Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation
Majid
Saidi
a,
Fereshteh
Samimi
a,
Dornaz
Karimipourfard
a,
Tarit
Nimmanwudipong
bc,
Bruce C.
Gates
*b and
Mohammad Reza
Rahimpour
*ab
aDepartment of Chemical Engineering, School of Chemical and Petroleum Engineering, Shiraz University, Shiraz 71345, Iran. E-mail: rahimpor@shirazu.ac.ir
bDepartment of Chemical Engineering and Materials Science, University of California, Davis, Davis CA 95616, USA. E-mail: bcgates@ucdavis.edu
cDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 11th November 2013
Abstract
The incentive for use of renewable resources to replace fossil sources is motivating extensive research on new and alternative fuels derived from biomass. Bio-oils derived from cellulosic biomass offer the prospect of becoming a major feedstock for production of fuels and chemicals, and lignin is a plentiful, underutilized component of cellulosic biomass. Lignin conversion requires depolymerization and removal of oxygen. Likely processes for lignin conversion involve depolymerization (e.g., by pyrolysis) and catalytic upgrading of the resultant bio-oils. A major goal of the upgrading is catalytic hydrodeoxygenation (HDO), which involves reactions with hydrogen that produce hydrocarbons and water. The aim of this review is to present a critical introduction to HDO chemistry focused on compounds derived from lignin, including a summary of HDO reactions and those that accompany them, with a comparison of catalysts addressing their activities, selectivities, and stabilities. The reactions are evaluated in terms of reaction pathways of compounds representative of lignin-derived bio-oils, including anisole, guaiacol, and phenol. The review includes recommendations for further research and an attempt to place HDO in a context of options for renewable fuels and chemicals, but it does not provide an economic assessment.
 Majid Saidi | Majid Saidi is working toward his PhD at Shiraz University in the research group of Professor Mohammad Reza Rahimpour. He holds a Bachelor of Science in HSE Engineering (2008) from the Petroleum University of Technology, Iran, and a Masters of Science in Chemical engineering (2011) from the University of Tehran. His undergraduate thesis was about the effects of heavy metals on biological wastewater treatment, and his graduate thesis is about optimization of continuous catalytic regeneration using a bee colony algorithm. His research interests include process modeling and optimization, chemical reaction engineering, catalytic upgrading of bio-oils, and environmental science. He wrote the book Heavy Metals Effects on Biological Wastewater Treatment. |
 Fereshteh Samimi | Fereshteh Samimi is a PhD student of the Department of Chemical Engineering, School of Chemical and Petroleum Engineering, Shiraz University. She received her Bachelor of Science in Chemical Engineering in 2011, working on the crystallization of calcium oxalate under the influence of potassium citrate. Her current research focuses on the development of ultraclean fuels, renewable energy, and process engineering; she is a member of Professor Mohammad Reza Rahimpour's research group. |
 Dornaz Karimipourfard | Dornaz Karimipourfard is a PhD student of the Department of Chemical Engineering, School of Chemical and Petroleum Engineering, Shiraz University. She completed her Bachelor of Science in Chemical Engineering in 2011, working on an investigation of various methods of soil decontamination. She now works on synthesis gas production via reforming reactions in Professor Mohammad Reza Rahimpour's research group. |
 Tarit Nimmanwudipong | Tarit Nimmanwudipong is a postdoctoral associate in the Department of Chemical Engineering at the Massachusetts Institute of Technology, working in the research group of Professor Yuriy Román-Leshkov. Working under the mentorship of Bruce C. Gates and David E. Block, he obtained his M.S. and Ph.D. degrees in chemical engineering from the University of California, Davis (2012), focusing on heterogeneous catalysis with a designated emphasis in biotechnology. He investigated reaction networks of compounds derived from lignocellulosic biomass and bio-oils upgrading processes. He obtained his B. Eng. (2006) from Chulalongkorn University, Thailand, and received a Fulbright Open Competition Scholarship (2008) to pursue his graduate studies. His research interests include renewable energy, biomass conversion, catalytic materials synthesis, and chemical reaction engineering. |
 Bruce C. Gates | Bruce C. Gates teaches in the Department of Chemical Engineering and Materials Science at the University of California, Davis. His research group is active in catalysis, with a focus on supported metal complex and metal cluster catalysts that are essentially molecular and lend themselves to design and in-depth characterization by spectroscopy and atomic-resolution microscopy. The group also works on catalysts for biomass conversion. Gates is a co-author of the textbook Chemistry of Catalytic Processes and author of Catalytic Chemistry, and he edits Advances in Catalysis. He serves on DOE's Basic Energy Sciences Advisory Committee and the North American Catalysis Society's Board of Directors. |
 Mohammad Reza Rahimpour | Mohammad Reza Rahimpour is a professor in the Department of Chemical Engineering at Shiraz University. He has spent sabbatical leaves at the University of Sydney and the University of Newcastle (Australia). His research group is active in fuel processing technology, with a focus on catalytic conversion of natural gas, petroleum, and bio-oils to fuels. His group also works on catalysts for chemical looping. He has authored more than 200 peer-reviewed papers and more than 100 conference presentations. He is a visiting Professor in the Department of Chemical Engineering and Materials Science at the University of California, Davis, working in the group of Professor Bruce C. Gates, where they are engaged in research on the catalytic conversion of compounds found in bio-oils derived from lignin. |
Broader contextThe goal of replacing fossil fuels has focused attention on lignocellulosic biomass as a renewable source—and especially on lignin, a major, underutilized component of biomass. Lignin is highly aromatic, with a polymeric structure characterized by ether linkages and hydroxy and methoxy groups. Prospective processes for converting lignin to fuels involve pyrolysis to produce bio-oils and subsequent refining to give fuels. The bio-oils themselves are poor fuels; their high oxygen contents make them poorly compatible with today's infrastructure for hydrocarbon fuels. Consequently, extensive effort has been devoted recently to methods for converting lignin-derived bio-oils to remove the oxygen, typically as water, methanol, CO, or CO2. The best conversions in prospect are catalytic, with H2 as a co-reactant. Catalysts such as noble metals (e.g., platinum) and metal sulfides (e.g., molybdenum sulfide) catalyze the reactions of compounds in bio-oils (typified by phenol, anisole, and guaiacol) to give water and hydrocarbons. However, numerous reactions occur, not all of them leading to oxygen removal, and much work remains to identify optimal catalysts that are active, stable, and selective for producing good fuel components without consuming too much expensive H2. The available data provide a good starting point for future research by indicating what catalyst components and what reaction conditions favor the production of desired products, such as hydrocarbons, but extensive work is needed. |
Introduction
To begin to meet the needs for renewable fuels, many researchers have turned their attention recently to a massive resource that today finds few applications: lignin.1 Fast pyrolysis of biomass, a process that is relatively well developed, gives products called bio-oils.2 Bio-oils derived from lignin are poor fuels, being unstable and having undesirable physical properties, and so they require upgrading. Fig. 1 illustrates a candidate biomass-to-biofuel conversion cycle. A central goal of the upgrading is to convert the oxygen-rich, high-molecular-weight lignin into hydrocarbons that are compatible with today's petroleum-derived fuels. Thus, a potentially valuable processing goal is to convert lignin to bio-oils and to subject the bio-oils to hydrodeoxygenation (HDO), a chemical conversion that takes place at high H2 partial pressures (as high as 100–200 bar) and high temperatures (570–670 K) to remove oxygen primarily in the form of water. | ||
| Fig. 1 Renewable fuels from biomass: simplified representation of conversion cycle. | ||
HDO of bio-oils gives hydrocarbons and oxygen-containing organic compounds in addition to water, and the classes of reactions include decarboxylation, hydrogenation, hydrogenolysis, hydrocracking, and dehydration (Fig. 2). Oxygen-containing products, in addition to water, include CO2, formed in decarboxylation reactions, CO, and methanol. Like bio-oils themselves, the organic products of HDO of bio-oils still include oxygen-containing compounds unlike the hydrocarbons in fossil fuels, and researchers are working both on producing hydrocarbon products from bio-oil conversion and on opportunities for blending bio-oils that contain oxygen-containing compounds with fossil fuels for use in an infrastructure like today's.
 | ||
| Fig. 2 Reactions that may take place in a hydrodeoxygenation process. | ||
Here we review recent work on the catalytic HDO of bio-oils derived from lignin, first presenting short summaries of the biomass feedstocks and their conversion into bio-oils. Interest in this subject has been growing rapidly, as shown (Fig. 3) by the frequency of publications and the number of reviews that have appeared. Consequently, we summarize the earlier work only briefly and focus instead on recent work. In the following sections, we summarize information about biomass, lignin-derived bio-oils, and processes for bio-oils upgrading, then focusing in more detail on the reactions and reaction networks characterizing HDO of compounds in bio-oils and on potential bio-oils HDO processes. We conclude with a statement of research opportunities.
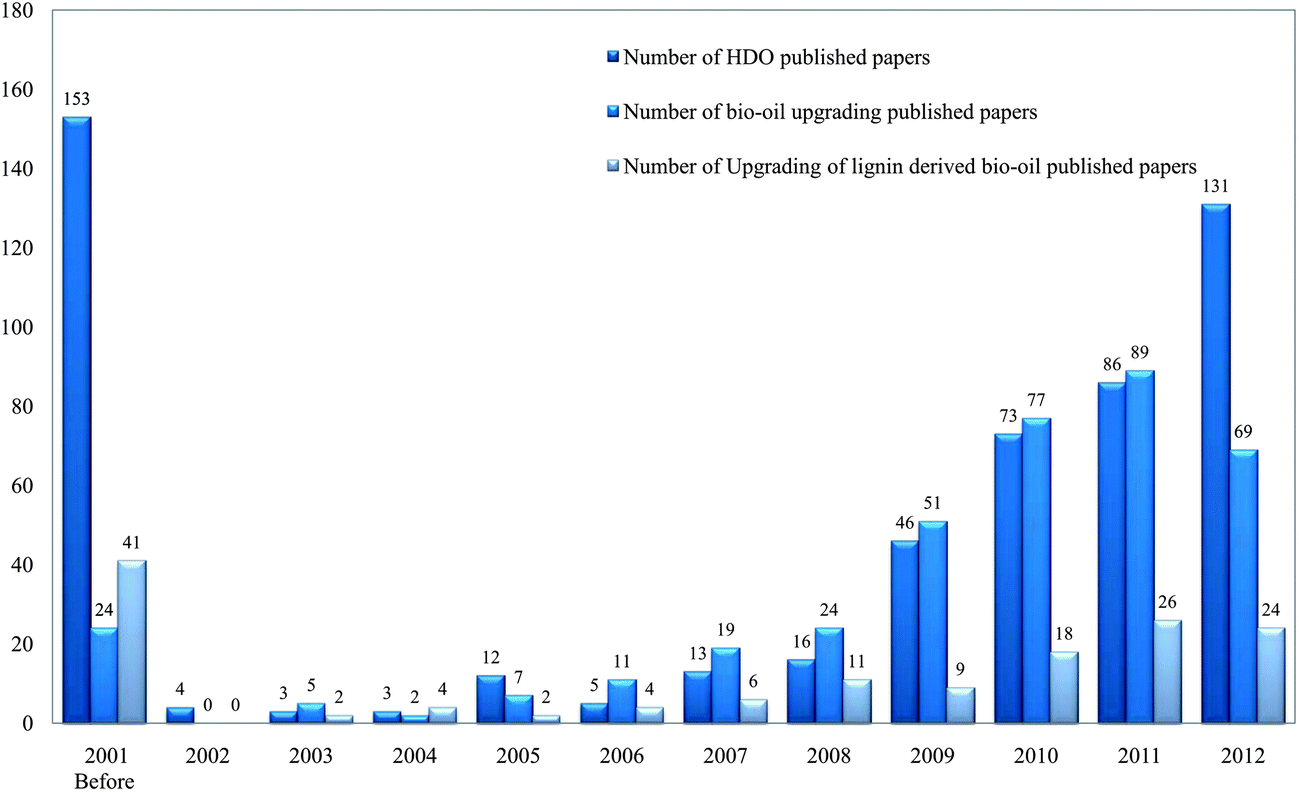 | ||
| Fig. 3 Number of papers published on the subject of bio-oils upgrading by year. | ||
Biomass and lignin
Biomass offers the prospect of playing a key role as a renewable energy resource, because it can be converted into products to replace fossil fuels directly, for example, biogas and synthetic gas instead of natural gas; liquid biofuels such as biodiesel and bioethanol instead of diesel and gasoline (petrol); and solids such as pellets and briquettes instead of coal.3 Lignocellulosic biomass is the most abundant source for candidate biofuels. It consists (Fig. 4) of hemicellulose (typically 25–35% of the total), cellulose (40–50%), and lignin (15–20%), with the composition depending on the plant species.4–7 Cellulose is a linear homopolymer of glucose units, and hemicellulose is a mixture of various polymerized monosaccharides including glucose, mannose, galactose, xylose, arabinose, 4-O-methyl glucuronic acid, and galacturonic acid residues.5 Lignin consists of aromatic units linked in large polymeric structures. In nature, lignin acts as an essential resin to strengthen lignocellulose matrices by filling the spaces between cellulose and hemi-cellulose, lending structural integrity to plants.7,8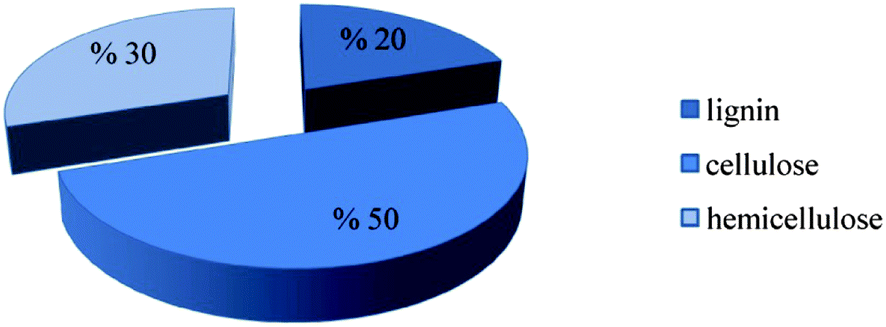 | ||
| Fig. 4 Typical composition of lignocelluloses.5 | ||
Our focus here is on lignin because it is an underutilized resource available in massive quantities. Lignin residue materials are available from biomass-to-ethanol processes and other biorefineries. Other sources of lignin are agricultural products, municipal wastes, and byproducts of pulp and paper processing.9–11 The enormous quantities and potential economic benefit of lignin processing provide a compelling motivation for further work on its conversion. However, the history of research on lignin conversion is marked with little success in terms of technology, and today lignin is used only in industries such as those producing low-grade fuels and those generating electricity via direct combustion.12 If efficient, economical methods for lignin conversion could be developed, lignin could become a major source of renewable energy.8,13
Lignin is markedly different in structure and composition from hemicellulose and cellulose, being highly aromatic and containing less oxygen. Of the biomass components, lignin is the one most similar to petroleum. Two important criteria for evaluation of biomass in our context are the hydrogen-to-carbon (H/C) and oxygen-to-carbon (O/C) atomic ratios. For comparison, such H/C ratios of crude oil are typically between about 1.60 and 2.10 and the O/C ratios between 0 and 0.03. In contrast, wood typically has O/C and H/C ratios greater than 0.61 and 1.4, respectively. In comparison with wood, lignin has lower O/C and H/C ratios, typically about 0.32 < O/C < 0.46 and 1.1 < H/C < 1.3. The low oxygen content relative to that of wood makes lignin a potentially attractive source of fuels.14,15
The structure of lignin is complex and not fully resolved; it is known to be amorphous and polyaromatic and to incorporate numerous ether linkages, OH, and methoxy groups. Lignin is regarded as a cross-linked macromolecule composed of three types of monolignol, including p-coumaryl alcohol, coniferyl alcohol, and synapyl alcohol, with the proportions depending on the source. Lignin from softwoods is mostly made up of coniferyl alcohol-derived components, but lignin from hardwoods consists of mixtures of coniferyl- and syringyl-derived structures.
In fast pyrolysis, lignin is converted substantially into compounds such as phenol, anisole, guaiacol, cresol, syringol, etc., as summarized below.1,2,16,17
Biomass conversion processes
Most of the reported work on biomass conversion to fuels has focused on feedstocks other than lignin; much effort has been devoted to triglycerides and to cellulose and hemicellulose and the sugars formed from them by hydrolysis.1 Crops such as corn, sugar cane, soybeans and palm oil and rapeseed oil are among the most widely investigated feedstocks. Processes in operation today produce bioethanol and biodiesel from food-grade biomass.16,17 But there are limitations of this technology related to the increasing need for food and concerns about the relatively low overall energy efficiencies of the processes.14,15,18,19 Therefore, in recent years, researchers have focused on developing new methods of producing bio-oils from sources such as agricultural wastes, wood, and plants such as switchgrass and miscanthus that do not compete much with food production and do not consume too much water.The potential processes that may emerge from research on lignocellulosic feedstocks are at a relatively early stage of development and not yet economical, but they offer tantalizing potential based on the expectation that research will lead to increasing efficiencies for the production of feedstocks that can be converted into fuels that are economically competitive and compatible with the existing infrastructure.16,17 It is highly likely that any economic biomass conversion processes will involve catalysis—biocatalysis and/or catalysis by solids for conversion of fluid products formed from biomass conversion processes such as pyrolysis, liquefaction, catalytic pyrolysis, or catalytic liquefaction. The products of such processes include light gases and solids such as chars in addition to the liquids (bio-oils) that are the focus here.
Catalytic upgrading of bio-oils
The bio-oils that can be formed from lignocellulosic biomass require massive chemical transformations to be useful as fuels in today's petroleum-based infrastructure. Bio-oils derived from lignin have such high oxygen contents that oxygen removal is an essential processing goal.20 Thus there is a rapidly growing literature related to the catalytic removal of oxygen from bio-oils, and especially from compounds in bio-oils. Removal of oxygen from bio-oils by reactions with H2 that form water and deoxygenated compounds are called HDO reactions. There are numerous reports of catalytic HDO of lignin-derived bio-oils and compounds found in bio-oils, including numerous reports of the conversions of such compounds with catalysts that are not selective for HDO.21–23 A premise underlying this review is that HDO may provide economical routes for the production of fuels from bio-oils, and we emphasize that this viewpoint reflects a prospect rather than a technological reality.Catalytic HDO is superficially similar to the well-established catalytic hydroprocessing carried out in petroleum refining—hydrodesulfurization (HDS) and hydrodenitrogenation (HDN)—but in petroleum refining, these give liquid products which are single phases, largely hydrocarbons, and are well suited to further processing or direct application as fuels.
There is already a substantial literature of catalytic HDO and related topics, much of it motivated by early work in coal conversion to liquid fuels. Little of this work has been focused on petroleum, because there is only little oxygen in petroleum (typically less than a few tenths of a weight percent), and the oxygen is readily removed in processes that have the goals of HDS and HSN.
In 2000, the literature of kinetics and reaction networks of HDO was reviewed by Furimsky.24 In 2004, Czernik and Bridgwater9 reviewed developments in the applications of bio-oils, and in 2006 Mohan et al.25 discussed pyrolysis of wood/biomass for bio-oils. A 2007 review by Elliott26 gives a historical perspective on developments in catalytic hydroprocessing of bio-oils. In a 2007 review, Zhang et al.27 focused on the properties of compounds and compositions of biomass pyrolysis oil. In 2010, Venderbosch and Prins28 and Oasmaa et al.29 reviewed processes for fast pyrolysis, and in 2011 Mortensen et al.30 considered catalytic upgrading of bio-oils to produce engine fuels, and Choudhary and Phillips31 reviewed applications of catalytic HDO from an industrial perspective. In 2012, Bu et al.32 considered catalytic HDO of lignin-derived phenols. In 2013, He and Wang33 considered recent advances in HDO of pyrolysis bio-oils with various catalysts, and Laskar et al.34 reviewed catalytic processing of lignin derivatives to give hydrocarbon fuels. Furimsky35 has just contributed a review of hydroprocessing catalysis with a focus on biomass conversion.
Much of the early work on HDO of whole bio-oils emerged from the group of Elliott,36 who worked primarily with hydroprocessing catalysts of the types used to remove sulfur and nitrogen from petroleum. Elliott's work demonstrated the need for severe reaction conditions for bio-oils hydroprocessing relative to those for petroleum hydroprocessing; these researchers encountered severe reactor plugging resulting from carbonaceous deposits on the catalysts. Their experiments indicated that a stabilization step would be of value preceding catalytic hydroprocessing.37–40 Gagnon and Kaliaguine41 investigated the effect of a hydrogenation pretreatment, and Sheu et al.42 suggested models based on an empirical function of temperature and pressure for overall removal of oxygen and compositional changes during hydroprocessing. In the early 1990s, Oasmaa and Boocock43 reported the reduction of the oxygen content of peat pyrolysate oils from 22 to 3 wt% resulting from catalytic HDO.
The early work demonstrated that hydroprocessing of bio-oils was feasible, although not economical, requiring severe reaction conditions and leaving open the question of whether economical processes were likely to emerge. The early work largely ignored issues of fundamental understanding of the chemistry of bio-oils conversion, identification of active, selective, and stable catalysts, and a foundation for estimation of process economics.
The recent emphasis on renewable fuels has focused interest on these issues, and the field is moving forward much faster than before, although extensive work remains before economical bio-oils processing is likely to be achieved. The review presented here addresses this topic. It is not comprehensive, and the reader is referred to the literature cited in the preceding paragraphs in this section for more foundational information. We consider here only reactions catalyzed by solids, although there are early results suggesting the possible benefits of soluble transition metal complex catalysts for biomass conversion.44
Lignin- and other biomass-derived bio-oils
Increasing attention is being paid to catalytic upgrading of lignin-derived bio-oils for production of aromatic compounds such as phenolics as well as for the production of hydrocarbons that could be used as fuels. The high concentrations of oxygen in lignin-derived bio-oils (typically, 35–40 wt%) make their conversion into fuels a processing challenge that is markedly different from those of petroleum refining.8,9,22,45Processes for conversion of lignin to phenolic compounds and biofuels are based on depolymerization reactions, which may be pyrolitic and (in part) catalytic, and several investigations have provided information about potential commercial processes.46 Pyrolysis involves anaerobic thermal decomposition for degrading of biomass into mixtures of small molecules, which can be used to form chemicals and fuels. At the high temperatures of pyrolysis (650–800 K), vapor-phase products react and condense to form liquid mixtures. To obtain relatively high liquid product yields and to decrease char formation, high temperatures and short reactor residence times are favorable. Lignin degradation processes such as fast pyrolysis produce oxygen-containing compounds including guaiacol (typically, 39%), syringyl (16%), and hydroxyphenyl derivatives (45%) (Fig. 5). Pyrolysis of softwood lignin mainly yields guaiacol derivatives, whereas pyrolysis of hardwood lignin produces both guaiacol and syringyl derivatives.47–60
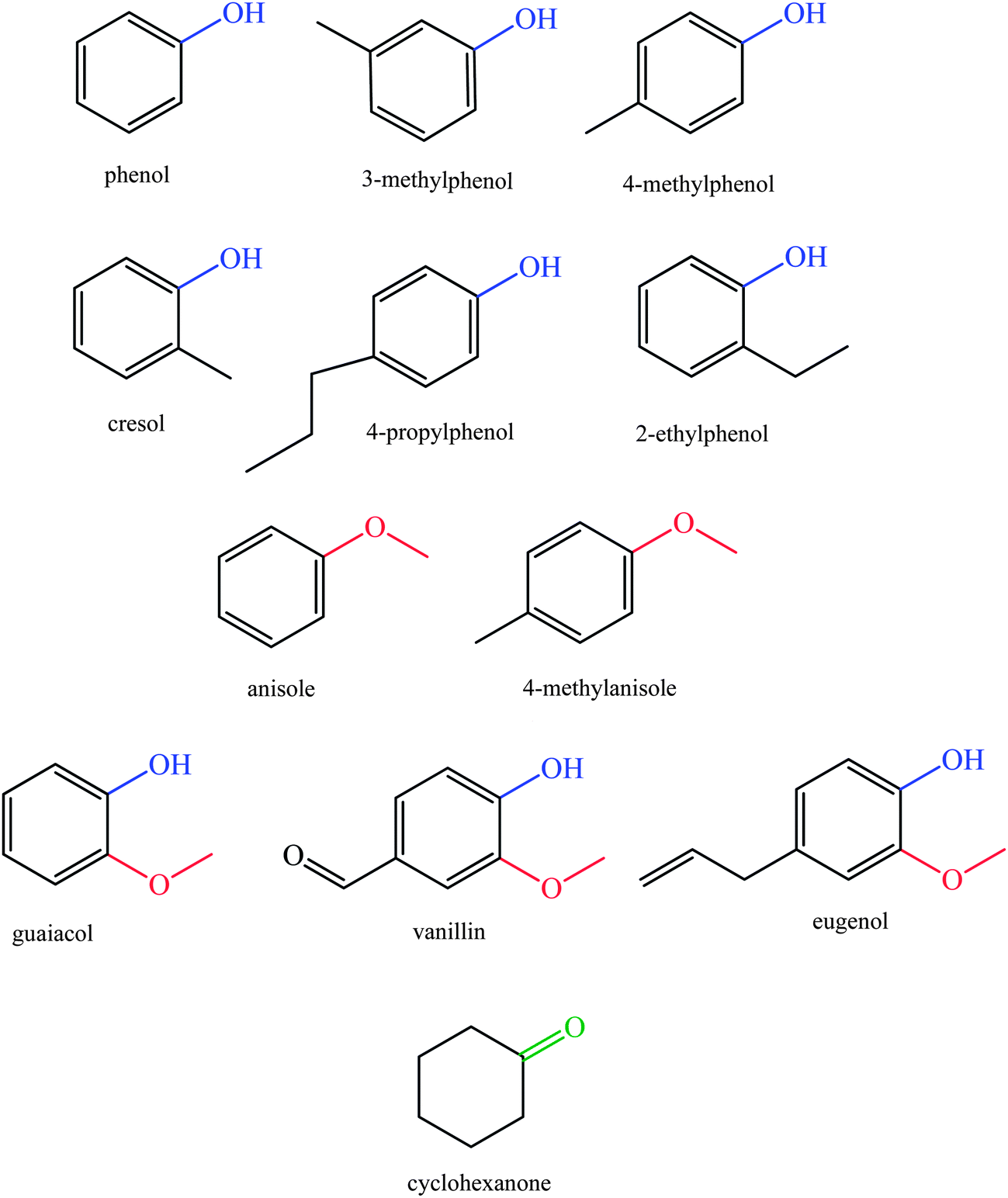 | ||
| Fig. 5 Compounds in lignin-derived bio-oils. | ||
Lignin can alternatively be depolymerized in an aqueous catalyst solution. Water is the solvent, but methanol, ethanol, or combinations thereof can also be used. Catalysts used in aqueous lignin depolymerization include NaOH, KOH, Ca(OH)2, and Mg(OH)2. Often, for preventing precipitation and reactor plugging, continuous stirred reactors are used. The reactions are typically carried out in the ranges of 500–625 K and 70–160 bar with liquid hourly space velocities in the range of 0.5 to 6 h−1.
Methods of catalytic upgrading of bio-oils
Bio-oils obtained from fast pyrolysis have high oxygen contents—typically more than 10 wt% and even as much as 50 wt%. The oxygen-containing compounds are responsible for chemical and thermal instability of the liquids and a strong tendency to polymerize. Low heating values, high viscosities, the acidic nature of the oils (which causes corrosion of vessels and pipes), and the high water contents are all major disadvantages of bio-oils as potential fuels.48,58,61–63 These limitations make bio-oils essentially unusable for almost all of today's fuel applications.Therefore, the (partially) depolymerized products formed by pyrolysis must be refined. The major prospective refining processes have the goal of forming stabilized deoxygenated products. Lignin-derived compounds can in prospect be converted to components that are good liquid fuels, such as high-octane-number alkylbenzenes.
Thus, in the processing step following pyrolysis, the partially depolymerized product may be subjected to a catalytic process for removal of oxygen. This step can in prospect convert compounds such as methoxyphenols and benzenediols to hydrocarbons that are valuable fuel components. The desired reactions involve formation of hydrocarbons and water in addition to other compounds that can be removed from the hydrocarbons; the reactions typically involve H2 as a reactant and are classified as HDO reactions.
Catalysts that are active for these conversions include noble metals and metal sulfides supported on metal oxides or carbon. The reactions typically take place at temperatures exceeding 475 K and at H2 partial pressures from 35–140 bar.64–66 For example, in the conversion of lignin to naphthenic kerosene, the HDO reaction temperature is about 475 to 675 K, and suitable catalysts are alumina-supported sulfides of molybdenum or tungsten, among others.67–70
Much of the research on catalytic HDO is based on the premise that the products should be usable in today's infrastructure built for hydrocarbon fuels. The lignin-derived products might be blended with petroleum-derived products, and they might also be refined as co-feeds with them. Another practical goal is to minimize the yields of light gases, char, and coke formed in the processing.71
The most commonly investigated methods for bio-oils upgrading (Fig. 6) involve physical, chemical, and catalytic processing. Physical upgrading of bio-oils involves filtration, emulsification, and solvent extraction. In emulsification and solvent extraction, the bio-oils are mixed with diesel oil and solvents, respectively, to extract the compounds with lower oxygen contents. Chemical upgrading includes aqueous-phase processing such as reforming and gasification, mild cracking, esterification, etc.
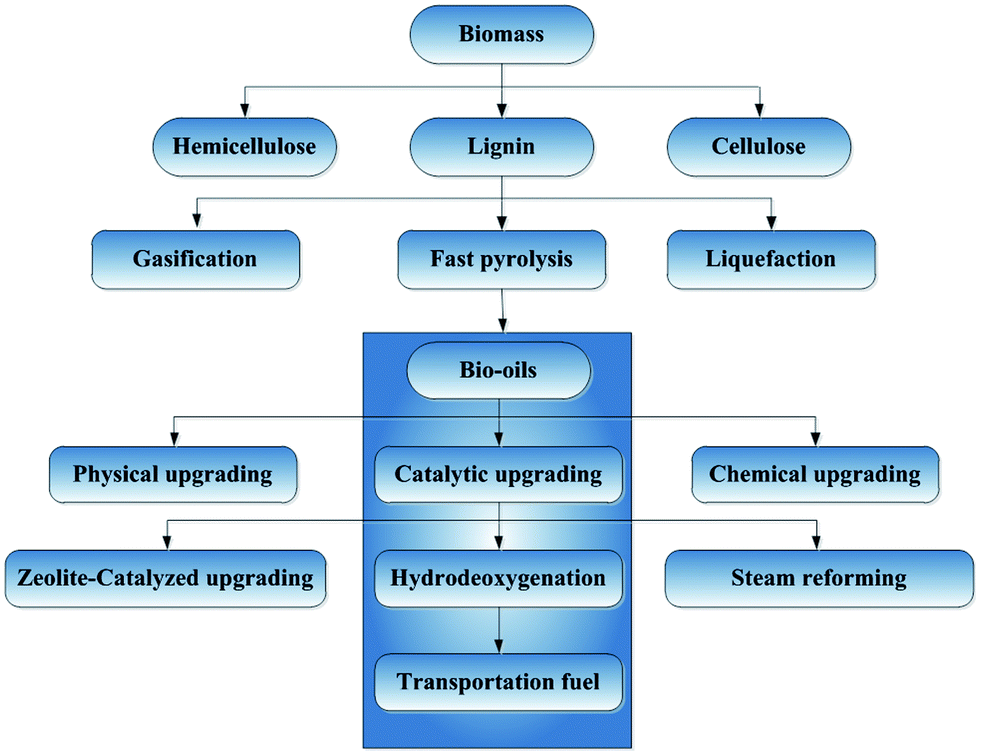 | ||
| Fig. 6 Simplified schematic flow diagram for candidate process for conversion of biomass to bio-fuels. | ||
Catalytic upgrading routes that have been investigated include steam reforming, zeolite-catalyzed cracking, and HDO.63,72 Catalytic steam reforming of pyrolysis oils is a recently explored method to produce H2. Cracking is related to the “fluid catalytic cracking” of petroleum fractions (which takes place with zeolite-containing catalysts in fluidized-bed reactors) and leads to the removal of oxygen; the process is operated at atmospheric pressure and high temperatures (575–875 K); no H2 is required, which is advantageous for processing costs.58,73 However, a major disadvantage of cracking is the high yields of carbonaceous deposits (coke) formed on the catalysts, which deactivate them within seconds and require regeneration by burning of the deposits.74 Zeolite-catalyzed cracking is not nearly as well developed as HDO in our context.48 HDO is considered in detail in the following sections.
Yan et al.75,76 investigated the degradation of lignin catalyzed by noble metals in a two-step process. The first step included the catalytic cleavage of C–O–C bonds without breaking of C–C bonds under relatively mild conditions, and the second step was hydrogenation of species to remove the aromaticity followed by hydrogenolysis of the C–O bonds to produce compounds classified as C9 and C14–18. The transformation of the phenolic compounds into desirable fuels in the second step is challenging.
HDO with conventional metal sulfide catalysts is problematic because of the rapid catalyst deactivation by coke, water, and other components. Catalytic reactions in the presence of aqueous-phase reactants offer prospects for the conversion of phenolic compounds that may help to overcome some of the limitations of more conventional catalytic hydroprocessing, but high operating temperatures are needed. Recently ionic liquids (ILs) have been proposed as promising reaction media (and catalysts) for biofuel production, and these offer some of the advantages of the water-containing reactants. Yan et al.76 examined the HDO of lignin-derived phenols into alkanes using nanoparticle catalysts dispersed in Brønsted acidic ILs. Their results indicate that in the HDO of phenol, high conversions with excellent selectivity to cyclohexane could be obtained with [bmim][BF4] or [bmim][TF2N]. They concluded that in comparison with metal sulfide or mineral acid/supported metal catalysts in water, upgrading of the lignin derivatives to alkanes catalyzed by nanoparticles combined with Brønsted acidic ILs is an efficient and less energy consuming process.
Jongerius et al.77 considered a process in which lignin was depolymerized to form monomers, dimers, and oligomers by liquid-phase reforming catalyzed by Pt/γ-Al2O3 followed by HDO of the extracted lignin-derived bio-oils catalyzed by CoMo/Al2O3 and Mo2C/CNF. The authors carried out the HDO at 573–648 K and a high H2 partial pressure (55 bar), observing that the latter catalyst outperformed the former and yielded more oxygen-free products such as benzene and toluene.
HDO reactions and those accompanying them
Because most of the reported work on catalytic HDO has been carried out with solid catalysts and under conditions resembling those of conventional catalytic hydroprocessing, our review is focused on such work. Thus, in the following sections, the reactions, catalysts, and potential processes for bio-oils HDO are considered, and results obtained with prototypical catalysts and individual compounds that are representative of lignin-derived bio-oils provide a foundation for the discussion. For example, phenol itself has been used often as a representative of phenolic compounds. Experiments with phenol and related compounds have been carried out to determine the reactions taking place and to elucidate what catalyst functions are engaged in these conversions. The approach mirrors that applied in years past to help elucidate the hydroprocessing reactions involved in the conversion of petroleum and of coal.74 We believe that this approach offers the prospect of being similarly valuable for improving bio-oils upgrading process.Reaction networks
Several reaction networks have been proposed for HDO and the reactions that accompany HDO. Understanding of these reaction networks has emerged from investigations of various individual compounds as reactants—one can refer to them as model compounds—with various catalysts.More than one pathway has been inferred for oxygen removal by reactions with H2 from compounds such as guaiacol, a compound that a number of researchers have considered to be a prototype. One pathway is direct hydrodeoxygenation (direct deoxygenation, DDO), which leads to the formation of aromatics and water. Another is hydrogenation of the phenolic ring followed by dehydration to form cyclohexene derivatives and rehydrogenation to produce cyclohexane derivatives.73,78–80 Furthermore, removal of methoxy groups from phenolic compounds can proceed by demethylation (DME) and by demethoxylation (DMO), which involve C–O bond breaking and respectively produce CH4 and methanol.78
Details follow in this section, showing that the catalyst type and reaction conditions are important in determining the relative importance of the competing reaction pathways.73 We illustrate with results determined in investigations of three compounds, phenol, anisole (methoxybenzene), and guaiacol (2-methoxyphenol) reacting with H2. These compounds were chosen to illustrate the main characteristics of the reaction networks because each of them is abundant in lignin-derived bio-oils, each is regarded as a good model compound representing lignin-derived bio-oils, and the data characterizing these compounds are more fundamental and complete than those characterizing the reactions of other candidate compounds to represent the bio-oils.
Because understanding of the mechanisms (details of the elementary steps of bond breaking and bond forming) of catalytic HDO reactions is still not well developed, we do not address the mechanisms here.
Conversion of phenol
Phenol is the simplest of the widely investigated compounds representative of bio-oils, incorporating the aromatic ring and hydroxyl group that are typical of lignin and lignin-derived compounds. Here we summarize the reactions characterizing HDO of phenol, with an emphasis on reactions catalyzed by metal sulfides (which have been used commonly) and noble metals. Regardless of which of these catalyst types is used, the major routes for HDO of phenol are hydrogenolysis and hydrogenation.81–84Fig. 7 illustrates the conversion of gas-phase phenol in the presence of a metal sulfide catalyst taking place by two pathways.49,79,85–87 Pathway (1) involves direct hydrogenolysis to give benzene and water by removal of a hydroxyl group. Subsequent hydrogenation gives cyclohexene, which can be hydrogenated to cyclohexane. Alternatively, in Pathway (2), phenol is converted to cyclohexanone, which is hydrogenated to form cyclohexanol. In most reported experiments characterizing these reactions, conversions have been high and the intermediate cyclohexanol has not been observed. Pathway (1) is typically dominant and is markedly favored at higher temperatures. Mass balance data indicate the hydrogen consumptions.62,72,79,80,82,87–89
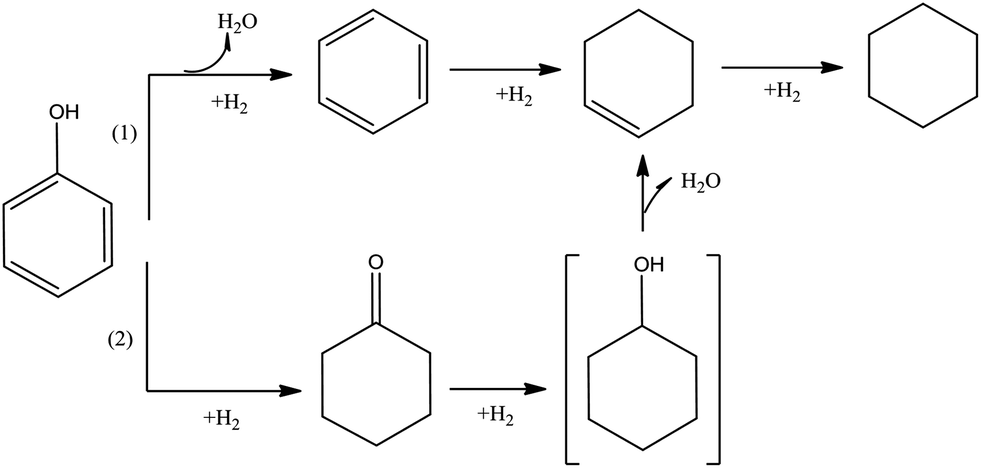 | ||
| Fig. 7 Reaction network characterizing phenol conversion catalyzed by metal sulfides and noble metals.50,79,85–88 | ||
When the metal sulfide catalysts are replaced by noble metals on supports, similar chemistry is observed, but the hydrogenation activity is typically higher. For example, Senol et al.62 concluded that as a consequence of the high hydrogenation activity of a NiMo catalyst, the formation of cyclohexane from phenol was predominant. In contrast, in HDO of phenol catalyzed by a La–Ni–W–B catalyst, reaction Pathway (1) was not observed, and hydrogenation of phenol by the second pathway was dominant.62,85,86
Conversion of anisole
Anisole is representative of bio-oil compounds that incorporate C–O–C moieties—which are prevalent in lignin as well as lignin-derived bio-oils. In the HDO of anisole, four reaction pathways—demethylation, hydrogenation, hydrogenolysis, and methyl group transfer (transalkylation)—occur, not all of them leading to the removal of oxygen. Because the Cmethyl–O bond is weaker than the Caromatic–O bond, demethylation via the rupture of the Cmethyl–O bond is typically favored kinetically.90Experiments with various catalysts (details are given below) have led to the linking of the reaction pathways with specific catalyst functions. A noble metal such as platinum catalyzes demethylation, HDO, and hydrogenation in sequence, giving phenol. Phenol, consistent with results summarized above, is converted to benzene by breaking of the Caromatic–O bond and to cyclohexane by hydrogenation of the aromatic ring, which is followed by hydrogenolysis of the Calicylic–O bond.62,73,90 When the solid catalyst incorporates an acidic function (such as γ-Al2O3 as the support), methyl group transfer reaction are kinetically significant.73
Fig. 8 is a scheme representing the reactions of anisole conversion that is consistent with the results of numerous researchers.52,58,73,90,91 Pathway (1) is demethylation, which takes place on the metal surface. Another reaction (Pathway 2) involves direct deoxygenation, leading to methanol and benzene.58 On catalysts that incorporate both metal and acid functions, in addition to Pathway (1), methyl group transfer and HDO are catalyzed by acidic sites via Pathway (3). Thus, the combination of a noble metal and an acidic support gives a higher catalytic activity than the metal on a nonacidic support, but not all the reactions involve oxygen removal.73
 | ||
| Fig. 8 Reaction network characterizing anisole conversion catalyzed by acidic and non-acidic supported metal catalysts.52,58,73,90,91 | ||
Conversion of guaiacol
Guaiacol appears to be the consensus preferred choice as a prototype compound to represent lignin-derived bio-oils—because it incorporates two types of C–O bond that represent both lignin and many of its derivatives. The moieties are hydroxy (Csp2OH) and methoxy (Csp2OCH3). Reactions of guaiacol have been used to resolve the roles of catalyst functions.92 Early experiments indicated consecutive transformations to produce catechol, methylated products, and deoxygenated compounds such as benzene and cyclohexane.78 Demethylation (or methyl group transfer) occurs by two pathways. One is hydrogenolysis of the O–CH3 bond, which leads to the formation of catechol and methane, and the other is demethoxylation, which occurs by cleavage of the Caromatic–OCH3 bond, leading to phenol and methanol. In the conversion of the phenolic OH group, one pathway includes hydrogenolysis of the Caromatic–OH bond and one includes hydrogenation of the aromatic ring.61The reaction pathways depend substantially on the catalyst composition. For example, the HDO of guaiacol catalyzed by sulfided CoMo and NiMo begins with demethylation/demethoxylation and dehydroxylation as the primary and kinetically significant reactions, followed by hydrogenation of the benzene ring. But when the catalysts are supported rhodium or supported palladium, hydrogenation of the benzene ring is followed by demethylation/demethoxylation.62,74,81,85,87,93–99
Because the acidic sites on catalyst surfaces are active for deoxygenation reactions, the deoxygenation activity is enhanced when more acidic sites are introduced. For example, when the conversion is catalyzed by noble metals supported on nitric-acid-treated carbon black, 2-methoxycyclohexanol is formed selectively by hydrogenation of the benzene ring, but when the support is SiO2–Al2O3, which incorporates acidic sites, the major product is cyclohexane.95
To elucidate more precisely and quantitatively which catalyst functions catalyze the various reactions, Nimmanwudipong et al.54 used a supported platinum catalyst, Pt/γ-Al2O3, under mild enough conditions to provide a detailed characterization of intermediate products. They showed that deoxygenation by removal of the methoxy group is more rapid than hydroxy group removal under their conditions. The main products of the reactions were phenol, catechol, and 3-methylcatechol. When the support was changed from the acidic γ-Al2O3 to the basic MgO, the products formed via transalkylation reactions (on acidic sites) were not formed in kinetically significant amounts.100 Phenol and catechol were then the major products, along with the unexpected product cyclopentanone.100
Results of other authors confirm that the acidic sites catalyze transalkylation and metal and metal sulfide sites catalyze the other classes of reactions.73
Summary reaction network synthesizing data from a number of investigations
A figure summarizing the reaction pathways occurring in the conversion catalyzed by platinum supported on γ-Al2O3 (Fig. 9) is representative of the results presented above for guaiacol, anisole, phenol, and other reaction intermediates that were mentioned (such as cyclohexanone and methylanisole), and it is based on data obtained in experiments in which guaiacol, anisole, and phenol each was used as a feed (with H2); data were also obtained when some of the intermediates were used as feeds. At the risk of overgeneralizing, we suggest that this reaction network is qualitatively representative of observations with most supported metal catalysts54,55,95,97,101,102 and even most supported metal sulfide catalysts; details of the performance of various catalyst types are given in a following section. The summary shown in Fig. 9 includes the several documented reaction classes, each of which is coded with a different type of arrow in the figure.103 | ||
| Fig. 9 Reaction network for the conversion of lignin-derived compounds (each compound shown in red was used as a reactant) with H2 catalyzed by Pt/γ-Al2O3 at 573 K and 1.40 bar. HDO, hydrogenolysis, and hydrogenation (or dehydrogenation) reactions are represented by dashed green, blue, and black arrows, respectively. Transalkylation reactions are represented by solid black arrows.103 | ||
Comparison of catalysts
Research has been done with various HDO catalysts, both in attempts to elucidate the roles of various catalyst functions and to seek improved catalysts—those with good combinations of activity, selectivity, and resistance to deactivation.The catalytic species that accelerate the HDO reactions include metals, metal sulfides, metal phosphides, metal carbides, and metal nitrides. These have been used on various supports, and in general the acidic supports catalyze methyl group transfers and also lead to catalyst deactivation by carbonaceous deposits (coke) that form on their surfaces. Noble metals such as platinum offer high activities for HDO reactions, but they are expensive.48,97,104–108 Furthermore, these metals are highly active for hydrogenation of aromatic rings, and consequently catalysts incorporating them are characterized by high H2 consumptions,109 which is an economic disadvantage and raises the question of the availability and cost of the H2. Consequently, many investigators have worked with supported non-noble transition metals which are less expensive than noble metals.90,101,110
Various supports have been investigated, notably those that offer prospects of being resistant to coke formation, including carbon, ZrO2, SiO2, and MgO,62,68,111–113 and these are discussed in a following section.
In the next paragraphs, the performance of various catalysts for conversion of lignin-derived compounds is summarized; details are given in Tables 1–4.
| Catalyst | Support | Reactor configuration | Operating conditions | Lignin-derived compound | Major products | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| T (K) | P (bar) | t (h) | WHSV (h−1) | ||||||
| Fe | SiO2 | Fixed bed | 623–723 | 1 | 0.5 | 0.67–9.09 | GUA | Ben, Tol | 114 and 115 |
| Co | Kieselguhr | Fixed bed | 623–723 | 1 | — | 1.22–2 | GUA | — | 114 |
| CoMo | γ-Al2O3 | Batch | 613 or 628 | 70 | — | — | 4MP | Tol, MCYHA | 89 |
| CoMo | γ-Al2O3 | Flow reactor | 523 | 15 | — | — | Ph | Ben | 62 |
| CoMo | γ-Al2O3 | Batch | 523 | 75 | 2 | — | Ph | — | 62 |
| CoMo | Al2O3 | Fixed bed | 613 | 70 | — | — | 2EP | — | 141 |
| CoMo | Al2O3 | Batch | 573 | 50 | 4.17 | — | 4MP | Tol | 96 |
| CoMo | Al2O3 | Batch | 598 | 69 | 1.68 | — | 4MG | Tol, Cr, MC | 152 |
| CoMo | Al2O3 | Batch | 573 | 69 | 5.74 | — | 4MC | Tol, Cr, MCYHA | 152 |
| CoMo | Al2O3 | Batch | 573 | 69 | 4 | — | EUG | PCYHA, PP, PG, PC | 152 |
| CoMo | Al2O3 | Batch | 573 | 69 | 4.24 | — | Van | MCYHA, Cr, MC | 152 |
| CoMo | Al2O3 | Batch | 523–598 | 34 | 6.67–10 | — | ANI | Ph, Ben, CYHA | 142 |
| CoMo | Al2O3 | Batch | 523 | 34 | 20 | — | GUA | CAT, Ph, Ben, CYHA | 142 |
| CoMo | Al2O3 | Flow reactor | 548–623 | 50 | — | — | o-MEP | ph, dioxygen compounds | 153 |
| CoMo | Al2O3 | Flow reactor | 548–623 | 50 | — | — | m-MEP | ph, dioxygen compounds | 153 |
| CoMo | Al2O3 | Batch | 553 | 70 | 2.5 | — | GUA | Ph. CAT | 67 |
| CoMo | Al2O3 | Fixed bed | 573 | 50 | — | — | ANI | ph, 2MP, Ben | 154 |
| CoMo | Al2O3 | Fixed bed | 573 | 50 | — | — | ANI | ph, 2MP, Ben | 98 |
| CoMo | Al2O3 | Flow reactor | 523 | 15 | — | — | Ph | Ben | 98 |
| CoMo | Al2O3 | Flow reactor | 573 | 15 | — | — | Ph | Ben, CYHA | 98 |
| CoMo | Al2O3 | Flow reactor | 523 | 15 | — | — | ANI | 2MP, Xol, ph, Ben | 98 |
| CoMo | Al2O3 | Flow reactor | 573 | 15 | — | — | ANI | Tol, ph, Ben, 2MP | 98 |
| CoMo | Al2O3 | Batch | 573 | 55 | 4 | — | GUA, Ph | Ben, Tol, Xy | 77 |
| CoMo | C | Batch | 553 | 70 | — | — | GUA | Ph, Ben, CYHA | 155 |
| Co–Mo–B | — | Batch | 548 | 40 | 10 | — | Ph | CYHA | 156 |
| Mo | Al2O3 | Fixed bed | 613 | 70 | — | — | 2EP | — | 141 |
| Mo | γ-Al2O3 | Fixed bed | 573 | 50 | — | 2 | ANI | Ph, Cr, 2,6-DMP | 117 |
| MoO2 | — | Batch | 623 | 44 | 5 | — | 4MP | Tol | 109 |
| MoO3 | — | Batch | 623 | 44 | 5 | — | 4MP | Tol | 109 |
| MoO3 | — | Packed-bed | 673 | 1 | 1 | — | CYHAONE, ANI | Ben | 157 |
| Mo2N | — | Batch | 573 | 50 | — | — | GUA | Ph | 102 |
| Mo2N | C | Batch | 573 | 50 | 6 | — | GUA | Ph | 94 |
| Mo2C | CNF | Batch | 573–648 | 55 | 4 | — | GUA | Ph | 119 |
| NiMo | Al2O3 | Fixed bed | 613 | 70 | — | — | 2EP | — | 141 |
| NiMo | Al2O3 | Fixed bed | 573 | 50 | — | — | ANI | Ph, 2MP | 154 |
| NiMo | Al2O3 | Fixed bed | 573 | 50 | — | — | ANI | Ph, 2MP, CYHA | 154 |
| NiMo | Al2O3 | Batch | 573 | 70 | 2.5 | — | GUA | Ph, CAT | 67 |
| NiMo | γ-Al2O3 | Flow reactor | 523 | 15 | — | — | Ph | CYHA | 62 |
| NiMo | γ-Al2O3 | Batch | 523 | 75 | 2 | — | Ph | — | 62 |
| NiMo | SiO2–Al2O3 | Fixed bed | 573 | 50 | — | — | Ph | C6 hydrocarbons | 146 |
| NiMo | SiO2–Al2O3 | Fixed bed | 598 | 50 | — | — | Ph | C6 hydrocarbons | 146 |
| NiMo | SiO2–Al2O3 | Fixed bed | 598 | 50 | — | — | 2MP | Ph, C7 hydrocarbons | 146 |
| Ni | SiO2 | Batch | 593 | 170 | 1 | — | GUA | CYHA, CYHAONE | 61 |
| Ni | SiO2 | Batch | 593 | 170 | — | — | GUA | CYHA, CYHAONE | 132 |
| Ni | SiO2 | Fixed bed | 423 | 1 | 0–4 | — | Ph | CYHAONE, CYHAOL, Ben | 130 |
| Ni | SiO2 | Fixed bed | 423 | 1 | 0–4 | — | CYHAONE | CYHAOL, Ph, Ben | 130 |
| Ni | SiO2 | Fixed bed | 573 | 50 | — | 2 | ANI | Ph, Ben | 117 |
| Ni | γ-Al2O3 | Fixed bed | 573 | 50 | — | 2 | ANI | Ph, Ben | 117 |
| Ni | SBA-15 | Flow reactor | 563–583 | 3 | — | 20.4 and 81.6 | ANI | Ben | 135 |
| Al-SBA-15 | |||||||||
| γ-Al2O3 | |||||||||
| Microporous carbon | |||||||||
| TiO2 | |||||||||
| CeO2 | |||||||||
| Ni | SiO2–ZrO2 | Batch | 573–613 | 50 | 5 | — | GUA | CYHA, MCYHA, PCYHA | 133 |
| Ni | ZrO2–SiO2 | Batch | 523–613 | 50 | — | — | Ph, GUA | CYHA | 158 |
| Ni–W | AC | Fixed bed | 423–573 | 15 | 0–6 | 0.5 | Ph | — | 85 |
| Ni–W–B | — | Batch | 498 or 523 | 40 | 4 | — | Ph | CYHAOL, CYHE, CYHA | 86 |
| Ni–W–B | — | Batch | 498 | 40 | 7 | — | Ph | — | 122 |
| Ni–W–B | — | Batch | 498 | 40 | — | — | Ph | — | 123 |
| Ni–Cu | Al2O3 | Batch | 593 | 170 | 1 | — | GUA | CYHA, CYHAONE | 61 |
| Ni–Cu | SiO2 | Batch | 593 | 170 | 1 | — | GUA | CYHA, CYHAONE | 61 |
| Ni–Cu | SiO2 | Batch | 593 | 170 | — | — | GUA | CYHA, CYHAONE | 132 |
| Ni–Cu | SiO2–ZrO2 | Batch | 593 | 170 | — | — | GUA | CYHA, CYHAONE | 132 |
| Ni–Cu | CeO2–ZrO2 | Batch | 593 | 170 | 1 | — | GUA | CYHA, CYHAONE | 61 |
| Ni–Cu | CeO2–ZrO2 | Batch | 593 | 170 | — | — | GUA | CYHA, CYHAONE | 132 |
| Ni–Cu | CeO2 | Fixed bed | 593 | 10 | 1 | — | ANI | CYHA | 156 |
| Ni–Cu | ZrO2–SiO2–La2O3 | Batch | 593 | 170 | 1 | — | GUA | CYHA, CYHAONE | 61 |
| Ni–Cu | ZrO2–SiO2–La2O3 | Batch | 593 | 170 | — | — | GUA | CYHA, CYHAONE | 132 |
| Ni–Cu | δ-Al2O3 | Packed-bed | 573 | 10 | 4 | 3–6 | ANI | Ben, Tol, CYHA, MCYHA | 118 |
| Ni and NiCu | ZrO2–SiO2 | Batch | 523–613 | 50 | 1–8 | — | GUA | CYHA, MCYHA, Ben | 134 |
| Sn | Inconel | Quartz tube | 673 | 1 | 3 | — | GUA | Ph | 58 |
| Ga | H-Beta zeolite | Packed-bed | 673–823 | 1 | 2–22 | — | 3MP | Tol, Ben, Xy | 159 |
| V | Al2O3 | Flow reactor | 623 | — | — | — | GUA | Methylated phenol | 160 |
| W2C | CNF | Batch | 573–648 | 55 | 4 | — | GUA | Ph | 119 |
| Pd, Pt, Ru, Fe, Cu, Pd–Fe | C | Fixed bed | 350 and 450 | 1 | 2 | — | GUA | Ph | 161 |
| CuCr2O4·CuO | — | Batch | 423–548 | 50 | 0–20 | — | Ph | CYHAOL | 116 |
| ANI | Ben, CYHA, CYHAOL, MCYHA | ||||||||
| Cr | MCYHA | ||||||||
| GUA | CYHAOL, MCYHDIOL, CYHA | ||||||||
| VA | 2ME4MEPH | ||||||||
| Catalyst | Support | Reactor configuration | Operating conditions | Lignin-derived compound | Major products | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| T (K) | P (bar) | t (h) | WHSV (h−1) | ||||||
| Pt | Al2O3 | Fixed bed | 473 | 40 | — | — | GUA | CYHA | 162 |
| Pt | Al2O3 | Fixed bed | 473 | 40 | — | — | Cr | — | 162 |
| Pt | Al2O3 | Packed bed | 573 | 1.4 | — | 17.6–437 | ANI | Ph, 2MP, Ben | 52 and 53 |
| Pt | Al2O3 | Packed bed | 573 | 1.4 | 0–6.67 | 0–4 | 4MA | 4MP, Tol | 147 |
| Pt | γ-Al2O3 | Tubular | 573 | 1.4 | 0–3 | 20 | GUA | Ph, CAT | 55 |
| Pt | γ-Al2O3 | Tubular | 573 | 1.4 | 0–4.17 | 7.9 | EUG | 4-PG | 56 |
| Pt | γ-Al2O3 | Packed bed | 573 | 1.4 | 0–6.67 | 0–4 | 4MA | 2,4,6-TMP | 147 |
| Pt | γ-Al2O3 | Packed bed | 523–623 | 1.4 | 0–6 | 56 | CYHAONE | Ph | 51 |
| Pt | MZ-5 | Fixed bed | 473 | 40 | — | — | GUA | — | 163 |
| Pt | MZ-5 | Fixed bed | 473 | 40 | — | — | Cr | MCYHA, alkylated cycloalkanes | 163 |
| Pt | SiO2–Al2O3 | Packed bed | 573 | 1.4 | — | 1–4 | 4MA | 4MP, Tol | 147 |
| Pt | SiO2 | Fixed bed | 673 | 1 | 0–5 | 2 | ANI | Ben | 73 |
| Pt | AC(N) | Batch | 553 | 40 | 2 | — | 4PP | — | 163 |
| Pt | H Beta zeolite | Fixed bed | 673 | 1 | 0–5 | 12 | ANI | Ben, Tol, Xy | 73 |
| Pt | HY zeolite | Fixed bed | 473–523 | 40 | — | 5–20 | Ph | CYHA | 49 |
| Pt | Inconel | Quartz tube | 673 | 1 | 3 | — | GUA | Ph | 58 |
| Pt–Sn | Inconel | Quartz tube | 673 | 1 | 0–3 | — | GUA | Ph, Ben | 58 |
| Pt–Sn | Inconel | Quartz tube | 673 | 1 | 0.75, 2.08 | 0.32 | ANI | Ph, Ben | 58 |
| Pt–Rh | ZrO2 | Batch | 573 | 80 | 3 | — | GUA | — | 47 and 164 |
| Rh | — | Batch | 400–700 | 50 | 1 | — | GUA | 2MECYHAOL, 2EXYHAONE | 97 |
| Rh | ZrO2 | Batch | 573 | 80 | 3 | — | GUA | Ben | 47 and 164 |
| Rh | SiO2 | Fixed bed | 573 | 10 | — | — | ANI | — | 156 |
| Ru | C | Batch | 473 | 69 | — | — | GUA | 2MECYHAOL, CYHAOL | 45 |
| Ru, Mo | C | Packed bed | 623–673 | 40 | 0.067 | — | GUA | Ben, Ph | 165 |
| Pd | C | Fixed bed | 353 | 5 | 7 | — | Ph | — | 50 |
| Pd | C, HZSM-5 | Batch | 473 | 50 | 2 | — | GUA | Cycloalkanes | 166 |
| Pd | C, HZSM-5 | Batch | 433 | 50 | 2 | — | Ph | Cycloalkanes | 166 |
| Pd | C, H3PO4 | Batch | 423 | 50 | 0–3.34 | — | ANI | — | 87 |
| Pd | C, H3PO4 | Batch | 473 | 50 | 0–3.34 | — | CAT | — | 87 |
| Pd | C, H3PO4 | Batch | 473 | 50 | 0.5 | Ph | CYHA | 87 | |
| Pt, Rh, Pd, Ru | Al2O3, SiO2–Al2O3, nitric-acid-treated carbon black | Batch | 523 | — | 2 | — | GUA | — | 95 |
| Catalyst | Support | Reactor configuration | Operating conditions | Lignin-derived compounds | Major products | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| T (K) | P (bar) | t (h) | WHSV (h−1) | ||||||
| CoMoWS | SBA-15 | Fixed bed | 583 | 30 | 0.84–4.17 | 24.5 | ANI | 91 | |
| CoMoWS | SBA-16 | Fixed bed | 583 | 30 | 0.84–4.17 | 24.5 | ANI | Ph, Cr, Xol | 91 |
| CoMoS | — | Fixed bed | 573 | 40 | — | — | GUA | Ben | 92 |
| CoMoS | — | Batch | 673 | 50 | 1 | — | GUA | Ben, Ph, CYHE, CYHA | 97 |
| CoMoS | — | Batch | 553 | 70 | — | — | GUA | — | 68 |
| CoMoS | — | Batch | 573 | 50 | 3.34 | — | 4MP | Tol, CYHA | 96 |
| CoMoS | γ-Al2O3 | Fixed bed | 573 | 40 | — | — | GUA | Ben | 92 |
| CoMoS | γ-Al2O3 | Batch | 523 | 75 | 0.08–5 | — | Ph | Ben | 88 |
| CoMoS | γ-Al2O3 | Flow reactor | 523 | 15 | — | — | Ph | Ben | 62 |
| CoMoS | γ-Al2O3 | Batch | 523 | 75 | 2 | — | Ph | — | 62 |
| CoMoS | Al2O3 | Fixed bed | 573 | 40 | — | — | GUA | — | 113 |
| CoMoS | Al2O3 | Trickle bed | 573 | 40 | — | — | GUA | — | 78 |
| CoMoS | Al2O3 | Batch | 573 | 80 | 1–5 | — | GUA | Ben | 48 |
| CoMoS | Al2O3 | Batch | 573 | 50 | — | — | GUA | Ph, CAT | 120 |
| CoMoS | Al2O3 | Batch | 573 | 50 | — | — | ANI | Ph | 120 |
| CoMoS | Al2O3 | Fixed bed | 573 | 28.5 | — | — | Ph | Aromatics, CYHA | 79 |
| CoMoS | Al2O3 | Batch | 573 | 50 | 4 | — | Ph | Ben | 120 |
| CoMoS | TiO2 | Fixed bed | 573 | 40 | — | — | GUA | — | 113 |
| CoMoS | ZrO2 | Fixed bed | 573 | 40 | — | — | GUA | — | 105 |
| CoMoS | C | Batch | 553 | 70 | 2 | — | GUA | — | 113 |
| CoMoS | Al2O3 | Fixed bed | 623 | 1 | — | 2EP, GUA | — | 145 | |
| NiMoS | — | Batch | 400–700 | 50 | 1 | — | GUA | Ben, Ph, CYHE, CYHA | 97 |
| NiMoS | — | Parr | 623 | 28 | 1 | — | Ph | — | 79 |
| NiMoS | γ-Al2O3 | Batch | 523 | 75 | 0.08–5 | — | Ph | CYHA | 88 |
| NiMoS | γ-Al2O3 | Flow reactor | 523 | 15 | — | — | Ph | CYHA | 69 |
| NiMoS | γ-Al2O3 | Batch | 523 | 75 | 2 | — | Ph | — | 69 |
| NiMoS | γ-Al2O3 | Batch | 723 | 28 | 1 | — | CAT | Ph, Ben, CYHA | 167 |
| NiMoS | γ-Al2O3 | Batch | 723 | 28 | 1 | — | GUA | Ben, Tol | 167 |
| NiMoS | γ-Al2O3 | Batch | 723 | 28 | 1 | — | Syr | Ben, Tol | 167 |
| MoS2 | — | Fixed bed | 573 | 40 | — | — | GUA | CYHE | 92 |
| MoS2 | — | Fixed bed | 573 | 50 | — | 1 | ANI | Ph, Ben | 117 |
| MoS2 | — | Batch | 593–643 | 28 | — | — | 4MP | Tol, Ph, 2,4-Xol | 81 |
| MoS2 | — | Batch | 623 | 44 | 5 | — | 4MP | Tol | 101 |
| MoS2 | — | Parr | 623 | 28 | 1 | — | Ph | Ben | 79 |
| MoS2 | — | Batch | 593–643 | 28 | 6 | — | 4MEP | Ben, Ph, ANI, MPh | 82 |
| MoS2 | C | Batch | 573 | 50 | 4.17 | GUA | Ph, CAT | 121 | |
| MoS2 | γ-Al2O3 | Fixed bed | 573 | 40 | — | — | GUA | CYHE, MCYHE | 92 |
| MoS2 | γ-Al2O3 | Fixed bed | 573 | 50 | — | 2 | ANI | Ph, Cr, 2,6-DMP | 117 |
| ReS2 | SiO2 | Batch | 573 | 50 | 4.4 | — | GUA | PH, Ben, CYHE, CYHA | 168 |
| γ-Al2O3 | PH, CAT | ||||||||
| Catalyst | Support | Reactor configuration | Operating conditions | Reactant | Major products | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| T (K) | P (bar) | t (h) | WHSV (h−1) | ||||||
| Ni2P | SiO2 | Packed bed | 573 | 1 | 0.33 | — | GUA | Ben | 101 |
| Ni2P | SiO2 | Fixed bed | 573 | 15 | 0–10 | 10 | ANI | Ph, Ben, CYHA | 90 |
| NiMoP | SiO2 | Fixed bed | 573 | 15 | 0–10 | 10 | ANI | Ph, Ben, CYHA | 90 |
| MoP | SiO2 | Packed bed | 573 | 1 | 0.33 | — | GUA | Ben | 101 |
| MoP | SiO2 | Fixed bed | 573 | 15 | 0–10 | 10 | ANI | Ph, Ben, CYHA | 90 |
| MoP | none | Batch | 573 | 44 | 5 | — | 4MP | Tol, MCYHA | 127 |
| Co2P | SiO2 | Packed bed | 573 | 1 | 0.33 | — | GUA | Ben | 101 |
| Fe2P | SiO2 | Packed bed | 573 | 1 | 0.33 | — | GUA | Ph | 101 |
| WP | SiO2 | Packed bed | 573 | 1 | 0.33 | — | GUA | Ph | 101 |
Metals, including bimetallics
Both mono- and bi-metallic catalysts have been tested for upgrading of lignin-derived bio-oils, and for appropriate accessibility of their surfaces (and corresponding high activity), the metals are dispersed on porous supports (Tables 1 and 2).In the upgrading reactions, there is generally a competition between HDO and hydrogenation of aromatic rings. Metals including iron, cobalt, nickel, ruthenium, palladium, rhodium, platinum, and iridium show activity. For example, Olcese et al.114 used iron supported on silica (Fe/SiO2) for HDO of guaiacol, finding it to have good selectivity for formation of benzene, toluene, and xylene without much aromatic ring hydrogenation. Fe/SiO2 was found to have a lower activity but higher selectivity for HDO than cobalt catalysts. Moreover, Fe/SiO2 produced less methane than cobalt-containing catalysts even at high conversions.114
An investigation115 of the effects of individual gases present in a pyrolysis mixture (H2, CO, CO2, H2O, CH4) on the selectivity of a Fe/SiO2 catalyst led to the conclusion that although CO has a strong positive effect on guaiacol HDO by reducing the rate of catalyst deactivation, CO2 has a strong negative effect on the initial reaction rate because of the resultant carburization of iron and coke formation. Water hinders the overall catalytic conversion and enhances the production of phenols rather than benzene as a consequence of the hydrolysis of adsorbed precursors. CH4 was found not to have a significant effect on the guaiacol conversion.115
In an investigation of the HDO of substituted phenols (including anisole, guaiacol, o-cresol, benzyl alcohol, catechol, and vanillyl alcohol) catalyzed by copper chromite at 423–548 K and 50 bar H2 partial pressure, Deutsch and Shanks116 concluded that copper catalyzed hydrogenation and hydrogenolysis reactions and Cr2O4, as a weak acid, catalyzed dehydration and transalkylation reactions. The authors concluded that in the conversion of anisole, demethoxylation to give benzene was the dominant reaction pathway, in contrast to the demethylation and transalkylation observed with conventional hydroprocessing catalysts. Thus, the authors inferred that their catalyst is characterized by unique selectivity and reaction networks for the removal of hydroxyl and methoxy groups relative to the widely investigated HDO catalysts.
Gutierrez et al.48 reported that the activity for guaiacol HDO decreased in the following order:
| Rh/ZrO2 > Co-MoS2/Al2O3 > Pd/ZrO2 > Pt/ZrO2 | (1) |
A number of investigators have reported experiments with bimetallic catalysts. For example, in the conversion of guaiacol, the combinations of rhodium with platinum and with palladium led to much better results than were observed with mono-metallic platinum and palladium catalysts.48 Huuska117 considered the effect of catalyst composition on the hydrogenolysis of anisole with catalysts containing molybdenum or nickel (Tables 5 and 6). When the catalysts were supported on an acidic oxide, the main products were phenol and methyl-substituted phenols, consistent with the rapid methyl group transfer catalyzed by the acidic sites. When the support was not acidic, only limited methyl group transfer occurred, and the main products were phenol and cyclic hydrocarbons. In related work, Ardiyanti et al.118 investigated catalytic hydroprocessing of anisole using Ni–Cu catalysts with various Ni/Cu ratios supported on δ-Al2O3. They observed lower conversions with the monometallic nickel than with the bimetallic catalyst. The higher activity of the bimetallic catalysts may be attributed to the incorporation of less nickel in the bulk Al2O3 when the copper is present and as a result less formation of nickel spinel than in the monometallic nickel catalyst.118
| Catalyst | Surface area (m2 g−l) | Bulk density (g cm−3) | Catalyst age (h) | Anisole conversion (%) |
|---|---|---|---|---|
| γ-Al2O3 | 100 | 0.8 | 4 | 22.5 |
| SiO2–Al2O3 | 100 | 0.6 | 4 | 10.2 |
| MoS2 (60% Mo) | 6.9 | 1.7 | 5 | 50.0 |
| Ni(11%)/SiO2 | 280 | 0.6 | 5 | 19.4 |
| Mo(6.7%)/γ-Al2O3 | 160 | 0.9 | 6 | 74.5 |
| Ni(11%)/γ-Al2O3 | 65 | 1.0 | 6 | 42.1 |
| Ni(0.5%)/γ-Al2O3 | 100 | 0.9 | 7 | 90.1 |
| Catalyst | Product | Selectivity, % |
|---|---|---|
| a Reaction conditions: experiments were carried out in a fixed-bed flow reactor fed with H2 and neat anisole containing CS2 to maintain catalysts in a sulfided state; catalysts D–G were presulfided; catalyst E was not. The temperature was 573 K; the H2 partial pressure 5 mPa; and the LHSV 2 h−1, except when the catalyst was C, for which the LHSV was 1 h−1. The data were collected after the each catalyst had been onstream for several hours. See the original reference for details regarding the mass balance closures and the calculation of the selectivities. | ||
| A, γ-Al2O3 | Phenol | 46.8 |
| o-Cresol | 27.9 | |
| 2,6-Dimethylphenol | 9.7 | |
| Other oxygen-containing compounds | 13.1 | |
| Total oxygen-containing compounds | 97.5 | |
| Benzene | 1.9 | |
| Other hydrocarbons | 0.6 | |
| Total hydrocarbons | 2.5 | |
| B, SiO2–Al2O3 | Phenol | 51.1 |
| o-Cresol | 12.9 | |
| 2,6-Dimethylphenol | 1.8 | |
| Other oxygen-containing compounds | 28.7 | |
| Total oxygen-containing compounds | 94.5 | |
| Benzene | 1.3 | |
| Other hydrocarbons | 4.1 | |
| Total hydrocarbons | 5.4 | |
| C, MoS2 (60% Mo) | Phenol | 63.1 |
| o-Cresol | 1.9 | |
| 2,6-Dimethylphenol | — | |
| Other oxygen-containing compounds | 0.4 | |
| Total oxygen-containing compounds | 65.4 | |
| Benzene | 27.8 | |
| Other hydrocarbons | 6.8 | |
| Total hydrocarbons | 34.6 | |
| D, Ni(11%)/SiO2 | Phenol | 53.8 |
| o-Cresol | 0.8 | |
| 2,6-Dimethylphenol | — | |
| Other oxygen-containing compounds | 5.1 | |
| Total oxygen-containing compounds | 59.7 | |
| Benzene | 31.4 | |
| Other hydrocarbons | 9.0 | |
| Total hydrocarbons | 40.4 | |
| E, Mo(6.7%)/γ-Al2O3 | Phenol | 47.0 |
| o-Cresol | 29.5 | |
| 2,6-Dimethylphenol | 14.8 | |
| Other oxygen-containing compounds | 1.8 | |
| Total oxygen-containing compounds | 93.1 | |
| Benzene | 3.0 | |
| Other hydrocarbons | 3.8 | |
| Total hydrocarbons | 6.8 | |
| F, Ni(11%)/γ-Al2O3 | Phenol | 77.1 |
| o-Cresol | 8.6 | |
| 2,6-Dimethylphenol | — | |
| Other oxygen-containing compounds | 0.6 | |
| Total oxygen-containing compounds | 86.3 | |
| Benzene | 8.3 | |
| Other hydrocarbons | 5.4 | |
| Total hydrocarbons | 13.7 | |
| G, Ni(0.5%)/γ-Al2O3 | Phenol | 40.4 |
| o-Cresol | 35.0 | |
| 2,6-Dimethylphenol | 18.6 | |
| Other oxygen-containing compounds | 5.0 | |
| Total oxygen-containing compounds | 99.0 | |
| Benzene | — | |
| Other hydrocarbons | 1.0 | |
| Total hydrocarbons | 1.0 | |
Jongerius et al.119 investigated the HDO of guaiacol catalyzed by carbon nanofiber-supported transition metal carbides (W2C/CNF and Mo2C/CNF) and obtained higher selectivity and conversion to phenolic compounds than with CoMo/Al2O3. According to their results, Mo2C/CNF has higher activity, selectivity, and stability than W2C/CNF and yields more completely deoxygenated aromatic products. The authors observed that higher temperatures and longer reaction times gave higher yields of the fully deoxygenated products benzene and toluene.
Metal sulfides
Metal sulfide catalysts are widely used in hydroprocessing technology to remove sulfur and nitrogen from petroleum. Many such catalysts have been tested for HDO. These include MoS2 and supported catalysts that incorporate cobalt and molybdenum or nickel and molybdenum (abbreviated as CoMoS and NiMoS).48,68,78–81,88,91,92,96,97,111,113,117,120,121 The supported catalysts typically incorporate clusters of MoS2 that include cobalt or nickel ions. These structures are maintained in the sulfide form in petroleum refining operations because the reaction environments contain H2 and H2S, which is formed in HDS reactions from components in petroleum such as thiols and thiophenic compounds. Because sulfur is almost absent from biomass, the supported metal sulfides would not be stable in the presence of the oxygen-containing compounds, and the catalysts would be converted into oxide forms—unless a sulfur source such as H2S or a thiol were added to the feed,89,122,123 with the detriment (a) that some sulfur would then be present in the product, which upon further processing with a noble metal catalyst would be poisoned by the sulfur-containing compounds or (b) that the product, upon combustion, would form sulfur oxides that pollute the atmosphere.Table 3 is a summary of results characterizing HDO catalyzed by metal sulfides. HDO of phenolic compounds catalyzed by sulfided CoMo/Al2O3 was investigated by Furimsky et al.124 Presulfiding of the catalyst had a pronounced effect on the conversion of phenols to benzene and cyclohexane. Vuori et al.125 observed a benefit of sulfiding of such catalysts in the conversion of guaiacol. Lin et al.97 inferred that demethylation, demethoxylation, and deoxygenation reactions following saturation of the aromatic ring in guaiacol accounted for the product distributions observed with sulfided CoMo/Al2O3 and NiMo/Al2O3 catalysts.
Ferrari et al.126 investigated the effect of H2S partial pressure on HDO of a mixture of compounds including guaiacol. They observed that increasing the H2S partial pressure led to inhibition of the hydrogenolysis and hydrogenation pathways. Gutierrez et al.48 reported that in guaiacol HDO with a conventional sulfided CoMo/Al2O3 catalyst, the products were contaminated with sulfur and the catalyst underwent deactivation associated with carbonaceous deposits.
Metal phosphides
Transition metal phosphides have been investigated widely as catalysts for petroleum hydroprocessing and drawn interest as candidate catalysts for bio-oils upgrading.90,101,109,127 Results are summarized in Table 4. These catalysts have relatively high activities in our context, with the reactions characterized by relatively low activation energies. Zhao et al.101 indicated that at high conversions of guaiacol, the major products were phenol, benzene, and methoxybenzene, whereas at low conversions, the main product was catechol. These authors observed higher HDO conversions with metal phosphide catalysts than with sulfided CoMo/Al2O3 catalysts, which underwent rapid deactivation. However, in the conversion of guaiacol, more coke formed on the phosphide than on the sulfide catalyst, consistent with the tendency of the Lewis acid sites (or Brønsted acid sites formed from them) of phosphide catalysts to favor the demethylation pathway. The authors reported the following decreasing order of activities for guaiacol HDO:| Ni2P > Co2P > Fe2P > WP > MoP | (2) |
Whiffen et al.109,127 considered HDO of 4-methylphenol on a MoP catalyst and compared it with that on other molybdenum-containing low-surface-area catalysts. They found the following decreasing order of activity measured by the conversion of 4-methylphenol:
| MoP > MoS2 > MoO2 > MoO3 | (3) |
The activation energy of the reaction increased in the reverse order.
Li et al.90 investigated the conversion of anisole catalyzed by Ni2P/SiO2, MoP/SiO2, and NiMoP/SiO2 with various Ni/Mo ratios. The reaction sequence was demethylation of anisole, hydrogenolysis of phenol, and hydrogenation of benzene. As the Ni/Mo ratio increased, the activity and stability of the phosphide catalyst increased. The nickel phosphide-containing catalysts, especially Ni2P/SiO2, were found to have higher activities than conventional NiMo/γ-Al2O3 catalysts.90 Yang et al.72 investigated the effect of phosphorus as an additive in a CoMo/MgO catalyst, finding that its addition gave a much more active catalyst for phenol conversion because of reduced coke formation.
Metal nitrides
Metal nitrides have also drawn attention as hydroprocessing catalysts and been investigated for HDO. Because of electronegativity differences between the metal and nitrogen atoms, nitrides can offer both acidic and basic sites. The potential advantages of metal nitride catalysts include their resistance to oxidation, ease of preparation, and low cost. Also, because of unique catalytic reaction pathways, hydrogen consumption may be minimized.In HDO reactions on nitride catalysts, nitrogen may be released from the bulk lattice and create surface sites for adsorption and catalysis.128 There is only little reported work on metal nitrides such as Mo2N as HDO catalysts.102,129 Ghampson et al.93,94 used Mo2N to catalyze guaiacol HDO, observing a high activity and a significant conversion of guaiacol to phenol when the catalyst was γ-Mo2N but not when it was β-Mo2N0.78 or other molybdenum-containing compounds such as MoO2 or molybdenum metal. Furthermore, their experiments showed that the bimetallic nitride catalyst CoMoN gave higher yields of deoxygenated products than the monometallic nitride catalyst, but the overall activity of the monometallic nitride catalyst was higher than that of the bimetallic.
Supports
A major role of a support is to stabilize the active catalytic species in a highly dispersed form, and one of the most commonly applied supports in HDO catalysts is alumina. The acidic character of a transition alumina influences the performance of the catalyst, being active for reactions such as transalkylation. Data characterizing the conversion of guaiacol are illustrative.68,92 A major disadvantage of the acidic support is the high yields of coke that are associated with the acidic surface sites. Weak Lewis acid sites have been inferred to lead to coke formation during HDO.68A number of recent investigations have focused on developing new supports for decreasing H2 consumption, increasing selectivity for direct oxygen removal, and minimizing coke formation. Echeandia et al.85 used Ni–W catalysts supported on active carbon for HDO of phenol, observing that much less coke formed on the carbon than on the alumina. Silica and carbon supports give catalysts with relatively high selectivities in HDO, but alumina-supported catalysts have higher activities.130
Lee et al.95 investigated various supports (Al2O3, SiO2–Al2O3, and nitric-acid-treated carbon black) for noble metal catalysts. Fig. 10 shows that the main product was cyclohexane when the support was SiO2–Al2O3, but when the support was Al2O3 or nitric-acid-treated carbon black, the yield of cyclohexanol and 2-methoxycyclohexanol exceeded that of cyclohexane. This comparison indicates that deoxygenation activity of the SiO2–Al2O3 support is higher than those of the other supports, and the SiO2–Al2O3 might have catalyzed dehydration of the alcohols.
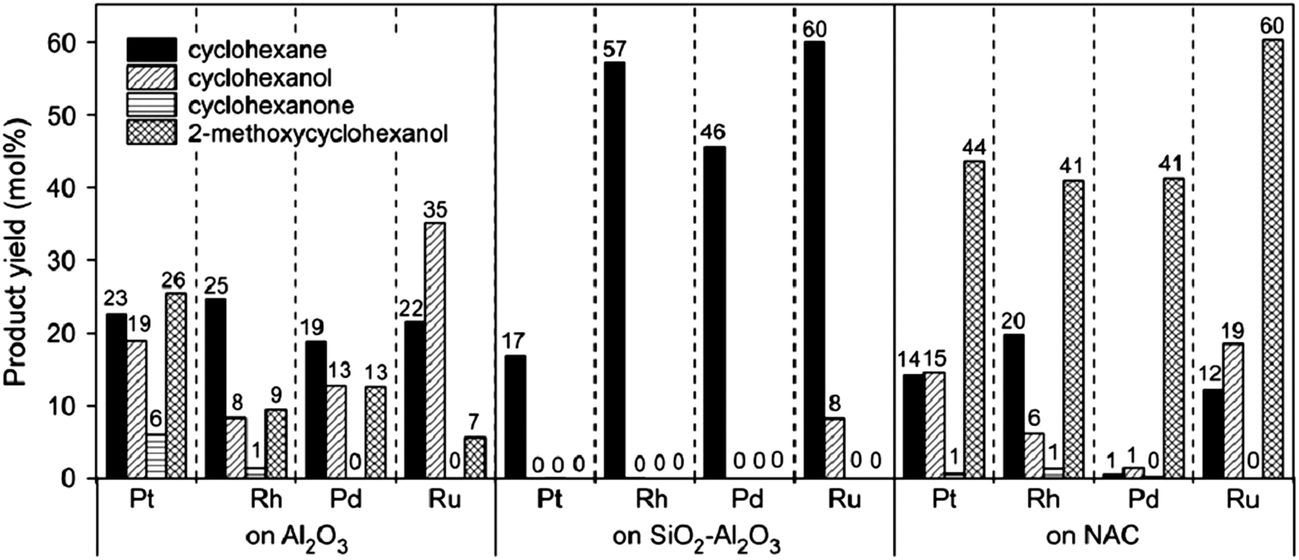 | ||
| Fig. 10 Product distributions in the HDO of guaiacol with platinum, rhodium, palladium, and ruthenium supported on alumina, silica-alumina, and nitric acid-treated carbon black.95 | ||
The acidic Al2O3 favors the formation of catechol via a demethylation route and subsequently the formation of methylphenols, but the more weakly acidic SiO2 favors the formation of phenol via a demethoxylation route. Sepúlveda et al.131 investigated the conversion of guaiacol catalyzed by ReS2 supported on Al2O3 and on SiO2, concluding that the ReS2/Al2O3 catalyst gives a higher initial rate of guaiacol conversion because of the direct formation of phenol via demethylation but that ReS2/SiO2 is more active for HDO because of the higher rate of formation of deoxygenated compounds. The main products observed with the ReS2/Al2O3 catalyst were phenol, catechol, and deoxygenated compounds such as benzene, cyclohexene, and cyclohexane, whereas with the ReS2/SiO2 catalyst, phenol and deoxygenated compounds were the major products.131
Loricera et al.91 investigated the HDO of anisole catalyzed by mesoporous silica (SBA-15 and SBA-16)-supported CoMoW catalysts, pointing out the potential advantages of these supports, including high surface areas and the opportunities to vary the framework compositions. Ghampson et al.93 compared the activities of Al2O3- and SBA-15-supported molybdenum nitride catalysts for HDO of guaiacol on the basis of reaction rates and phenol/catechol yields. They observed that the SBA-15-supported catalysts gave higher phenol/catechol yields than the Al2O3-supported catalysts because of the direct transformation of guaiacol to phenol. The lower production of catechol with the SBA-15 support is a potential advantage in minimizing coke formation and pore blocking in upgrading of mixtures of compounds in lignin-derived bio-oils.93
Bui et al.92,113 compared various supports for MoS2 and CoMoS catalysts in guaiacol HDO (Fig. 11); the supports were γ-Al2O3, ZrO2, and TiO2. ZrO2 gave a catalyst with a high activity and a higher selectivity for hydrogenolysis than the others. As expected, methyl group transfer reactions were kinetically significant when the support was γ-Al2O3, and the activity for formation of deoxygenated compounds was relatively low.48,113,132 An investigation of the conversion of lignin-derived phenolic compounds to hydrocarbons catalyzed by Ni/SiO2–ZrO2133,134 demonstrated high conversions and selectivities to hydrocarbons with high octane numbers. According to the authors, Ni/SiO2–ZrO2 catalysts have appropriate recyclability and anti-coking performance in the HDO reaction of model phenolic compounds because of the amphoteric character of the SiO2–ZrO2 support.
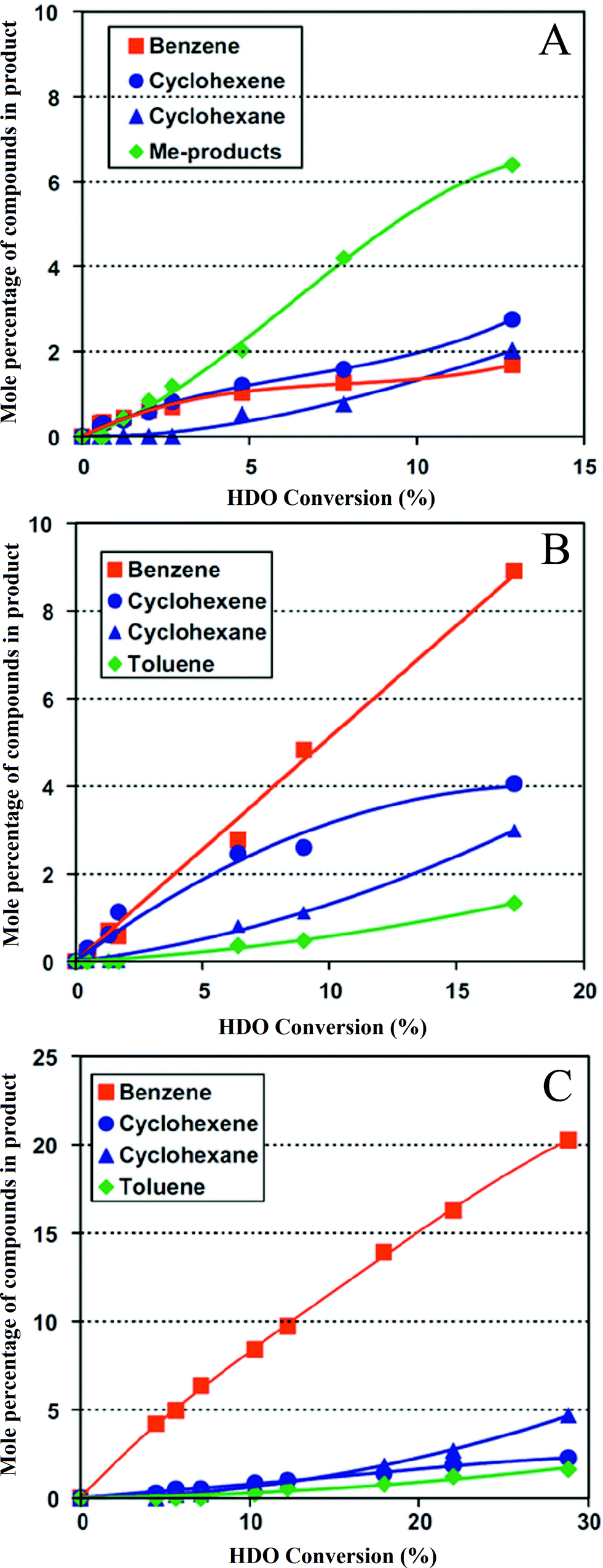 | ||
| Fig. 11 (A) Conversion of guaiacol catalyzed by cobalt-promoted MoS/Al2O3.113 (B) Conversion of guaiacol catalyzed by cobalt-promoted MoS/TiO2.113 (C) Conversion of guaiacol catalyzed by cobalt-promoted MoS/ZrO2.113 | ||
Yang et al.135 investigated the influence of supports on the selectivity of anisole HDO for aromatic products with a series of nickel-containing catalysts on supports including SBA-15, Al-SBA-15, γ-Al2O3, microporous carbon, TiO2, and cerium oxide. The data show that Ni/C catalysts not only give high conversions similar to those observed with Ni/SBA-15, Ni–Al-SBA-15, and Ni/γ-Al2O3, but that they lead to relatively low yields of oxygen-containing compounds in comparison with the other catalysts. The results indicate that, like Ni/C, the nickel catalysts supported on reducible oxides such as TiO2 and CeO2 are characterized by significant selectivities for formation of aromatic compounds. The data also demonstrate a trend of increasing selectivity for benzene at the expenses of cyclohexane for the series Ni/C, Ni/Al-SBA-15, and Ni/γ-Al2O3; the authors inferred that carbon deposition on nickel sites led to a moderate loss of hydrogenation activity, as a result enhancing aromatic production.135
Nimmanwudipong et al.54,55 investigated the conversion of guaiacol catalyzed by Pt/Al2O3 in the presence of H2, comparing the performance of this catalyst with that of the acidic HY zeolite. They observed that the two catalysts were similarly selective for HDO and transalkylation reactions but that the oxygen removal reactions were limited when the catalyst was the zeolite.
A few researchers have worked on catalysts supported on the basic MgO.100,136–140 Yang et al.72 used MgO-supported catalysts for HDO of phenolic compounds, concluding that they were highly active for conversion of phenol to benzene and cyclohexyl-aromatics. Nimmanwudipong et al.100 used Pt/MgO to catalyze the reactions of guaiacol, finding that the basic support gave a catalyst that was more stable than the acidic γ-Al2O3, presumably because the MgO led to less coke formation on the surface; the two catalysts were similar in activity.
Promoters
The metal sulfide catalysts used in petroleum hydroprocessing are much improved by the presence of promoters, typically cobalt or nickel in the presence of MoS2 supported on Al2O3; the promoter is present as ions in the layer structure of MoS2.80 Romero et al.141 investigated the effect of cobalt and nickel promoters on the performance of sulfided molybdenum catalysts used for HDO of 2-ethylphenol; each promoter increased the activity by about 1.7 times under their conditions. Yoosuk et al.80 showed that Ni–Mo sulfides were more active than nickel or molybdenum sulfides alone, and the maximum activity was obtained at a Ni/(Ni + Mo) atomic ratio of 0.3. Bui et al.92,113 considered the effect of cobalt as a promoter of MoS2 catalysts for HDO of guaiacol (Fig. 12), finding that it improved performance by increasing the rate of direct deoxygenation. These authors observed that the major deoxygenated products formed in the presence of the promoted catalyst were methyl-substituted aromatic compounds such as toluene, whereas methylcyclohexene was a major product when the catalyst was unpromoted.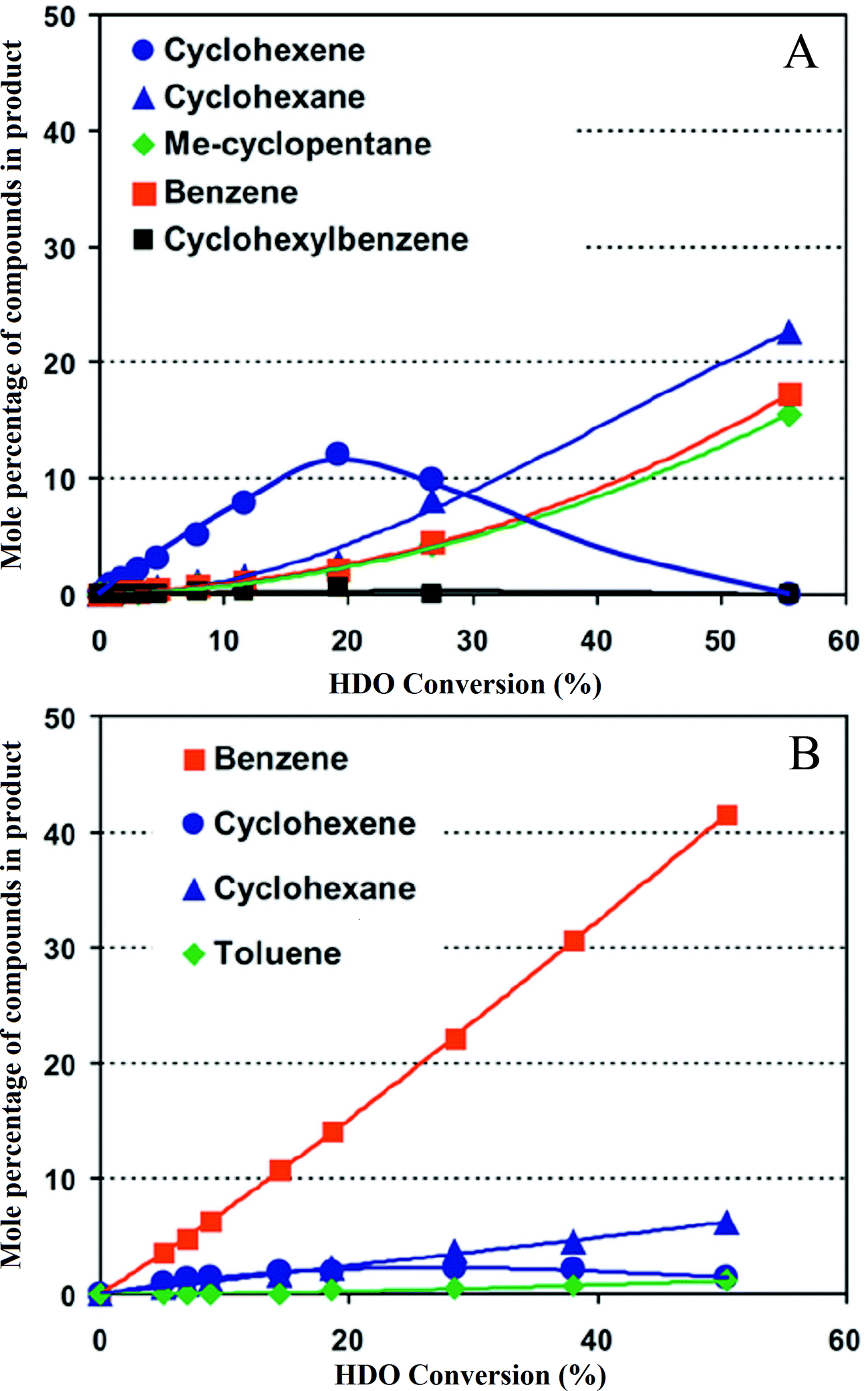 | ||
| Fig. 12 (A) Conversion of guaiacol at 573 K catalyzed by MoS2 (ref. 92) these data represent performance of the unpromoted catalyst. (B) Conversion of guaiacol at 573 K catalyzed by cobalt-promoted MoS.92 A comparison with (A) shows the influence of the promoter. | ||
One might tentatively suggest that the promotion of sulfided catalysts may affect HDO more or less as it affects HDS, although work is needed to test the suggestion. The roles of promoters in non-sulfide HDO catalysts remain to be elucidated.
Catalyst deactivation
Catalyst deactivation in HDO processes is caused by deposition of carbonaceous material (coke), sintering and loss of surface area of catalytic components such as metals, and poisoning of catalytic sites by compounds such as those containing nitrogen and phosphorus, which are present in biomass.112 Oxide-containing catalysts are not stable in liquid water at high temperatures, and water produced in HDO can destroy catalysts.Catalyst deactivation typically influences the rates of HDO and accompanying reactions such as transalkylation differently because they often take place on different components of the catalyst (e.g., metal particles and acidic supports).73
The major cause of deactivation of HDO catalysts as stated in published reports is evidently associated with deposits of coke or related materials; besides covering catalytic sites, the deposits may block pores, especially those of small-pored materials such as zeolites. Coke formation occurs through polymerization and polycondensation reactions and takes place at rates that depend strongly on the nature of catalyst, the nature of the bio-oil compounds, and the operating conditions. For example, Nimmanwudipong et al.55 in an investigation of guaiacol conversion found that the conversion declined by about 50% within only an hour on stream when the catalyst support was HY zeolite, whereas the deactivation was much slower with Pt/γ-Al2O3 under similar conditions (Fig. 13), presumably because the blocking of zeolite pores was much more pronounced than the blocking of the larger γ-Al2O3 pores.
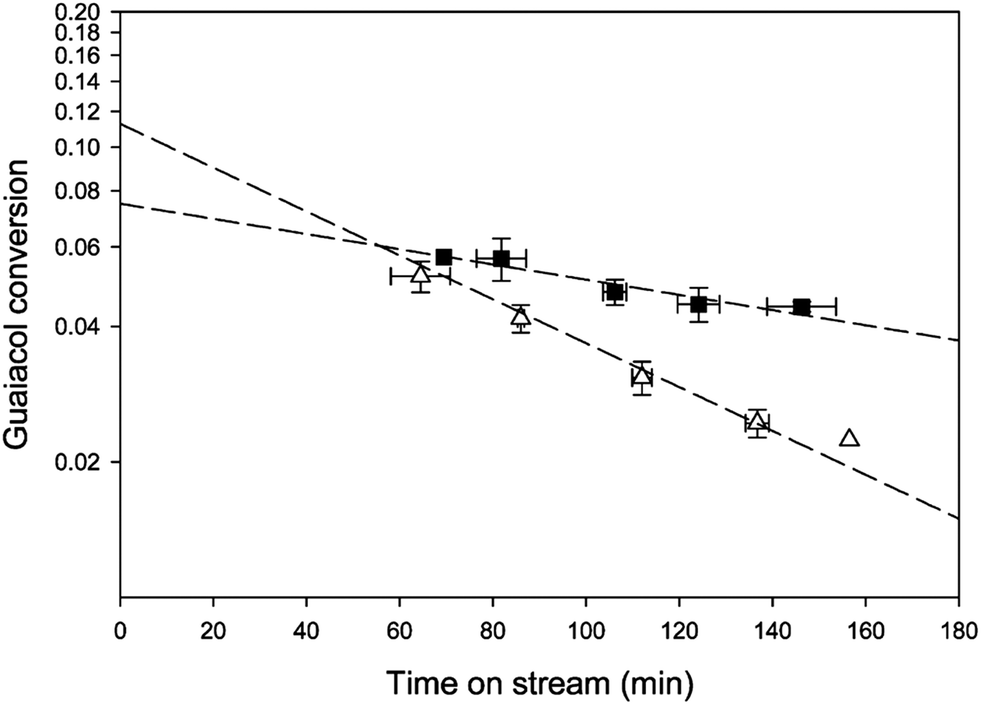 | ||
| Fig. 13 Conversion of guaiacol catalyzed by (■) Pt/γ-Al2O3 and (△) HY zeolite at 573 K.55 | ||
Because of their high concentrations of unsaturated hydrocarbon moieties, lignin-derived bio-oils are strongly prone to coke formation. These reactants typically interact strongly with surface catalytic sites, likely facilitating coke formation. During hydrotreating reactions, it is observed that adsorption of phenolic components on the support hinders the access of other compounds to the active sites.14 Oxygen-containing compounds with more than one oxygen atom, such as catechol, have been inferred to be more prone to coke formation than phenol.67,101,142 As mentioned above, although the acidity (Lewis and Brønsted) increases HDO activity, it also fosters catalyst deactivation by coke formation.30 Thus, one might expect that an optimum in strength and number of acidic sites may maximize catalyst performance in HDO, but this suggestion is still preliminary. One also expects that coke formation can be mitigated by lowering the temperature and reactant-catalyst contact time and by increasing the H2 partial pressure, especially when metals are present to catalyze dissociation of H2 and thereby facilitate hydrogenation of coke precursors.73,91,92 Because higher temperatures increase the rates of HDO reactions, it may be useful to use staged reactors operating with different catalysts and different temperatures to find the best trade-offs between rates of desired catalytic reactions and rates of catalyst deactivation.143 Catalyst staging is common in petroleum hydroprocessing. More data are needed to test these hypotheses and provide more thorough quantitative results.
Catalyst poisons such as compounds of nitrogen and even oxygen-containing compounds (e.g., water, CO) may compete with the reactants for catalytic sites in HDO. Adsorption of poisons on Lewis and Brønsted acidic sites depends on reaction conditions.112 Water and other oxygen-containing compounds have an inhibiting effect on conventional metal sulfide and metal phosphide catalysts (e.g., NiMo/Al2O3 and CoMo/Al2O3).90,144 Sulfide catalysts may be deactivated as the sulfide structures are converted to oxides. Addition of sulfur-containing compound such H2S to the feed can minimize such catalyst deactivation, but at the potential cost of formation of undesirable sulfur-containing products.48,85,90,144 Water also causes the oxidation of phosphide catalysts, leading to the formation of metal oxides and metal phosphates.90 Odebunmi and Ollis83 observed a reduction of catalytic activity caused by water during HDO of m-cresol, and they regenerated the catalyst by resulfiding it. Laurent and Delmon67,89 observed that water caused only weak inhibition in HDO of phenols relative to that observed with compounds such as H2S and NH3, but when a high enough concentration of H2O–H2S was present in the feed, the detrimental effect of water was more significant. Nevertheless, water may have a beneficial effect by mitigating the temperature increases resulting from the strong exothermicity of hydrotreating reactions and thereby minimize catalyst deactivation resulting from sintering.
In attempts to understand catalyst deactivation in HDO, Popov et al.145 investigated the interactions of guaiacol, phenol, and ethylphenols with a sulfided CoMo/Al2O3 catalyst. They concluded that the surface properties of the catalyst support and the basicity and nature of the oxygen-containing compound affect the interactions and the resultant poisoning. Phenate-type species on the Al2O3 support obstruct the accessibility to the metal sulfide crystal edge sites where catalysis occurs. The metal sulfide sites are also poisoned by oxygen-containing compounds.145
The data summarized in Table 7 provide information about carbon formation on various catalysts after catalytic reactions of guaiacol. The data indicate that mono-metallic catalysts are less resistant to coke formation than bimetallics, but one should be cautious about generalizing this statement. A generalization that is more strongly supported by data is that catalysts with more strongly acidic supports typically undergo deactivation faster than those with less strongly acidic supports, as exemplified by data characterizing NiMo supported on SiO2–Al2O3vs. γ-Al2O3.146 H-Beta zeolite was found to be characterized by a higher rate of deactivation than Pt/H-Beta zeolite (or Pt/SiO2), because dissociation of H2 on the metal regenerated acidic sites by hydrogenation of coke precursors and/or coke.73 The hydrophobic character of carbon surfaces corresponds to less rapid catalyst deactivation by water than occurs on acidic supports.85,92 But recall that acidic groups on supports, although they are responsible for coke formation, also may be desired because they help catalyze hydrogenolysis and hydrocracking.91
| Catalyst | Operating conditions | Carbon deposition (wt%) | Ref. | ||
|---|---|---|---|---|---|
| T (K) | P (bar) | t (h) | |||
| γ-Al2O3 | 553 | 70 | 3 | 10.3 | 68 |
| K–CoMoS–Al2O3 | 553 | 70 | 3 | 9.5 | 68 |
| Pt–CoMoS–Al2O3 | 553 | 70 | 3 | 9.1 | 68 |
| CoMoS–Al2O3 | 553 | 70 | 3 | 8.9 | 68 |
| CoMoS | 553 | 70 | 3 | 2.8 | 68 |
| CoMoS/SiO2 | 553 | 70 | 3 | 2.7 | 68 |
| Ni/SiO2 | 593 | 170 | 1 | 15.8 | 48 |
| NiCu/SiO2 | 593 | 170 | 1 | 2.3 | 48 |
| CoMoS–Al2O3 | 373 | 80 | 5 | 6.7 | 61 |
| Rh/ZrO2 | 373 | 80 | 5 | 1.8 | 61 |
| Pd/ZrO2 | 373 | 80 | 5 | 0.7 | 61 |
| Pt/ZrO2 | 373 | 80 | 5 | 0.6 | 61 |
| ZrO2 | 373 | 80 | 5 | 0.5 | 61 |
Kinetics
Because of the complexity and variability of structures of compounds in bio-oils and variations in molecular weights and concentration, little quantitative information characterizing the kinetics of bio-oils hydroprocessing is available. In HDO process, oxygen can be removed as CO2, CO, H2O, and other compounds, by a variety of reactions. Little information is available to resolve the reactions and characterize their rates separately.Most of the available kinetics data characterize reactions of individual oxygen-containing compounds in the presence of H2. A number of researchers have represented their data in terms of reactions that are first-order in the oxygen-containing compound.51–53,56,68,79,82,85,101,109,127,147,148 Summaries of some pseudo-first-order rate constants and apparent activation energies for conversion of lignin-derived bio-oil compounds are summarized in Tables 8–10.
| Reactant compound | Catalyst | Pseudo-first-order rate constant | Dimensions of rate constant | Ref. |
|---|---|---|---|---|
| a At 598, 623, 648 K. b At 598, 623, 648 K. c At 598, 623, 648 K. d At 598, 623, 648 K. | ||||
| ANI | Pt/Al2O3 | 19 (at 573 K) | L per g of catalyst per h | 52 and 53 |
| GUA | Pt/Al2O3 | 16.2 (at 573 K) | L per g of catalyst per h | 55 |
| GUA | γAl2O3 | 0.02 (at 553 K) | L per g of catalyst per h | 68 |
| GUA | CoMo/Al2O3 | 0.08 (at 553 K) | L per g of catalyst per h | 68 |
| GUA | CoMo/Al2O3 modified with Pt | 0.04 (at 553 K) | L per g of catalyst per h | 68 |
| GUA | CoMo/SiO2 | 0.02 (at 553 K) | L per g of catalyst per h | 68 |
| GUA | CoMo/C | 0.01 (at 553 K) | L per g of catalyst per h | 68 |
| GUA | CoMoS | 0.02 (at 553 K) | L per g of catalyst per h | 68 |
| 4MP | MoO3 | 4.54, 21.7, 29.3a | L h−1 molMo−1) | 109 |
| 4MP | MoO2 | 1.04, 3.17, 6.16b | L h−1 molMo−1 | 109 |
| 4MP | MoS2 | 1.04, 2.70, 6.19c | L h−1 molMo−1 | 109 |
| 4MP | MoP | 2.74, 9.43, 13.25d | L h−1 molMo−1 | 109 |
| CYHAONE | Pt/γ-Al2O3 | 90 (at 573 K) | L per g of catalyst per h | 51 |
| 4MA | Pt/γ-Al2O3 | 11 (at 573 K) | L per g of catalyst per h | 147 |
| 4MA | Pt/SiO2–Al2O3 | 31 (at 573 K) | L per g of catalyst per h | 147 |
| EUG | Pt/γ-Al2O3 | 12.4 (at 573 K) | L per g of catalyst per h | 56 |
| 4MP | CoMoS | 0.14, 0.03 (at 573 K) | L per g of catalyst per h | 96 |
| 2MP | CoMoS | 0.08, 0.01 (at 573 K) | L per g of catalyst per h | 96 |
| Reaction number (keyed to Fig. 14) | Reactant | Rate constanta | Reaction class |
|---|---|---|---|
| a Rate constants in L per g catalyst per h were determined for the conversion of reactant to a particular product; for example, phenol is formed in more than one reaction and the rate constant accounts for the formation of phenol in both of these reactions. | |||
| 1 | Guaiacol | 0.11 | Hydrodeoxygenation |
| 2 | Guaiacol | 4.4 | Hydrodeoxygenation |
| 3 | Guaiacol | 6.5 | Hydrogenolysis |
| 4 | Guaiacol | 1.8 | Transalkylation |
| 5 | Guaiacol | 0.50 | Bimolecular transalkylation |
| 6 | Guaiacol | 0.21 | Bimolecular transalkylation |
| 7 | Guaiacol | 0.26 | Bimolecular transalkylation |
| 8 | Anisole | 12 | Hydrogenolysis |
| 9 | Anisole | 0.86 | Hydrodeoxygenation |
| 10 | Anisole | 2.8 | Transalkylation |
| 11 | Anisole | 0.039 | Transalkylation |
| 12 | Anisole | 0.14 | Bimolecular transalkylation |
| 13 | 4-Methylanisole | 0.76 | Hydrodeoxygenation |
| 14 | 4-Methylanisole | 4.2 | Hydrogenolysis |
| 15 | 4-Methylanisole | 2.2 | Transalkylation |
| 16 | Cyclohexanone | 77 | Dehydrogenation |
| 17 | Cyclohexanone | 5.5 | Hydrogenation |
| 18 | Cyclohexanone | 0.05 | Hydrodeoxygenation |
Runnebaum et al.53 considered the reaction network of anisole conversion catalyzed by Pt/Al2O3 and presented pseudo-first-order rate constants for the removal of anisole, using selectivity-conversion data to determine the rate constants for formation of individual products. The rate constants for formation of phenol, 2-methylphenol, 2-methylanisole, and 4-methylphenol as primary products were found to be 12, 2.8, 0.14, and 0.039 L per g of catalyst per h, respectively, at 573 K and atmospheric pressure. Nimmanwudipong et al.55 developed an extensive reaction network for the conversion of guaiacol catalyzed by Pt/γ-Al2O3, reporting a set of pseudo-first-order rate constants for guaiacol conversion at 573 K (Table 10; this table is keyed to Fig. 14).
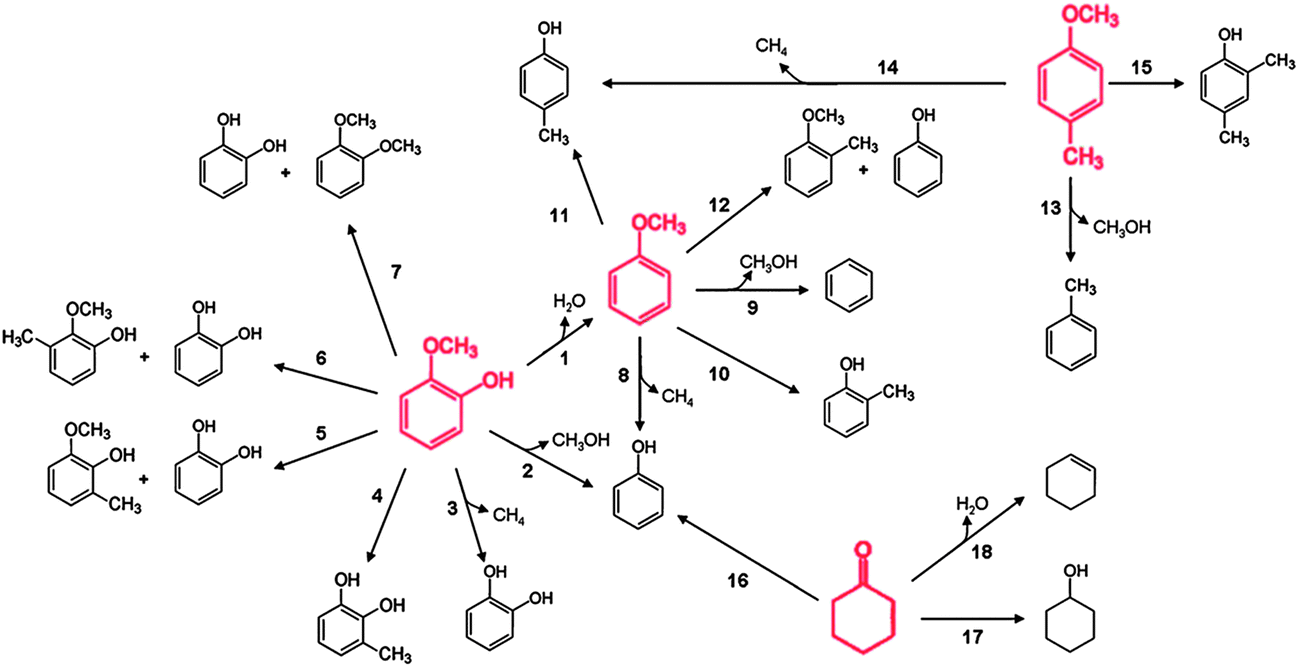 | ||
| Fig. 14 Reaction network accounting for formation of primary products determined from analysis of selectivity-conversion plots for the conversion of individual reactants (shown in red), guaiacol, anisole, 4-methylanisole, and cyclohexanone, catalyzed by Pt/Al2O3 in the presence of H2 at 573 K and atmospheric pressure. The reactant H2 is omitted for simplicity. Pseudo-first-order rate constants for the individual primary products formed in the conversion of the individual reactants catalyzed by Pt/Al2O3 are shown in Table 10; the numbers next to the arrows in this figure are keyed to the list in Table 10.103 | ||
Zhang et al.149 investigated the HDO of bio-oils in the presence of a Co–MoS2/Al2O3 catalyst in a batch reactor and temperatures between 633 and 663 K. They inferred an overall reaction order of 2.3 for HDO (and an order such as this may correspond to simultaneous first-order reactions of each of a group of oxygen-containing compounds). A weak dependence of rate on H2 partial pressure was observed in the range of 15 to 30 bar. In an investigation of guaiacol HDO on transition metal phosphide catalysts,101 activation energies in the range of 23–63 kJ mol−1 were observed.
Massoth et al.79 used Langmuir–Hinshelwood expressions to represent the kinetics of HDO of methyl-substituted phenols catalyzed by a sulfided CoMo/Al2O3 in a flow microreactor at 573 K and 2.85 MPa H2 partial pressure. The equations account for adsorption of the organic reactants on catalytic sites and for competitive direct deoxygenation and hydrogenation reactions; they do not account for adsorption of hydrogen or products and do not distinguish between adsorption of reactants on different catalytic sites for direct deoxygenation and hydrogenation. One may expect that, as more complete data become available to characterize HDO reactions, Langmuir–Hinshelwood expressions will be found to apply with some generality, but now there are too few data to justify any generalizations.
Bui et al.92,113 investigated the kinetics of HDO of guaiacol in the presence of CoMo catalysts in a fixed-bed reactor, obtaining data that they represented by first-order kinetics. Such an approximation was also made by Runnebaum et al.,103 as shown in the comparison of reactivities of various compounds shown in Table 10 for reactions catalyzed by Pt/Al2O3.
In sum, there is a lack of detailed, quantitative determinations of kinetics of HDO and a need for further research in this direction, with the expected benefits being quantitative indications of the relative strengths of adsorption of the various reactants and products on various catalysts and comparisons of the reactivities of various compounds with various catalysts. Such results would help elucidate patterns of competitive adsorption and reaction in catalytic HDO and lead to equations corresponding to Langmuir–Hinshelwood forms.
Reactor configurations and catalyst staging
Data indicating the types of reactors and operating conditions in investigations of HDO of lignin-derived bio-oils are summarized in Tables 1–4. Testing of catalysts for HDO of bio-oils has been carried out under a wide range of conditions, and, roughly speaking, one refers to mild HDO taking place at about 523 K and 100 bar and to severe HDO taking place at about 623 K and 200 bar. We emphasize that most of the experiments with individual compounds as reactants were carried out under milder conditions than these.When the reactors are staged, operation of the upstream reactor operating under the milder conditions may facilitate hydrogenation, and operation of the downstream reactor under more severe conditions may facilitate deoxygenation. This approach allows selection of the optimum catalyst for each of the stages with respect to the oil yield and degree of deoxygenation. Elliot et al.36,37 proposed such a configuration of reactors, with the upstream reactor operated at a temperature below 553 K, a pressure of about 140 bar, and a liquid hourly space velocity (LHSV) of 0.62. A deep deoxygenation step was performed in the downstream reactor in the presence of H2 at a total pressure of 142 bar, a temperature higher than 623 K, and a very low LHSV of 0.11. Oil yields between 30 and 55% and deoxygenation conversions up to 99% were achieved.
In early bio-oils HDO research, batch reactors were typically used, and two-step procedures were applied; in the first step (mild HDO), thermal decomposition reactions were significant, and overall conversions for oxygen removal were typically about 50%. In the second step, conversions for oxygen removal of more than 90% at about 623 K and 200 bar were typically achieved in reaction times of 2 h.
Most recent experiments characterizing bio-oils HDO have been carried out in continuous packed-bed flow reactors. Downflow has given better performance and less tendency to plug the reactors than upflow, and residence time distributions have favored higher conversions in downflow operation because of the optimized contacting of reactant and catalyst.27,150
Research opportunities
Investigations of HDO of lignin-derived bio-oils and bio-oil components for fuel production are still at an early stage, and much more information is needed to improve candidate catalysts and processes if commercialization is to become economical. Commercial applications do not seem to be close at hand absent public subsidies for research and development or for avoidance of non-renewable fuels. It is not yet evident what the most appropriate catalysts will be, as insufficient work has been done to identify the optimum combinations of catalyst functions, let alone the specific catalyst components.We reemphasize the usefulness of the approach of identifying catalyst functions for particular classes of reactions (determined with individual compounds) and quantification of the catalyst performance data as a basis for identifying improved catalysts. We also recommend investigations of the conversions of individual compounds in whole bio-oils by application of analysis methods like those used for investigations of petroleum conversion; the methods include high-resolution mass spectrometry and gas chromatography. These methods are expensive and time consuming but can yield a wealth of data that would not be characterized by the limitations of those determined with individual compounds and would correctly reflect competitive adsorption and reaction effects that cannot be determined in experiments with individual compounds as feeds.
The reported work reflects a strong emphasis on catalysts that have been well investigated for hydroprocessing of reactants that do not contain oxygen, and the implicit assumption that HDS, HDN, and hydrogenation catalysts may be good choices for HDO is not well tested. The oxygen-containing compounds present in biomass-derived feedstocks have their own reactivities that seemingly demand their own catalysts, and we suggest that these catalysts are yet to be discovered and may be much different from those used in hydroprocessing today. A specific goal of catalyst development might be to identify catalysts that function with minimal H2 consumption by allowing C–O bond breaking without substantial aromatic ring saturation. Rapid throughput catalyst testing is expected to have a place in future research on HDO.
The optimum catalysts for any application will depend on the processing goals, which could range from production of partially deoxygenated mixtures that could be co-fed with petroleum in petroleum refineries for production of fuels or fuel blending components—or production of chemicals, of which there are many that could be formed from bio-oils. It is still an open question which candidate processing goals may turn out to be practically important, and one realizes that projections of such goals will depend on the discovery of new catalysts. Process conception and catalyst discovery are intertwined (and one should not neglect the truism that catalysts are discovered for needed processes, and processes are almost never found for intriguing new catalysts).
Much remains to be learned about how candidate catalysts function over long times and how their activities, selectivities, and stabilities depend on composition, temperature, pressure, the ratio of H2 to feedstock, and the presence of minor components such as those containing phosphorus, metals, and other heteroatoms in the bio-oils. The effects of these components vary widely from one catalyst class to another. Systematic variations of these processing variables and long-term testing to determine catalyst lifetimes and performance are needed, but such testing is expensive and time consuming. It might be valuable to apply modified rapid-throughput catalyst testing methods to screen not just for initial activity and selectivity but to test for long-term catalyst performance.
Development of improved catalysts and processes would also benefit from fundamental investigations of reaction networks and kinetics expanded beyond compounds such as phenol, anisole, and guaiacol to larger compounds with more functional groups (such as those reported by Sergeev and Hartwig44,151 (Fig. 15)) and to reactions of mixtures of compounds that adsorb competitively on catalytic sites—one might begin with pairs of compounds and extend the work to increasingly complex mixtures. It would be valuable in all these experiments to use catalysts with prototypical functions (e.g., noble metals) and to explore a wide range of reaction conditions, especially pressure, as little is known about how H2 partial pressure affects catalyst performance. Investigations of reaction mechanism would also help in the identification of catalyst functions. Kinetics of HDO would also be helpful in providing a quantitative framework for interpreting data and elucidating reaction mechanisms.
 | ||
| Fig. 15 Compounds investigated by Sergeev and Hartwig with: (A) 4-O–5 linkages, (B) α-O–4 linkages, and (C) β-O–4 linkages in lignin.44,151 | ||
It might be also fruitful to explore reactions that produce H2 taking place simultaneously with or in concert with HDO to minimize costs of H2. And research with reactions in ionic liquids and supercritical fluids and even in plasmas is worthy of attention.
Conclusions
Lignin is a major component of biomass that is chemically similar to petroleum but differs in having high oxygen content. Lignin offers the prospect of being an important source of renewable fuels, but production of fuels from lignin requires the removal of much of the oxygen. Removal of the oxygen-containing compounds results from catalytic conversion with H2 of the compounds formed from lignin, for example, by pyrolysis. The reactions are called hydrodeoxygenation (HDO), and extensive recent research has been done on HDO of various biomass-derived compounds, including those derived from lignin and typified by phenol, anisole, and guaiacol. Depending on the catalyst, the reactions include direct removal of water, hydrogenation of aromatic rings, and migration of methyl groups in the bio-oils compounds. Catalysts with various functions, such as noble metals and metal sulfides typified by MoS2, catalyze HDO, but side reactions also occur, and the catalysts undergo deactivation by deposition of carbonaceous residues. Much work is needed to determine good catalysts and elucidate how various catalyst functions (such as metals and acidic supports) affect the conversions and product distributions. Recent work with individual compounds representative of bio-oils is helping to elucidate the chemistry, and one may anticipate systematic progress in the identification of catalysts and process conditions for HDO, although it is likely to be slow to determine optimum catalysts and processing conditions for whole bio-oils. Economical processes for conversion of lignin to fuels that are compatible with today's infrastructure are not close at hand, and much research is needed, as recommended in this review.Abbreviations
| ANI | Anisole |
| Ben | Benzene |
| CAT | Catechol |
| CNF | Carbon nanofiber |
| Cr | Cresol |
| CYHA | Cyclohexane |
| CYHAOL | Cyclohexanol |
| CYHAONE | Cyclohexanone |
| CYHE | Cyclohexene |
| DDO | Direct deoxygenation |
| DME | Demethylation |
| DMO | Demethoxylation |
| 2,6-DMP | 2,6-Dimethylphenol |
| 2EP | 2-Ethylphenol |
| EUG | Eugenol |
| 2EXYHAONE | 2-Ethoxycyclohexanone |
| GUA | Guaiacol |
| HDO | Hydrodeoxygenation |
| HYD | Hydrogenation |
| LHSV | Liquid hourly space velocity |
| MA | Methylanisole |
| 4MA | 4-Methylanisole |
| MCYHA | Methylcyclohexane |
| MCYHE | Methylcyclohexene |
| 2MECYHAOL | 2-Methoxycyclohexanol |
| 2ME4MEPH | 2-Methoxy-4-methylphenol |
| 4MEP | 4-Methoxyphenol |
| MCYHDIOL | 1-Methyl-1,2-cyclohexanediol |
| MP | Methylphenol |
| 4MP | 4-Methylphenol |
| P | Pressure |
| Ph | Phenol |
| PCYHA | Propylcyclohexane |
| 4PG | 4-Propylguaiacol |
| 4PP | 4-Propylphenol |
| Syr | Syringol |
| t | Time |
| T | Temperature |
| 2,4,6-TMP | 2,4,6-Trimethylphenol |
| Tol | Toluene |
| VA | Vanillyl |
| WHSV | Weight hourly space velocity |
| Xol | Xylenol |
| 2,4-Xol | 2,4-Xylenol |
| Xy | Xylene. |
Acknowledgements
T. Nimmanwudipong was supported at the University of California, Davis, by a fellowship from Chevron.References
- F. K. Forson, E. K. Oduro and E. Hammond-Donkoh, Renewable Energy, 2004, 29, 1135–1145 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- A. Németh, M.Sc. thesis, Wageningen University Rabobank, 2009.
- J. Akhtar and N. A. S. Amin, Renewable Sustainable Energy Rev., 2011, 15, 1615–1624 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- M. V. Olarte, Ph.D. thesis, Georgia Institute of Technology, Atlanta, 2011.
- L. Ma, T. Wang, Q. Liu, X. Zhang, W. Ma and Q. Zhang, Biotechnol. Adv., 2012, 30, 859–873 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch and Y. Y. Lee, Bioresour. Technol., 2005, 96, 1959–1966 CrossRef CAS PubMed.
- H. L. Chum and R. P. Overend, Prepr. Symp. – Am. Chem. Soc., Div. Fuel Chem., 2004, 49, 798–799 CAS.
- D. Ferdous, A. K. Dalai, S. K. Bej and R. W. Thring, Can. J. Chem. Eng., 2010, 79, 913–922 CrossRef.
- J. B. Binder, M. J. Gray, J. F. White and Z. C. Zhang, Biomass Bioenergy, 2009, 33, 1122–1130 CrossRef CAS PubMed.
- J. Holmgren, R. Marinageli, P. Nair, D. C. Elliott and R. Bain, Hydrocarbon Process., 2008, 95–103 CAS.
- R. H. Venderbosch, A. R. Ardiyanti, J. Wildschut, A. Oasmaa and H. J. Heeres, J. Chem. Technol. Biotechnol., 2010, 85, 674–686 CrossRef CAS.
- L. Lin, Z. Cunshan, S. Vittayapadung, S. Xiangqian and D. Mingdong, Appl. Energy, 2011, 88, 1020–1031 CrossRef PubMed.
- M. F. Demirbas, Appl. Energy, 2011, 88, 17–28 CrossRef PubMed.
- H. Wenzel, Breaking the biomass bottleneck of the fossil free society, Technical report, Concito, 2010 Search PubMed.
- P. McKendry, Bioresour. Technol., 2002, 83, 37–46 CrossRef CAS.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68–72 CrossRef CAS PubMed.
- P. Grange, E. Laurent, R. Maggi, A. Centeno and B. Delmon, Catal. Today, 1996, 29, 297–301 CrossRef CAS.
- C. Perego and A. Bosetti, Microporous Mesoporous Mater., 2011, 144, 28–39 CrossRef CAS PubMed.
- J. G. Rogers and J. G. Brammer, Biomass Bioenergy, 2009, 33, 1367–1375 CrossRef PubMed.
- E. Furimsky, Appl. Catal., A, 2000, 199, 144–190 CrossRef.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- Q. Zhang, J. Chang, T. Wang and Y. Xu, Energy Convers. Manage., 2007, 48, 87–92 CrossRef CAS PubMed.
- R. H. Venderbosch and W. Prins, Biofuels, Bioprod. Biorefin., 2010, 4, 178–208 CrossRef CAS.
- A. Oasmaa, Y. Solantausta, V. Arpiainen, E. Kuoppala and K. Sipil, Energy Fuels, 2010, 24, 1380–1388 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- T. V. Choudhary and C. B. Phillips, Appl. Catal., A, 2011, 397, 1–12 CrossRef CAS PubMed.
- Q. Bu, H. Lei, A. H. Zacher, L. Wang, S. Ren, J. Liang, Y. Wei, Y. Liu, J. Tang, Q. Zhang and R. Ruan, Bioresour. Technol., 2012, 124, 470–477 CrossRef CAS PubMed.
- Z. He and X. Wang, Catal. Sustainable Energy Prod., 2013, 28–52 CAS.
- D. D. Laskar, B. Yang, H. Wang and J. Lee, Biofuels, Bioprod. Biorefin., 2013, 7, 602–626 CrossRef CAS.
- E. Furimsky, Catal. Today, 2013, 217, 13–56 CrossRef CAS PubMed.
- D. C. Elliott and E. G. Baker, Biotechnol. Bioeng. Symp., 1984, 14, 159–174 CAS.
- D. C. Elliott and G. F. Schiefelbein, Abstr. Pap. Am. Chem. Soc., 1989, 34, 1160–1166 CAS.
- D. C. Elliott and G. G. Neuenschwander, Liquid fuels by low-severity hydrotreating of biocrude, in Developments in thermochemical biomass conversion, ed. A. V. Bridgwater and D. G. Boocock, Blackie, London, 1996, vol. 1, pp. 611–621 Search PubMed.
- D. C. Elliott, K. L. Peterson, D. S. Muzatko, E. V. Alderson, T. R. Hart and G. G. Neuenschwander, Appl. Biochem. Biotechnol., 2004, 113–116, 807–825 CrossRef CAS.
- A. H. Zacher, D. Elliott and D. Santosa, AIChE Annual Meeting, Lake City, 2010 Search PubMed.
- J. Gagnon and S. Kaliaguine, Ind. Eng. Chem. Res., 1988, 27, 1783–1788 CrossRef CAS.
- Y. H. E. Sheu, R. G. Anthony and E. J. Soltes, Fuel Process. Technol., 1988, 19, 31–50 CrossRef CAS.
- A. Oasmaa and D. G. B. Boocock, Can. J. Chem. Eng., 1992, 70, 294–300 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- D. C. Elliott and T. R. Hart, Energy Fuels, 2009, 23, 631–637 CrossRef CAS.
- D. Meier, J. Berns, O. Faix, U. Balfanz and W. Baldauf, Biomass Bioenergy, 1994, 7, 99–105 CrossRef CAS.
- G. Telysheva, G. Dobele, D. Meier, T. Dizhbite, G. Rossinska and V. Jurkjane, J. Anal. Appl. Pyrolysis, 2007, 79, 52–60 CrossRef CAS PubMed.
- A. Gutierrez, R. K. Kaila, M. L. Honkela, R. Siloor and A. O. I. Krause, Catal. Today, 2009, 147, 239–246 CrossRef CAS PubMed.
- D. Y. Hong, S. J. Miller, P. K. Agrawal and C. W. Jones, Chem. Commun., 2010, 46, 1038–1040 RSC.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. B. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- T. Nimmanwudipong, R. C. Runnebaum, D. E. Block and B. C. Gates, Catal. Lett., 2011, 141, 1072–1078 CrossRef CAS PubMed.
- R. C. Runnebaum, T. Nimmanwudipong, D. E. Block and B. C. Gates, Catal. Lett., 2011, 141, 817–820 CrossRef CAS PubMed.
- R. C. Runnebaum, R. J. Lobo-Lapidus, T. Nimmanwudipong, D. E. Block and B. C. Gates, Energy Fuels, 2011, 25, 4776–4785 CrossRef CAS.
- T. Nimmanwudipong, R. C. Runnebaum, D. E. Block and B. C. Gates, Catal. Lett., 2011, 141, 779–783 CrossRef CAS.
- T. Nimmanwudipong, R. C. Runnebaum, D. E. Block and B. C. Gates, Energy Fuels, 2011, 25, 3417–3427 CrossRef CAS.
- T. Nimmanwudipong, R. C. Runnebaum, S. E. Ebeler, D. E. Block and B. C. Gates, Catal. Lett., 2012, 142, 151–160 CrossRef CAS.
- X. Zhu, R. G. Mallison and D. E. Resasco, Appl. Catal., A, 2010, 379, 172–181 CrossRef CAS PubMed.
- M. A. Gonzalez-Borja and D. E. Resasco, Energy Fuels, 2011, 25, 4155–4162 CrossRef CAS.
- E. Dorrestijn, L. J. J. Laarhoven, I. W. C. E. Arends and P. Mulder, J. Anal. Appl. Pyrolysis, 2000, 54, 153–192 CrossRef CAS.
- A. V. Bridgwater and M. L. Cottam, Energy Fuels, 1992, 6, 113–120 CrossRef CAS.
- M. V. Bykova, D. Y. Ermakov, V. V. Kaichev, O. A. Bulavchenko, A. A. Saraev, M. Y. Lebedev and V. A. Yakovlev, Appl. Catal., B, 2012, 113–114, 296–307 CrossRef CAS PubMed.
- O. I. Senol, E.-M. Ryymin, T.-R. Viljava and A. O. I. Krause, J. Mol. Catal. A: Chem., 2007, 277, 107–112 CrossRef CAS PubMed.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS PubMed.
- R. W. Thring, S. P. Katikaneni and N. N. Bakhshi, Fuel Process. Technol., 2000, 62, 17–30 CrossRef CAS.
- S. M. Kadangode, Ph.D. thesis, University of Utah, Salt Lake City, 2001.
- D. K. Johnson, E. Chornet, W. Zmierczak and J. Shabtai, Prepr. Symp. – Am. Chem. Soc., Div. Fuel Chem., 2002, 47, 380–381 CAS.
- E. Laurent and B. Delmon, Appl. Catal., A, 1994, 109, 77–96 CrossRef CAS.
- A. Centeno, E. Laurent and B. Delmon, J. Catal., 1995, 154, 288–298 CrossRef CAS.
- C. E. Burgess, D. J. Clifford and J. R. Horvath, Prepr. Symp. – Am. Chem. Soc., Div. Fuel Chem., 2002, 47, 376–379 CAS.
- R. W. Thring, E. Chornet and R. P. Overend, Can. J. Chem. Eng., 1993, 71, 107–115 CrossRef CAS.
- L. Liguori and T. Barth, J. Anal. Appl. Pyrolysis, 2011, 92, 477–484 CrossRef CAS PubMed.
- Y. Yang, A. Gilbert and C. Xu, Appl. Catal., A, 2009, 360, 242–249 CrossRef CAS PubMed.
- X. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2011, 281, 21–29 CrossRef CAS PubMed.
- M. J. Girgis and B. C. Gates, Ind. Eng. Chem. Res., 1991, 30, 2021–2058 CrossRef CAS.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. J. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549–5553 CrossRef CAS PubMed.
- A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, Green Chem., 2013, 15, 3049–3056 RSC.
- V. N. Bui, G. Toussaint, D. Laurenti, C. Mirodatos and C. Geantet, Catal. Today, 2009, 143, 172–178 CrossRef CAS PubMed.
- F. E. Massoth, P. Politzer, M. C. Concha, J. S. Murray, J. Jakowski and J. Simons, J. Phys. Chem. B, 2006, 110, 14283–14291 CrossRef CAS PubMed.
- B. Yoosuk, D. Tumnantong and P. Prasassarakich, Fuel, 2012, 91, 246–252 CrossRef CAS PubMed.
- T. R. Viljava and A. O. I. Krause, Stud. Surf. Sci. Catal., 1997, 106, 343–352 CrossRef CAS.
- Y. Yunquan, L. He'an, T. Gangsheng, K. J. Smith and T. C. Thian, Chin. J. Chem. Eng., 2008, 16, 733–739 CrossRef.
- E. O. Odebunmi and D. F. Ollis, J. Catal., 1983, 80, 76–89 CrossRef CAS.
- E. Furimsky and F. E. Massoth, Catal. Rev. Sci. Eng., 2005, 47, 297–489 CAS.
- S. Echeandia, P. L. Arias, V. L. Barrio, B. Pawelec and J. L. G. Fierro, Appl. Catal., B, 2010, 101, 1–12 CrossRef CAS PubMed.
- W. Wang, Y. Yang, H. Luo, H. Peng and F. Wang, Ind. Eng. Chem. Res., 2011, 50, 10936–10942 CrossRef CAS.
- C. Zhao, J. He, A. A. Lemonidou, X. Li and J. A. Lercher, J. Catal., 2011, 280, 8–16 CrossRef CAS PubMed.
- E. M. Turpeinen, Ph.D. thesis, Aalto University, 2011.
- E. Laurent and B. Delmon, Ind. Eng. Chem. Res., 1993, 32, 2516–2524 CrossRef CAS.
- K. Li, R. Wang and J. Chen, Energy Fuels, 2011, 25, 854–863 CrossRef CAS.
- C. V. Loricera, B. Pawelec, A. Infantes-Molina, M. C. Alvarez-Galvan, R. Huirache-Acuña, R. Nava and J. L. G. Fierro, Catal. Today, 2011, 172, 103–110 CrossRef CAS PubMed.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS PubMed.
- I. T. Ghampson, C. Sepúlveda, R. Garcia, B. G. Frederick, M. C. Wheeler, N. Escalona and W. J. DeSisto, Appl. Catal., A, 2012, 413–441, 478–484 Search PubMed.
- I. T. Ghampson, C. Sepúlveda, R. Garcia, L. R. Radovic, J. L. García Fierro, W. J. DeSisto and N. Escalona, Appl. Catal., A, 2012, 439–440, 111–124 CrossRef CAS PubMed.
- C. R. Lee, J. S. Yoon, Y. W. Suh, J. W. Choi, J. M. Ha, D. J. Suh and Y. K. Park, Catal. Commun., 2012, 17, 54–58 CrossRef CAS PubMed.
- B. S. Gevert, J. E. Otterstedt and F. E. Massoth, Appl. Catal., 1987, 31, 119–131 CrossRef CAS.
- Y. C. Lin, C. L. Li, H. P. Wan, H. T. Lee and C. F. Liu, Energy Fuels, 2011, 25, 890–896 CrossRef CAS.
- T. R. Viljava, R. S. Komulainen and A. O. I. Krause, Catal. Today, 2000, 60, 83–92 CrossRef CAS.
- E. M. Ryymin, M. L. Honkela, T. R. Viljava and A. O. I. Krause, Appl. Catal., A, 2010, 389, 114–121 CrossRef CAS PubMed.
- T. Nimmanwudipong, C. Aydin, J. Lu, R. C. Runnebaum, K. C. Brodwater, N. D. Browning, D. E. Block and B. C. Gates, Catal. Lett., 2012, 142, 1190–1196 CrossRef CAS PubMed.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Appl. Catal., A, 2011, 391, 305–310 CrossRef CAS PubMed.
- E. Furimsky, J. A. Mikhlin, D. Q. Jones, T. Adley and H. Baikowitz, Can. J. Chem. Eng., 1986, 64, 982–985 CrossRef CAS.
- R. C. Runnebaum, T. Nimmanwudipong, D. E. Block and B. C. Gates, Catal. Sci. Technol., 2012, 2, 113–118 CAS.
- D. Prochazkova, P. Zamostny, M. Bejblova, L. Cerveny and J. Cejka, Appl. Catal., A, 2007, 332, 56–64 CrossRef CAS PubMed.
- D. C. Elliott and T. R. Hart, Energy Fuels, 2008, 23, 631–637 CrossRef.
- J. Wildschut, F. H. Mahfud, R. H. Venderbosch and H. J. Heeres, Ind. Eng. Chem. Res., 2009, 48, 10324–10334 CrossRef CAS.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 121, 4047–4050 CrossRef.
- T. T. Pham, L. L. Lobban, D. E. Resasco and R. G. Mallinson, J. Catal., 2009, 266, 9–14 CrossRef CAS PubMed.
- V. M. L. Whiffen and K. J. Smith, Energy Fuels, 2010, 24, 4728–4737 CrossRef CAS.
- S. T. Oyama, X. Wang, Y. K. Lee and W. J. Chun, J. Catal., 2004, 221, 263–273 CrossRef CAS.
- G. de la Puente, A. Gil, J. J. Pis and P. Grange, Langmuir, 1999, 15, 5800–5806 CrossRef CAS.
- E. Furimsky and F. E. Massoth, Catal. Today, 1999, 52, 381–495 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Delichere and C. Geantet, Appl. Catal., B, 2011, 101, 246–255 CrossRef CAS PubMed.
- R. N. Olcese, M. Bettahar, D. Petitjean, B. Malaman, F. Giovanella and A. Dufour, Appl. Catal., B, 2012, 115–116, 63–73 CrossRef CAS PubMed.
- R. Olcesea, M. M. Bettahar, B. Malamanc, J. Ghanbajac, L. Tibavizco, D. Petitjean and A. Dufour, Appl. Catal., B, 2013, 129, 528–538 CrossRef PubMed.
- K. L. Deutsch and B. H. Shanks, Appl. Catal., A, 2012, 447–448, 144–150 CrossRef CAS PubMed.
- M. K. Huuska, Polyhedron, 1986, 5, 233–236 CrossRef CAS.
- A. R. Ardiyanti, S. A. Khromova, R. H. Venderbosch, V. A. Yakovlev and H. J. Heeres, Appl. Catal., B, 2012, 117–118, 105–117 CrossRef CAS PubMed.
- A. L. Jongerius, R. W. Gosselink, J. Dijkstra, J. H. Bitter, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2013, 5, 2964–2972 CrossRef CAS.
- A. L. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315–323 CrossRef CAS PubMed.
- P. E. Ruiz, B. G. Frederick, W. J. De Sisto, R. N. Austin, L. R. Radovic, K. Leiva, R. García, N. Escalona and M. C. Wheeler, Catal. Commun., 2012, 27, 44–48 CrossRef CAS PubMed.
- W. Wang, Y. Yang, J. Bao and Z. Chen, J. Fuel Chem. Technol., 2009, 37, 701–706 CrossRef.
- W. Wang, Y. Yang, J. Bao and H. Luo, Catal. Commun., 2009, 11, 100–105 CrossRef CAS PubMed.
- E. Furimsky, J. A. Mikhlin, D. Q. Jones, T. Adley and H. Baikowitz, Can. J. Chem. Eng., 1986, 64, 982–985 CrossRef CAS.
- A. Vuori, A. Helenius and J. B. Son Bredenberg, Appl. Catal., 1989, 52, 41–56 CrossRef CAS.
- M. Ferrari, R. Maggi, B. Delmon and P. Grange, J. Catal., 2001, 198, 47–55 CrossRef CAS.
- V. M. L. Whiffen, K. J. Smith and S. K. Straus, Appl. Catal., A, 2012, 419–420, 111–125 CrossRef CAS PubMed.
- S. T. Oyama, Catal. Today, 1992, 15, 179–200 CrossRef CAS.
- D. Sajkowski and S. T. Oyama, in ACS Div. Petr. Prepr. 199th ACS Meeting, Boston, 1990 Search PubMed.
- E. J. Shin and M. A. Keane, Ind. Eng. Chem. Res., 2000, 39, 883–892 CrossRef CAS.
- C. Sepúlveda, N. Escalona, R. García, D. Laurenti and M. Vrinat, Catal. Today, 2012, 195, 101–105 CrossRef PubMed.
- M. V. Bykova, O. A. Bulavchenko, D. Y. Ermakov, M. Y. Lebedev, V. A. Yakovlev and V. N. Parmon, Catal. Ind., 2011, 3, 15–22 CrossRef.
- X. Zhang, Q. Zhang, T. Wang, L. Ma, Y. Yu and L. Chen, Bioresour. Technol., 2013, 134, 73–80 CrossRef PubMed.
- X. Zhang, T. Wang, L. Ma, Q. Zhang, Y. Yu and Q. Liu, Catal. Commun., 2013, 33, 15–19 CrossRef CAS PubMed.
- Y. Yang, C. Ochoa-Hernández, V. A. O'Shea, P. Pizarro, J. M. Coronado and D. P. Serrano, Appl. Catal., B: Environ., 2014, 145, 91–100 CrossRef CAS PubMed.
- M. Zdražil, Catal. Today, 2003, 86, 151–171 CrossRef.
- H. Shimada, T. Sato, Y. Yoshimura, J. Haraishi and A. Nishijima, J. Catal., 1988, 110, 275–284 CrossRef CAS.
- M. J. Ledoux, A. Peter, E. A. Blekkan and F. Luck, Appl. Catal., A, 1995, 133, 321–333 CrossRef CAS.
- T. Klicpera and M. Zdražil, J. Mater. Chem., 2000, 10, 1603–1608 RSC.
- T. Klicpera and M. Zdražil, Appl. Catal., A, 2001, 216, 41–50 CrossRef CAS.
- Y. Romero, F. Richard and S. Brunet, Appl. Catal., B, 2010, 98, 213–223 CrossRef CAS PubMed.
- S. J. Hurff and M. T. Klein, Ind. Eng. Chem. Fundam., 1983, 22, 426–430 CAS.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- M. Badawi, J. Paul, S. Cristol and E. Payen, Catal. Commun., 2011, 12, 901–905 CrossRef CAS PubMed.
- A. Popov, E. Kondratieva, L. Mariey, J. M. Goupil, J. E. Fallah, J. P. Gilson, A. Travert and F. Maugé, J. Catal., 2013, 297, 176–186 CrossRef CAS PubMed.
- J. B. Son Bredenberg, M. Huuska, J. Räty and M. Korpio, J. Catal., 1982, 77, 242–247 CrossRef.
- R. C. Runnebaum, T. Nimmanwudipong, R. R. Limbo, D. E. Block and B. C. Gates, Catal. Lett., 2012, 142, 7–15 CrossRef CAS.
- Y. Q. Yang, C. T. Tye and K. J. Smith, Catal. Commun., 2008, 9, 1364–1368 CrossRef CAS PubMed.
- S. Zhang, Y. Yan, R. Zhengwei and L. Tingchen, Energy Sources, 2003, 25, 57–65 CrossRef.
- W. Baldauf, U. Balfanz and M. Rupp, Biomass Bioenergy, 1994, 7, 237–244 CrossRef CAS.
- J. C. Hicks and J. Phys, Chem. Lett., 2011, 2, 2280–2287 CAS.
- F. P. Petrocelli and M. T. Klein, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24, 635–641 CrossRef CAS.
- J. B. S. Bredenberg, M. Huuska and P. Toropainen, J. Catal., 1989, 120, 401–408 CrossRef CAS.
- M. Huuska and J. Rintala, J. Catal., 1985, 94, 230–238 CrossRef CAS.
- M. Ferrari, B. Delmon and P. Grange, Carbon, 2002, 40, 497–511 CrossRef CAS.
- V. A. Yakovlev, S. A. Khromova, O. V. Sherstyuk, V. O. Dundich, D. Y. Ermakov, V. M. Novopashina, M. Y. Lebedev, O. Bulavchenko and V. N. Parmon, Catal. Today, 2009, 144, 362–366 CrossRef CAS PubMed.
- T. Prasomsri, T. Nimmanwudipong and Y. R. Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 CAS.
- X. Zhang, T. Wang, L. Ma, Q. Zhang, X. Huang and Y. Yu, Appl. Energy, 2013, 112, 533–538 CrossRef CAS PubMed.
- A. Ausavasukhi, Y. Huang, A. T. To, T. Sooknoi and D. E. Resasco, J. Catal., 2012, 290, 90–100 CrossRef CAS PubMed.
- J. Filley and C. Roth, J. Mol. Catal. A: Chem., 1999, 139, 245–252 CrossRef CAS.
- J. Sun, A. M. Karim, H. Zhang, L. Kovarik, X. S. Li, A. J. Hensley, J. S. McEwen and Y. Wang, J. Catal., 2013, 306, 47–57 CrossRef CAS PubMed.
- Y. Wang, T. He, K. Liu, J. Wu and Y. Fang, Bioresour. Technol., 2012, 108, 280–284 CrossRef CAS PubMed.
- H. Ohta, H. Kobayashi, K. Hara and A. Fukuoka, Chem. Commun., 2011, 47, 12209–12211 RSC.
- A. Gutierrez, R. K. Kaila and A. O. I. Krause, Proc. 14th Int. Congr. Catal., Seoul, Korea, 2008 Search PubMed.
- J. Chang, T. Danuthai, S. Dewiyanti, C. Wang and A. Borgna, ChemCatChem, 2013, 5, 3041–3049 CrossRef CAS.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64–68 CrossRef CAS.
- R. K. M. R. Kallury, W. M. Restivo, T. T. Tidwell, D. G. Boocock, A. Crimi and J. Douglas, J. Catal., 1985, 96, 535–543 CrossRef CAS.
- C. Sepúlveda, N. Escalona, R. García, D. Laurenti and M. Vrinat, Catal. Today, 2012, 195, 101–105 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |