil also correlated with the amount of total carbon (plot not shown). The PCP concentration in the Georgia soil was 200–5000 mg kg−1 (ppm) of soil, with an average of ∼6.0% carbon content. In the Montana soil, PCP was the major contaminant but with a lower concentration of 18–368 mg kg−1 (ppm) of soil and correspondingly lower average carbon content of ∼2.0%. In the Washington sediment, there were multiple contaminants, with concentration ranges of: PAHs (1000–20000 μg kg−1 of soil (ppb)34), PCBs (60–3000 μg kg−1 of soil (ppb)34), and traces of PBDE (3–80 μg kg−1 of soil (ppb)34) resulted to an average total carbon content of ∼1.5%. Although it is not surprising, these results confirmed that the higher concentrations of the major organic pollutants present in soil and sediment corresponded to an elevation of the total carbon content of the soil.
The concentration of the EPFRs in the soils and sediments also correlated with the total carbon, using linear fitting, y = mx + b. As depicted in Fig. 5, there is a strong linear correlation between EPFR concentration and total carbon content, R2 = 0.9990, 0.93, and 0.85 for the Georgia, Montana, and Washington samples, respectively. The Georgia and Montana soils were both heavily contaminated with PCP and fell on the same trendline. When both data were combined in a single fit (plot not shown) a strong correlation of R2 = 0.98 was found. In contrast, the Washington sediment was primarily contaminated with PAHs, PCBs, and PBDEs, and followed a different trendline. The trends observed in Fig. 5 exhibited a faster rise in spins concentration for the Washington sediment than did the Georgia and Montana soils.
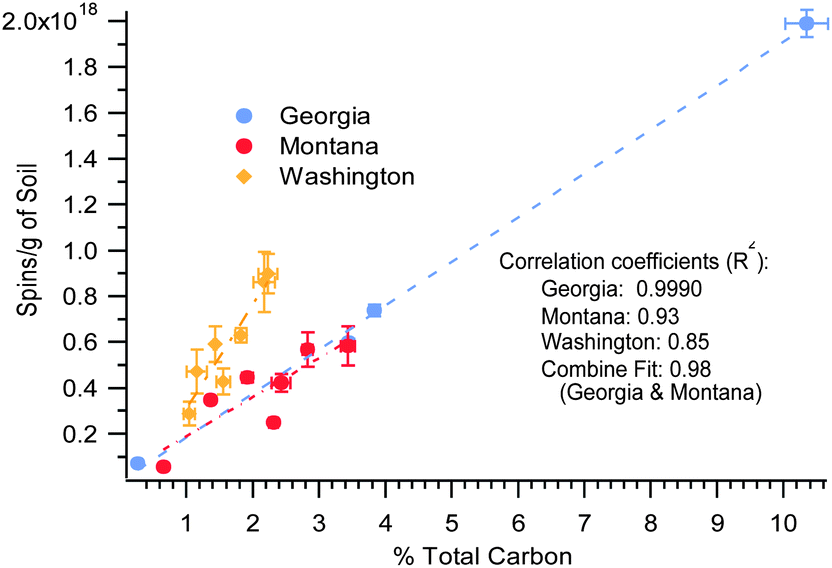 | ||
| Fig. 5 EPFR concentration versus % total carbon for the 3 Superfund. R2 = 0.98 for the combined data. | ||
The Washington sediment EPR signal can also be attributed from sources other than the contaminant. For example, the degradation processes of the adjacent aromatic sheets present in soil can cause separation of radicals and be responsible for a steeper enhancement of total spins concentration of the radicals.8 Additionally, large macromolecular/polyaromatic structures can undergo decomposition to radical-containing subunits. Considering that the contamination has been ongoing in the Washington sediments, these reactions and transformations may have occurred. In the case of PCP in Georgia and Montana, the pentachlorophenoxyl EPFR is in its most stable form and the only way for the spins concentration to increase is higher concentration availability of molecular PCP.
EPFRs formation
The formation of EPFRs in soils and sediments is initiated by the trapping of the organic molecular precursors in the soil or sediment matrix. Literature reports indicate contaminant sorption is facilitated by the three major soil components: inorganic (minerals – clay mineral phase and metals), organic (soil organic matter, SOM – humic acid, fulvic acid, and humin) and the biological components.6,35–37The inorganic fraction of soil, e.g. smectite clays, can easily undergo cation exchange and acquire transition metals sites akin to those previously shown to promote EPFR formation.13,14,17 Several studies have indicated sorption of phenols to Fe3+ and Cu2+ exchanged smectite clays that resulted in radical-cation formation by transferring an electron from the organic molecule to the metal and polymerization of the parent phenol.13,14,38 Other studies suggested organic radicals are formed when pentachlorophenol sorbs to boehmite – an aluminum oxide mineral.39 The humin organic fraction exhibited the highest radical concentration40 suggesting the radicals found in this fraction are stabilized by local effects, such as π stacking and hydrophobic associations.41 In addition, the biological component of soil, e.g., white rot fungi, may also produce radicals as it degrades various soil contaminants.42–44 Peroxidase and laccase enzymes are implicated in phenol degradation, yielding phenoxyl radicals in the presence of either hydrogen peroxide or oxygen, respectively.42–44
The presence of the transition metals and hazardous materials such as PCPs, PAHs, PCBs, or PBDEs in the contaminated soils may result in interaction between them, leading to the formation of the radicals as reported earlier for smectites, with the exception that our research indicated formation of a radical rather than a radical cation. We have previously demonstrated a mechanism of formation and stabilization of EPFRs by chemisorption and electron transfer of the molecular precursors to redox-active transition metals.32,45 In contrast, radical-cations are formed by electron-transfer from a physisorbed molecule to the soil substrate without prior chemisorption. We subsequently demonstrated that substituted aromatic species such as hydroquinone, catechol, chlorophenol, and chlorobenzenes, form EPFRs following exposure to Cu(II) and Fe(III) oxides at temperatures between 150 and 500 °C for less than 30 s, with half lives of up to 3 days in air.45–49 These reactions can also proceed at ambient temperature for soils, however, the reaction time is in years rather than in seconds.21
The pollutant to metal center electron transfer process is a plausible mechanism for EPFR formation in the Montana and the Washington soils and sediment, respectively. However, given the high organic content of the Georgia soil and because it will be covered by the organic carbon, a bare mineral surface may not be available. Soil organic matter has been reported to act as an electron conduit between the pollutant and the metal center; thus a direct contact of the pollutant to the metal center is not necessarily needed to facilitate the electron transfer process.50 Therefore, even when the mineral/clay surface is completely covered by organic matter, as can be assumed from the high organic content of the Georgia soil, the metal center can serve as the final electron sink. Partially catalyzed formation of radicals from PCP can also be facilitated by the metal center donating the acquired electron to the soil organic matter (SOM). The idea of SOM stabilizing the formed radical by local effects, such as π-stacking and hydrophobic associations, was supported by both mechanisms.51
Soils and sediments both contain the three main key components for the formation of EPFRs; the redox-active transition metal, the molecular precursor-organic pollutant, and the silica matrix support (for the case of soil and sediment: clay minerals can also be considered as a support matrix). Thus it is plausible to propose the same general mechanism of for soil and sediments, that involves the following three main steps: sorption of contaminant to a surface, followed by transfer of an electron from the sorbed contaminant to the redox-active transition metal center, and EPFRs formation.
Experimental
Site descriptions
Georgia. This Superfund site was a 4 acre wood treatment facility for railroad ties and poles from 1946–1991.52 Until the 1970's, the facility utilized creosote in the preservation process. PCP was added to the process in the 1980's and ultimately was used exclusively until the facility closed.52 In 1994, the EPA removed tanks containing 30723 gallons of PCP and creosote as part of the site remediation.52
Montana. Between 1946 and 1969, the site was operated as a wood treatment facility.52 Wood treating fluids such as creosote and PCP were disposed and spilled at the facility in several different locations. The first reports of contamination of domestic wells located within the vicinity of the facility were publicized in 1979, followed by EPA on-site investigation. In 1983 the site was included in the National Priorities List (NPL).52 In 1988, the EPA and the Montana Department of Environmental Quality (MDEQ) enacted final clean up methods for the affected media.52
Washington. The contaminated sediments were obtained from a 5.5 mile stretch of waterway that was added to National Priority List in 2001.52 The waterway was surrounded by industrial facilities and numerous sources of pollution. Industrial wastes from metal plating, slaughter houses, packing plants, carbide sludge, acid cleaning, caustic cleaning and spilled oil were discharged directly into the river, along with raw and treated sewage and storm water.52 Sediments present in the waterway contained a complex mixture of contaminants. Although recent source control efforts have substantially reduced contaminant inputs and some cleanup activities have been conducted on the waterway (led by the EPA and the Washington Department of Ecology), contamination of the waterway remains ongoing. The primary contaminants of concern in the waterway sediments include PCBs, PAHs, mercury and other metals, and phthalates.52
Soil sampling and preparations
The remediation technique that is common to the three Superfund sites is dredging and the placement of clean sand/gravel or capping.52 We do not believe that any remediation took place at the exact sampling locations of the soil and sediment samples; however, they did conduct some ground water treatment in the area for Georgia and Washington sites.52 For the Montana site, we know they use a bioreactor to clean up groundwater and used enzymes and nutrients for the land treatment units.52Georgia. The contaminated soils were randomly collected inside the perimeter of the once-standing complex at nine different locations (cf.Map 153). At each location, soil samples were collected from three different depths: top (0–10 cm), middle (>10–20 cm), and bottom (>20–30 cm). Background/non-contaminated soil samples were collected 152 m outside the contaminated area. To prevent outside contamination and safe transport back to the laboratory, all samples were placed in sealable plastic bags. The soil samples were dried in an oven for 12 h at 55 °C to remove water prior to chemical analyses and ground to a homogeneous powder and sieved through a USA Standard Testing Sieve No. 120 (125 μm opening) to eliminate any coarse-sized mineral and vegetative matter. Soil samples prepared in this way were referred to as the whole soil (WS).
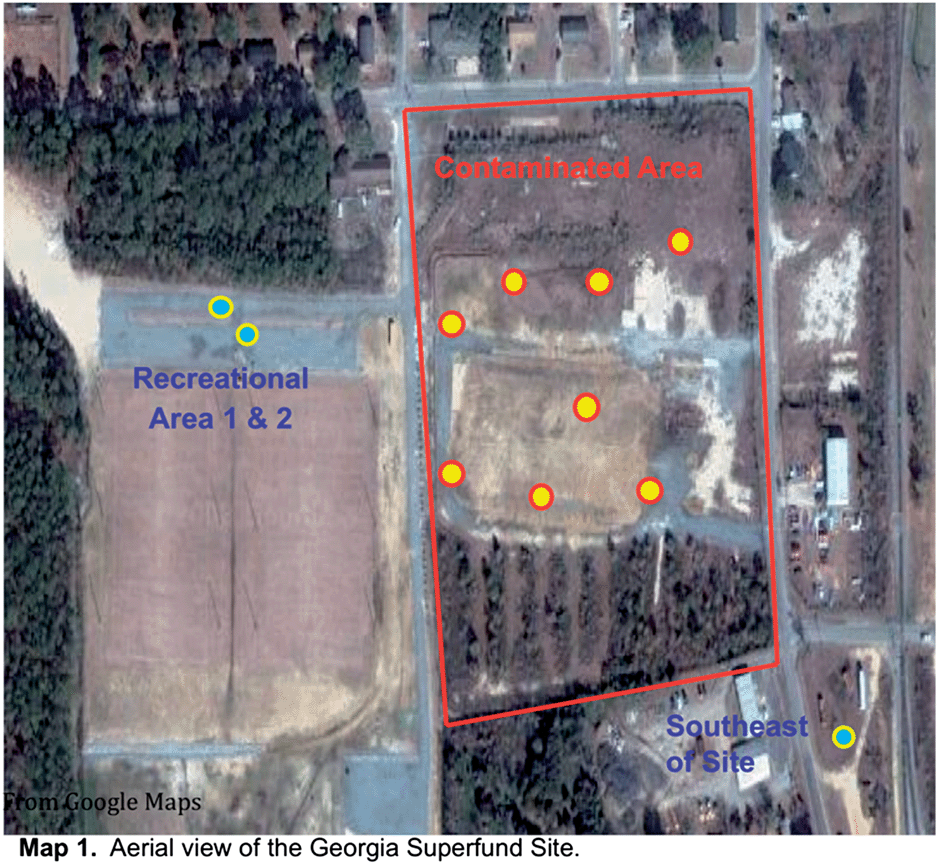 | ||
| Scheme 1 Aerial view of the Georgia Superfund site. | ||
Montana and Washington. The contaminated soils and sediments were collected and provided by collaborators from Texas A&M University, College Station TX. The soil samples were collected in 2005 and 2006 for the Montana site, and the sediment samples were collected in 2007 and 2008 for the Washington site.34 All samples were placed in sealable vials to prevent contamination and assure preservation until further use. The samples were dried and sieved identically to the Georgia soil samples.
Background samples
For this study the samples were collected as contaminated and non-contaminated whole soils/sediments in the same geographical location (for each Superfund sites). Contaminated samples were sampled inside the fence of the area of contamination and the non-contaminated were sampled just outside the fence of area of contamination. We rationally treated the non-contaminated samples as the pristine and unadulterated form of which the contaminated samples originally was prior to the contamination and was designated in this study as the blank or background samples with respect to the contaminated samples. Any difference between the blanks/backgrounds and the contaminated samples was deemed to be due to the contamination.Soil physical and chemical analyses
The contaminated and non-contaminated soil and sediment samples for each site were analyzed in four replicates for moisture, total carbon, and percent ash (includes the incombustible content or residue).54Moisture content (loss on drying). A 500 mg sample of whole soil/sediments was heated for 15 h at 100–110 °C and cooled in a dessicator prior to weighing.
Total carbon analysis (combustion–oxidation reaction). A 500 mg sample of whole soil/sediments was placed in a previously baked and cleaned sample boat. Samples were combusted in a Shimadzu model SSM-5000A furnace in conjunction with total carbon check analyzer under UHP oxygen flow at 900 °C, a temperature sufficient to completely oxidize most organic species. The calibration standards were prepared by weighing 5, 15, and 30 mg of glucose evenly spread out in the bottom of a previously baked and cleaned sample boat. Each standard and the sample were analyzed for a total run time of 7 min.
Percent ash. The samples from the total carbon analysis were cooled and weighed. % Ash = [(weight after combustion)/(weight before combustion)] × 100
Percent volatile. % Volatile = (100 − % total carbon − % moisture − % ash)
Elemental analysis by Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES). Briefly, the contaminated and non-contaminated samples from Georgia were digested with hot ICP grade nitric acid and were analyzed for the determination of selected redox-active transition metals and some heavy metals via Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES) spectrophotometer. We did not perform elemental analysis for the Montana and Washington soils due to insufficient amount available.
Electron paramagnetic resonance (EPR) spectroscopic analyses
The soil and sediment samples in four replicates were placed in suprasil EPR tubes and EPR spectra were collected at room temperature using a Bruker EMX – 10/2.7 EPR Spectrometer with X-band microwave frequency of 9.72 GHz, microwave power of 2.02 mW, spectral window of 1000 Gauss, and modulation amplitude of 4.00 Gauss.GC-MS analysis of pentachlorophenol (PCP)
The method of analysis was taken from our previous research.21 In summary, a 200–250 mg sample of whole soil/sediments in four replicates was placed in a scintillation vial, and 10 mL of 4-methyl-2-pentanone was added as the extracting solvent. After extraction, a 250 μL aliquot of the extracting solvent was placed in an amber vial, to which 250 μL of derivatizing agent, N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA), and 500 μL of extracting solvent, tert-butylmethyl ether (TBME) was added, making up a total volume of 1000 μL. The vial was capped using Teflon/silicone 11 mm crimp caps and shaken. The vial was then placed in a pre-heated heating block for 30 minutes at 76 °C (±5 °C), and subsequently cooled to room temperature for GC-MS analysis. Sample solutions were verified to contain pentachlorophenol concentration that fell within the range of the calibration curve.An Agilent 6890 Gas Chromatograph (GC) fitted with a 5973 Mass Selective Detector (MSD) in manual injection mode was used with the following parameters: column type – J&W DB5 MS 60 m × 0.25 mm i.d. × 0.25 μm, preceded by 5 m of 0.25 mm deactivated retention gap; splitless injection/250 °C; column temperature program – initial 60 °C hold for 6 minutes, ramp 10 °C min−1 to 180 °C, 15 °C min−1 to 300 °C, hold for 2 minutes; total run time – 28.0 minutes; carrier gas – Helium; transfer line temperature – 280 °C; injection volume – 1 μL; column flow – 1 μL min−1 (constant flow); solvent delay – 14 minutes; MS source temperature – 230 °C; MS quadrupole temperature – 150 °C; MS mode – SIM; and ion dwell time – 100 ms.
Conclusions
The results presented here support our previous report on detection of pentachlorophenoxyl radical from the Georgia Superfund site. The detection of EPFRs at elevated concentrations in an additional contaminated soil and sediment relative to their non-contaminated counterparts suggests EPFRs are not confined to combustion-generated particulate matter (PM) and are more widely present in the environment than originally suspected. The presence of these radicals in sites contaminated more than 10 years ago suggests the molecular precursors may be continually forming EPFRs and can have dramatic consequences to human health. Contaminated soils containing EPFRs can become airborne as dust leading to the inhalation exposure. It has been shown that humans exposure to agricultural dust result in adverse health effects.55 In the same way, it can be envisioned that upon inhalation and deposition of EPFRs containing soil dust will result in ROS generation based on the health effects data on EPFRs of 2-monochlorophenol and 1,2-dichlorobenzene,22,23,56–59 it is reasonable to believe that EPFRs associated with soils and sediments will also have significant impact on human health. Recognition of the presented findings, are of great relevance as they, question the long held belief that sorption of an organic pollutant is a method of mitigating its environmental impact. Thus, techniques for detecting, monitoring, and evaluating the health and environmental ramifications of EPFRs in soils and sediments should be of great interest. From a more environmental perspective this study also shows that more research is needed into the role of the inorganic, organic, and biological components of the soils and sediments in the formation of EPFRs, as well as their combined or synergistic interactions, in order to expand the previously proposed general mechanism of formation of EPFRs45–49 to geomatrices.Acknowledgements
Support for this research was provided by the NIEHS Superfund Research Program for LSU through grants 2P42ES013648-03, 3P42ES013648-02S1, 5P42ES013648-02, and for Texas A&M through grant P42ES04917; and the LSU Patrick F. Taylor Chair. We also thank Mr Scott Miller of EPA for providing access to the site in Georgia, and we acknowledge Mr Tom Ross of International Paper for access to the Montana site. The authors also wish to show their appreciation to members of US EPA, Region 10, including but not limited to Bruce Duncan, the EPA Region 10 Dive Team, and staff at the US EPA Manchester Lab. For the technical support and helpful comments on soil proximate analysis, we thank Dr Charisma Lattao.Notes and references
- D. J. Paustenbach, B. L. Finley and T. F. Long, Int. J. Toxicol., 1997, 16, 339–362 CrossRef CAS.
- J. K. Hawley, Risk Anal., 1985, 5(4), 289–301 CrossRef CAS.
- M. Vrijheid, Environ. Health Perspect., 2000, 108, 101–112 Search PubMed.
- N. J. Simcox, R. A. Fenske, S. A. Wolz, I.-C. Lee and D. A. Kalman, Environ. Health Perspect., 1995, 103(12), 1126–1134 CrossRef CAS.
- N. Senesi and E. Loffredo, The chemistry of soil organic matter, in Soil Physical Chemistry, ed. D. L. Sparks, CRC Press, Boca Raton, 1999, pp. 242–345 Search PubMed.
- K. M. Spark and R. S. Swift, Sci. Total Environ., 2002, 298, 147–161 CrossRef CAS.
- R. Sutton and G. Sposito, Environ. Sci. Technol., 2005, 39, 9009–9015 CrossRef CAS.
- F. Czechowski and A. Jezierski, Energy Fuels, 1997, 11, 951–964 CrossRef CAS.
- P. P. Falciglia, M. G. Giustra and F. G. A. Vagliasindi, J. Hazard. Mater., 2011, 185, 392–400 CrossRef CAS PubMed.
- C. T. Chiou, S. E. McGroddy and D. E. Kile, Environ. Sci. Technol., 1998, 32, 264–269 CrossRef CAS.
- W. J. J. Weber and W. Huang, Environ. Sci. Technol., 1996, 30, 881–888 CrossRef CAS.
- J. J. Pignatello and B. Xing, Environ. Sci. Technol., 1996, 30, 1–11 CrossRef CAS.
- S. A. Boyd and M. M. Mortland, Nature, 1985, 316, 532–535 CrossRef CAS.
- S. A. Boyd and M. M. Mortland, Environ. Sci. Technol., 1986, 20, 1056–1058 CrossRef CAS PubMed.
- K. Rana, S. A. Boyd, B. J. Teppen, H. Li, C. Liu and C. T. Johnston, Phys. Chem. Chem. Phys., 2009, 11, 2976–2985 RSC.
- C. Liu, H. Li, B. J. Teppen, C. T. Johnston and S. A. Boyd, Environ. Sci. Technol., 2009, 43, 2777–2783 CrossRef CAS.
- S. A. Boyd, S. Shaobai, J.-F. Lee and M. M. Mortland, Clays Clay Miner., 1988, 36(2), 125–130 CAS.
- N. Govindraj, M. M. Mortland and S. A. Boyd, Environ. Sci. Technol., 1987, 21, 1119–1123 CrossRef.
- F. Otto, G. Leupold, H. Parlar, R. Rosemann, M. Bahadir and H. Hopf, Anal. Chem., 1998, 70(14), 2831–2838 CrossRef CAS.
- Y. Dudal, A. R. Jacobson, R. Samson and L. Deschenes, Water Res., 2004, 38, 3147–3154 CrossRef CAS PubMed.
- A. L. N. dela Cruz, W. Gehling, S. Lomnicki, R. Cook and B. Dellinger, Environ. Sci. Technol., 2011, 45(15), 6356–6365 CrossRef CAS PubMed.
- S. Balakrishna, S. Lomnicki, K. M. McAvey, R. B. Cole, B. Dellinger and S. Cormier, Part. Fibre Toxicol., 2009, 6(11), 1–14 Search PubMed.
- L. Khatchatryan, E. Vejerano, S. Lomnicki and B. Dellinger, Environ. Sci. Technol., 2011, 45(19), 8559–8566 CrossRef PubMed.
- S. Qi, G. J. M. den Hartog and A. Bast, Environ. Toxicol. Pharmacol., 2008, 26, 96–101 CrossRef CAS PubMed.
- K. Flogeac, E. Guillon and M. Aplincourt, J. Colloid Interface Sci., 2005, 286, 596–601 CrossRef CAS PubMed.
- A. Jezierski, G. Skrzypek, P. Jezierski, D. Paul and M. O. Jedrysek, Spectrochim. Acta, Part A, 2008, 69(5), 1311–1316 CrossRef PubMed.
- K. Polewski, D. Slawinska, J. Slawinski and A. Pawlak, Geoderma, 2005, 126, 291–299 CrossRef CAS PubMed.
- A. Jezierski, F. Czechowski, M. Jerzykiewicz, Y. Chen and J. Drozd, Spectrochim. Acta, Part A, 2000, 56(2), 379–385 CrossRef.
- K. C. Christofiridis, S. Un and Y. Deligiannakis, J. Phys. Chem. A, 2007, 111(46), 11860–11866 CrossRef PubMed.
- A. Jezierski, F. Czechowski, M. Jerzykiewicz, I. Golonka, J. Drozd, E. Bylinska, Y. Chen and M. R. D. Seaward, Spectrochim. Acta, Part A, 2002, 58(6), 1293–1300 CrossRef.
- A. L. N. dela Cruz, R. Cook, S. Lomnicki and B. Dellinger, Environ. Sci. Technol., 2012, 46(11), 5971–5978 CrossRef PubMed.
- S. Lomnicki and B. Dellinger, J. Phys. Chem. A, 2003, 107, 4387–4395 CrossRef CAS.
- J. E. Wertz and J. R. Bolton, Electron Spin Resonance: Elementary Theory and Practical Applications, McGraw Hill, New York, 1972, pp. 192–222 Search PubMed.
- M. A. Kelley, Ph.D. Dissertation, Texas A&M University, College Station, Texas, USA, 2010.
- A. K. Pandey, S. D. Pandey, V. Misra and P. N. Viswanathan, Sci. Total Environ., 1999, 231, 125–133 CrossRef CAS.
- S. Fingler, V. Drevenkar and Z. Frobe, Arch. Environ. Contam. Toxicol., 2005, 48, 32–39 CrossRef CAS PubMed.
- R. Calvet, Environ. Health Perspect., 1989, 83, 145–177 CrossRef CAS.
- B. L. Sawhney, Clays Clay Miner., 1985, 33, 123–127 CAS.
- K. H. S. Kung and M. B. McBride, Environ. Sci. Technol., 1991, 25, 702–709 CrossRef CAS.
- S. C. Saab and L. Martin-Neto, J. Braz. Chem. Soc., 2004, 15(1), 34–37 CrossRef CAS PubMed.
- E. Giannakopoulos, M. Drosos and Y. Deligiannakis, J. Colloid Interface Sci., 2009, 336, 59–66 CrossRef CAS PubMed.
- J. A. Field and R. Sierra-Alvarez, Rev. Environ. Sci. Biotechnol., 2008, 7, 211–241 CrossRef CAS.
- D. P. Barr and S. D. Aust, Environ. Sci. Technol., 1994, 28, 78A–87A CAS.
- M. L. Rabinovich, A. V. Bolobova and L. G. Vasil'chenko, Appl. Biochem. Microbiol., 2004, 40, 1–17 CrossRef CAS.
- S. Lomnicki, H. Truong, E. Vejerano and B. Dellinger, Environ. Sci. Technol., 2008, 42, 4982–4988 CrossRef CAS.
- S. A. Cormier, S. Lomnicki, W. Backes and D. Dellinger, Environ. Health Perspect., 2006, 114(6), 810–817 CrossRef CAS.
- B. Dellinger, S. Lomnicki, L. Khatchatryan, Z. Maskos, R. W. Hall, J. Adounkpe, C. McFerrin and H. Truong, Proc. Combust. Inst., 2007, 31, 521–528 CrossRef PubMed.
- B. Dellinger, W. Pryor, R. Cueto, G. L. Squadrito, V. Hedge and W. A. Deutsch, Chem. Res. Toxicol., 2001, 14, 1371–1377 CrossRef CAS PubMed.
- B. Dellinger, S. Lomnicki, W. Pryor, R. Cueto, G. L. Squadrito and W. A. Deutsch, Proc. Combust. Inst., 2000, 28, 2675–2681 CrossRef CAS.
- M. Aeschbacher, M. Sanders and R. P. Schwarzenbach, Environ. Sci. Technol., 2010, 44, 87–93 CrossRef CAS PubMed.
- J. D. Coates, K. A. Cole, R. Chakraborty, S. M. O'Connor and L. A. Achenbach, Appl. Environ. Microbiol., 2002, 68, 2445–2452 CrossRef CAS.
- Superfund, United States Environmental Protection Agency (EPA) – Official Website, http:///www.epa.gov/superfund/, accessed 18 June 2010.
- GoogleMaps, Georgia Superfund Site, (2010), http://maps.google.com, accessed 18 June 2010.
- B. A. Schumacher, Methods for the determination of total organic carbon (TOC) in soils and sediments, Ecological Risk Assessment Support Center, 2002, pp. 1–23 Search PubMed.
- M. B. Schenker, K. E. Pinkerton, D. Mitchell, V. Vallyathan, B. Elvine-Kreis and F. H. Y. Green, Environ. Health Perspect., 2009, 117(6), 988–994 CrossRef PubMed.
- S. Mahne, G. C. Chuang, E. A. Pankey, L. Kiruri, P. J. Kadowitz, B. Dellinger and K. Varner, Am. J. Physiol.: Heart Circ. Physiol., 2012, 303(9), H1135–H1142 CrossRef CAS PubMed.
- P. T. Thevenot, J. Saravia, N. Jin, J. D. Giaimo, R. E. Chustz, S. Mahne, M. A. Kelley, V. Y. Hebert, B. Dellinger, T. Dugas, F. J. DeMayo and S. Cormier, Am. J. Respir. Cell Mol. Biol., 2013, 48(2), 188–197 CrossRef CAS PubMed.
- J. Saravia, G. I. Lee, S. Lomnicki, B. Dellinger and S. Cormier, J. Biochem. Mol. Toxicol., 2013, 27(1), 56–68 CrossRef CAS PubMed.
- S. Balakrishna, J. Saravia, P. T. Thevenot, T. Ahlert, S. Lomnicki, B. Dellinger and S. Cormier, Part. Fibre Toxicol., 2011, 8(11), 1–12 Search PubMed.
Footnote |
| † This manuscript is dedicated In memoriam to the late Prof. Kirby C. Donnelly, Ph.D., from the School of Rural Public Health, Texas A&M University: College Station, Texas, USA. |
| This journal is © The Royal Society of Chemistry 2014 |