Biomimetic di-manganese catalyst cage-isolated in a MOF: robust catalyst for water oxidation with Ce(IV), a non-O-donating oxidant†
Rebecca E.
Hansen
and
Siddhartha
Das
*
Dept. of Chem. and Biochem., Utah State University, 0300 Old Main Hill, Logan, UT, USA. E-mail: siddhartha.das@usu.edu; Fax: +1 435 797 3390; Tel: +1 435 797 2267
First published on 4th November 2013
Abstract
A biomimetic di-Mn catalyst catalyzes water oxidation with (NH4)2Ce(NO3)6 for >7 days with initial TOF of 40 h−1. Isolation of one molecule of catalyst in each pore of a Cr-based MOF prevents decomposition of the catalyst and allows sustained water oxidation even at pH 1.00.
Broader contextDeveloping a water oxidation catalyst (WOC) that is cheap (made of earth-abundant materials), robust (long-lasting) and efficient, all at the same time, is the chemical bottleneck for sunlight-driven water splitting. We report herein a WOC with manganese and chromium as metals; this molecular-material system is modular and well defined, but is heterogeneous in nature, which ensures its ease of use in practical applications. This molecular-material system catalyzes water oxidation with a non-oxygen-donating oxidant (cerium(IV) ammonium nitrate (CAN)), which is widely considered the step before the incorporation of a WOC into a photoelectrochemical cell. The rate of the catalysis is among the highest ones with earth-abundant metals using CAN, and is stable even after a week under highly corrosive conditions (pH = 1) vs. ≤2 hours for the best known system. The unique, but simple design concept (cage-isolation of one catalyst molecule in each segregated pore of a porous material) recently developed by our group seems to open the door for developing modular and tunable robust materials for solar water splitting in near future. |
1. Introduction
Light-driven water splitting is a multidimensional challenge requiring an orchestration of numerous processes, such as light absorption, energy transfer, electron and proton transfers, and catalysis, to name a few. An efficient catalyst for water splitting is a critical part of this challenge. Molecular, homogeneous catalysts with earth-abundant elements are very attractive because of their tunability, but these are generally not long lasting. On the other hand, materials are long lasting, but lack the predictability for efficient integration in multi-functional systems and hence, the crucial fine-tuning.Recently, we took the approach of developing materials with molecular level definition using metal–organic frameworks (MOF): a well-defined molecular-material system (MnTD⊂MIL-101(Cr) (Fig. 1)). This not only showed promising water oxidation rates, but also maintained the same rate for hours with K-oxone (KHSO5) as the sacrificial oxidant.1 It is constructed by synthesizing one molecule of a high-valent di-μ-oxo dimanganese catalyst, [(terpy)Mn(μ-O)2Mn(terpy)]3+ (Mn-terpy dimer: MnTD; terpy: 2,2′:6′,2′′-terpyridine), in each pore of a (μ3-O)Cr3(COO)6 containing MOF, MIL-101(Cr).2 Sustaining water oxidation with MnTD was a unique observation. However, use of K-oxone (KHSO5), an O-atom donating oxidant, leaves several concerns: (a) is the source of the dioxygen O-atoms H2O, or the peroxo bond of oxone (O3S–O–O)? Mechanistic investigations suggested that one O-atom of dioxygen was definitely from the peroxo bond of oxone. Exchange of the peroxo oxygen and oxygen of water molecules made it very difficult to reach a definitive conclusion regarding the actual mechanism of oxygen evolution.3–8 (b) Success with oxone does not guarantee the catalyst's application in electrochemical cells, where the Mn ions need to be oxidized via electron transfer, not by O-atom transfer.3,7 Therefore the question remains: can this well-defined manganese system, MnTD⊂MIL-101(Cr), catalyze water oxidation with non-oxygen-donating oxidants?
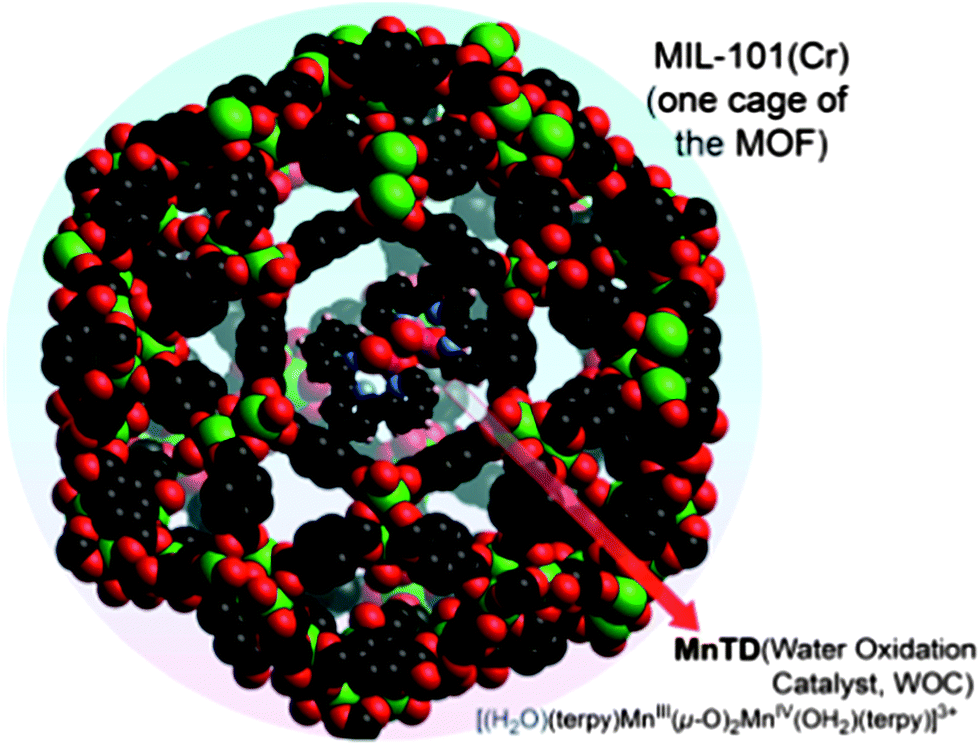 | ||
| Fig. 1 MnTD⊂MIL-101(Cr): high-valent di-Mn complex (MnTD) cage-isolated in pores of a MOF (MIL-101(Cr)). Only one cage of the extended framework, MIL-101(Cr), is shown; all protons on MOF and all the anions and solvent molecules omitted for clarity. The structure has been derived from individual crystal structures of MnTD and MIL-101(Cr). | ||
Cerium(IV) ammonium nitrate (CAN) is a non-oxygen-donating oxidant. Its mechanism of activating water-oxidation catalysts (WOC) via single electron transfers is known to be very close to that in photoelectrochemical cells.9 Historically, all the WOCs capable of water oxidation with CAN were also shown to be successful electrochemical water oxidation catalysts.9,10 CAN's requirement of harsh conditions (pH ∼ 1), requires the catalyst to be very robust and also capable of oxidizing water under thermodynamically more challenging conditions compared to photo- and/or electrochemical cells (where typical pH should be ∼7).
Among the first row transition metals, only a handful of Fe-based catalysts have been reported for their activity towards water oxidation with CAN.11,12 Although Mn is nature's choice for water oxidation in Photosystem II, to the best of our knowledge, there has been no report of a Mn-based catalyst with molecular level definition for water oxidation with CAN.13,14
Instability of molecular MnTD owing to very fast intermolecular reactions among two or more catalyst molecules prevented its application in catalytic water oxidation with CAN.15,16 The same reason has been attributed to the impossibility of electrochemical water oxidation with MnTD.17
Herein, we report the application of MnTD in water oxidation with CAN. MnTD⊂MIL-101(Cr) not only shows very high rates of water oxidation with CAN, but also maintains a high rate of water oxidation even after weeks of continuous performance. Therefore, MnTD⊂MIL-101(Cr), to the best of our knowledge, is the first Mn-based catalyst to catalyze water oxidation with CAN. The catalyst-construct is heterogeneous, but has molecular-level definition owing to its modularity; this leads us to an important step forward in developing well-defined material-based catalysts for photoelectrochemical water oxidation that can be tuned with the simplicity of homogeneous, molecular systems.
2. Results and analysis
2.1. Synthesis and characterization
We synthesized MnTD⊂MIL-101(Cr) following the same synthetic procedure as previously described by our group:1 diffusing in Mn(II) salt and the ligand, terpy, into the pores of MIL-101(Cr) followed by addition of stoichiometric amount of K-oxone (KHSO5) to assemble the high-valent MnTD. Pores of MIL-101(Cr) are big enough to accommodate one molecule of MnTD, but the apertures of the pores of MIL-101(Cr) is smaller than the dimensions of MnTD. Therefore diffusion of pre-assembled MnTD into the pores of MIL-101(Cr) has been found to be an ineffective strategy. As MnTD, in its molecular or homogeneous state, is very sensitive to heat, solvents and low pH, we could not employ the highly popular technique of synthesizing a MOF around a catalyst; MIL-101(Cr) necessitates heating at 155 °C and addition of HF. However, we found that our technique of assembling the MnTD inside MIL-101(Cr) is an effective method to obtain one molecule of MnTD per pore in MIL-101(Cr). Spectroscopic characterization (EPR, FTIR, UV-Vis, powder XRD) of Mn(III/IV) system and μ3-oxo trichromium unit, and the structure of MOF, as well as, detailed quantification of amount of MnTD inside MIL-101(Cr) (by UV-Vis, TGA and elemental analysis) have been performed following previously reported procedures (Fig. 2 and S3†).1 This ensures an average loading of one molecule of MnTD per pore of MIL-101(Cr), which amounts to 10 wt% MnTD with respect to total MnTD⊂MIL-101(Cr).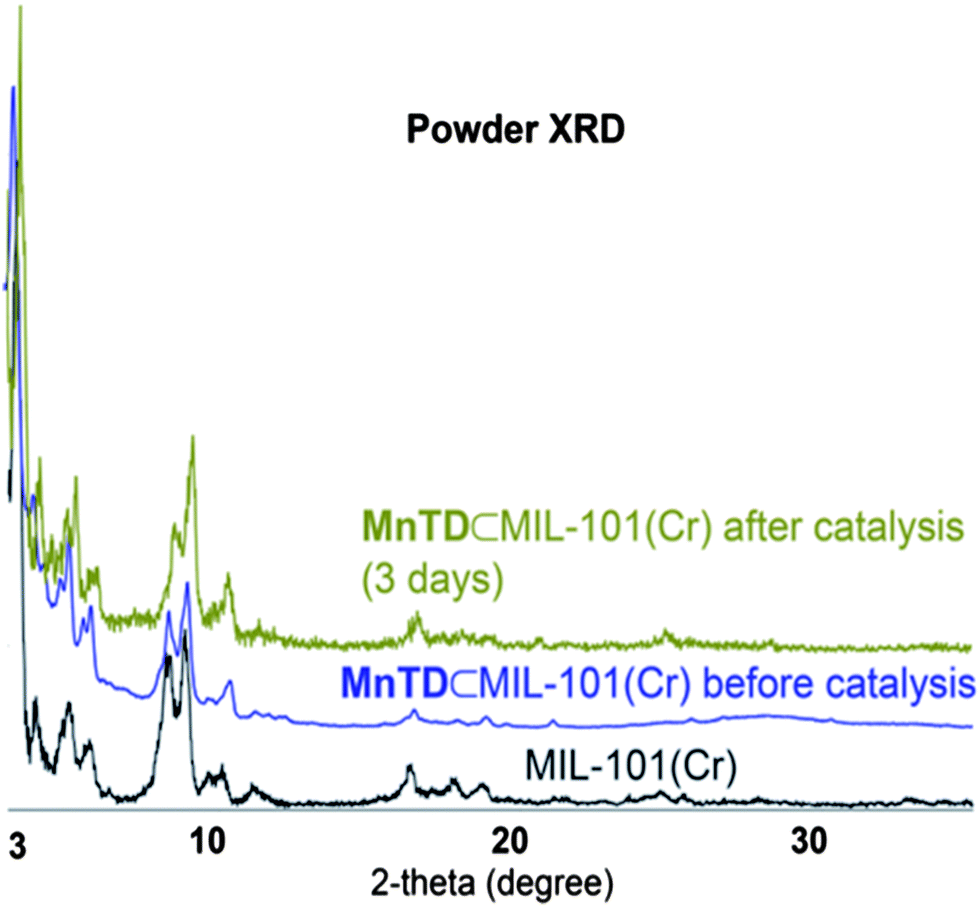 | ||
| Fig. 2 Powder XRD of MIL-101(Cr), and MnTD⊂MIL-101(Cr) before and after catalysis. The crystallinity of MIL-101(Cr) is maintained. | ||
2.2. Catalytic studies
Catalytic water oxidation was carried out in a double-walled glass chamber equipped with a circulating water bath that was maintained at 25 °C. A 6 mL dilute nitric acid solution (pH ∼ 1) (see ESI†) was taken in the chamber with the material of interest (MnTD, a suspension of MnTD⊂MIL-101(Cr), or MIL-101(Cr)), to which a solution of CAN was added. Evolved O2 was quantified by a Clark-type oxygen electrode fitted to the sample chamber by a Delrin collar to be airtight. Amount of molecular MnTD used for each catalysis is equivalent to the calculated amount of MnTD in MnTD⊂MIL-101(Cr) (1/10th of the total mass of MnTD⊂MIL-101(Cr)).Molecular MnTD and only MIL-101(Cr) did not show any detectable oxygen production with CAN. 4 mg of MnTD⊂MIL-101(Cr) (contains 0.4 μmol of MnTD) started O2 production at a very high rate. We recorded a TOF (mole of O2 per mole of MnTD in MnTD⊂MIL-101(Cr)) of >40 per hour, when 1.6 mmol of CAN (catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oxidant = 1:4000) was added (Fig. 3).
:oxidant = 1:4000) was added (Fig. 3).
 | ||
| Fig. 3 Oxygen evolution plots with (A) 4 mg MnTD⊂MIL-101(Cr), 4 mg MnO2, 4 mg MIL-101(Cr), no catalyst/material added (background) and (B) 0.4 μmol of MnTD (inset) in 6 mL pH = 1 solution (with HNO3) with 900 mg CAN (1.6 mmol). (C) O2 evolution with 4 mg MnTD⊂MIL-101(Cr) + 900 mg CAN after 7 days of continuous catalysis shows very slow rate of water oxidation; the same catalyst after solvent exchange (to remove any unwanted and soluble chemicals from pores) show revived rate of O2 evolution. | ||
We compared the rates of water oxidation that have been reported with first-row transition metal-based WOCs with CAN (Table 1). In fact, only two other first row transition metal-based WOCs showed such high rates with CAN; both are Fe-based catalysts with tetradentate N-containing ligands: Fe(TAML) (by Bernhard, Collins and coworkers; TAML: tetra-amido macrocyclic ligand)12 and Fe(tacn) (by Fillol, Costas and coworkers; tacn: tetraazocyclononane)11 – type complexes. Fe(TAML) does not show any O2 evolution after ∼12 seconds. Fe(tacn) type catalysts remain active for 10 min to 2 hours (depending on the ligand). To the best of our knowledge, there is no Mn-based biomimetic catalyst that shows any water oxidation activity with CAN; however, Åkermark and coworkers have reported a di-manganese catalyst that shows water oxidation with [Ru(bpy)3]3+, and can attain a maximum total turnovers of ∼25 in one hour. At this point the catalyst becomes inactive.18
| Catalysts | CAN:catalyst |
Initial TOF (h−1) (mol O2 per mol catalyst) | Lifetimes (min) | Ref. |
|---|---|---|---|---|
| [Fe(OTf)2(mcp)] | 10000 |
838 | 26 | 11 |
| [(Fe(mcp))2(μ-O)(μ-OH)](OTf)2 | 20000 |
105 | 120 | 11 |
| Fe(III)-TAML | ∼200 | 4680 | 0.2 | 12 |
| MnTD physically adsorbed on kaolin | 1400 | 0.35 | 480 | 24 |
| MnTD⊂FDU-12 | 130 | 6 | 430 | This work |
| MnTD⊂MIL-101(Cr) | 4000 | 40 | >10080(7 days) |
This work |
MnTD⊂MIL-101(Cr) continues to catalyze O2 evolution for days, at least. We left our MnTD⊂MIL-101(Cr) submerged in the catalytic medium for one week, and even after that we recorded significant amount of O2 production (Fig. 3c). We characterized the evolved gas with GC-MS: we see gradual increase in O2 concentration in the headspace, while the concentration of CO2 remains comparable to background.
The rate of the O2 production was, however, decreasing over time. This is in contrast to what we saw with K-oxone, where the water oxidation rate remains exactly the same as the initial rate even after hours.1 It should be noted that unlike oxone (HSO5−), Ce(IV), upon reduction to Ce(III), starts yielding insoluble cerium oxide materials.19 Over time this reduced CAN might start clogging the pores of MIL-101(Cr) and preventing fresh Ce(IV) from diffusing in. This would affect the rate of the catalysis adversely.
To test this hypothesis, the amount of CAN was lowered. Indeed, the TOF with 0.8 mmol CAN (450 mg) (catalyst:oxidant = 1:2000) was 44% after 3 hours, compared to the initial rate; whereas the TOF with 0.13 mmol CAN (75 mg) (catalyst:oxidant = 1:325) was 93%, after 3 hours, compared to the initial rate. After keeping 4 mg of MnTD⊂MIL-101(Cr) (MnTD: 0.4 μmol) submerged in a solution of 900 mg of CAN (1.6 mmol) for 7 days, when we noticed that the catalytic rate became very slow, we performed solvent exchange on MnTD⊂MIL-101(Cr), a common technique to get rid of impurities from the pores of MOFs. This involved washing the catalyst-particles several times with fresh diluted HNO3 (pH = 1), not containing any CAN. Upon this treatment and subsequent addition of a fresh 6 mL aqueous solution of 75 mg of CAN, the same catalyst particles started showing water oxidation with a higher rate than the rate observed before washing, but still lower than the initial catalytic rate (after solvent exchange, the TOF = 5 h−1). This is expected as, although solvent exchange probably cleared out the soluble reduced forms of CAN, we did not anticipate it to remove cerium oxide particles from inside the pore. For practical applications in photoelectrochemical water oxidation with MnTD⊂MIL-101(Cr), no chemical oxidant should be used; in that case, this complexity owing to the clogging of the pore would not be an issue.
2.3. Probing the catalytically active species
The characteristic 16-line EPR-signal from the mixed valent MnIII(μ-O)2MnIV disappears upon addition of CAN (Fig. S3†). This makes it difficult to follow the catalyst using readily available spectroscopic tools. However, Brudvig, Crabtree and coworkers have reported a detailed study following the degradation pathways of molecular MnTD.15 Generally, MnTD undergoes disproportionation reactions yielding catalytically inactive tetra-Mn(IV) complex and Mn(II) complex as the major products.15 If the disproportionation is faster than catalytic turnover, then the catalyst becomes deactivated. Mn(IV)-tetramers are EPR silent; but Mn(II) complexes have very strong characteristic 6-line EPR signal at 7 K. Cryogenic EPR (7 K) with MnTD⊂MIL-101(Cr) treated with CAN solution did not show any trace of Mn(II), ruling out the disproportionation pathways for high-valent MnTD. We did not observe any CO2 formation either (based on GC-MS), ruling out the decomposition pathways for MnTD. The FT-IR recorded after 7 days of catalysis showed the peak characteristic of high-valent Mn(μ-O)2Mn with terpyridine as a ligand (Fig. S2†).1,3,17Manganese oxides also show water oxidation under electrochemical conditions and are often suspected as the degradation product from high-valent Mn-complexes.13 Our control experiments with MnO2 showed negligible O2 production with CAN as the oxidant, which is consistent with previous studies with different forms of manganese oxides.13 The PXRD of the MnTD⊂MIL-101(Cr), recorded after 72 hours of catalysis, showed the maintenance of the crystal structure of MOF and did not show any peak characteristic of crystalline manganese oxides (Fig. 2).
We also carried out oxygenation of cis-stilbene using tetrabutylammonium oxone (TBAO) as oxidant with MnTD⊂MIL-101(Cr) to probe the nature of the manganese-complex inside the MOF. Catalysis using TBAO is not comparable to that with CAN. However, the product distribution from epoxidation/oxygenation of cis-stilbene is very sensitive to the catalytic mechanism. Therefore, we hoped that this study would provide us with an additional chemical handle to understand the catalytic species at the onset of catalysis (similar chemical handles can be applied, in future, in the speciation studies of molecular-material systems).20 With MnTD⊂MIL-101(Cr), the major products were cis-stilbene oxide, benzophenone and 1,2-diphenylethanone, but no trans-stilbene oxide was found (Table S1†). This matches the products obtained with molecular MnTD, including the relative ratio of these products, as analyzed by GC-MS. In contrast, bulk manganese oxides did not show any significant turnover in stilbene epoxidation under our reaction conditions.
The exact fate of MnTD, upon addition of CAN, remains unclear. Also, the catalytically active species that Brudvig and Crabtree specified as unknown remains elusive, and so is the mechanism of O–O bond formation.15 As there is only one open coordination site available on each Mn-ion and no O2 evolution takes place in absence of an oxidant, the O–O bond formation is unlikely (a) to follow a non-metal oxo based mechanism, as had been proposed by Milstein and coworkers with a Ru-based catalyst,21 or (b) to involve a MnV(O)(OH) intermediate. As the available coordination sites on two Mn-ions are trans to each other, a radical-coupled mechanism involving both metal sites would necessitate participation and/or breaking of the bridging oxo units. However, a nucleophilic attack by water (probably H-bonded to the μ-oxo) to an electrophilic oxygen on high-valent Mn(V)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O could provide dioxygen not requiring the disruption of bridging oxo.22–24
O could provide dioxygen not requiring the disruption of bridging oxo.22–24
Above experiments imply that (a) the catalytic species, at the onset of catalysis, has similarity with the molecular MnTD, (b) we are not obtaining Mn(II) species through disproportionation of high-valent di-manganese moiety, and (c) commonly known forms of crystalline manganese oxides are not forming. However, this does not rule out the presence of a new type of amorphous manganese oxide nanoparticles that are catalytically highly active. This itself would be an important topic of future research. Our efforts to conclusively identify the true nature of this active species are under way.
2.4. Importance of catalyst cage-isolation
We feel that individual isolation of MnTD molecules plays an important role in this remarkable catalytic activity. It should be noted that the lack of electrochemical water oxidation and catalytic water oxidation with MnTD with CAN have been attributed to the intermolecular reactions of MnTD molecules, resulting in a manganese-tetramer.4,14,17The importance of this cage-isolation of each molecule of a reactive catalyst was further investigated by synthesizing materials with bigger cages and incorporating more than one molecule of MnTD per cage. We synthesized FDU-12, mesoporous silica, which has pores of the diameter 18.6 nm and apertures of the diameter 4.2 nm (for MIL-101(Cr), pore diameter: 29 and 34 Å, aperture diameters: 12 and 15 Å).25,26 We made the apertures smaller by further silylation to prevent MnTD from diffusing out, and then assembled the MnTD from its precursors inside FDU-12 following a similar procedure as with MIL-101(Cr) (see ESI†); this technique yielded MnTD⊂FDU-12. Based on elemental analysis we found the MnTD⊂FDU-12 has ≫1 molecules of MnTD per pore of FDU-12 (MnTD constitutes 20 wt% of MnTD⊂FDU-12). With MnTD⊂FDU-12, with CAN as the oxidant, we observed a good rate in water oxidation (Fig. 4); this is very different from the molecular MnTD, in the homogeneous state, that does not show any catalysis (Fig. 3). However the rate of water oxidation was found to be only one fourth compared to that with MnTD⊂MIL-101(Cr). Also with MnTD⊂FDU-12, the catalysis almost stops after ∼7 hours. It should be noted that recently, Li, Yang and coworkers have reported FDU-12 and a similar mesoporous silica material, SBA-16, to incorporate Jacobsen's catalyst [CoIII(salen)] (for epoxidation) and water-oxidation catalyst, monomeric [RuII(bda)] (bda: 2,2′-bipyridine-6,6′-dicarboxylate) respectively.25,26 In both cases rate enhancements in respective catalysis were observed. In the case of water oxidation, the authors attributed the rate enhancement to the higher cooperativity between catalyst molecules and substrates in the limited spaces of the cages. In their case the total turnover of the water oxidation also improved compared to the molecular catalyst in the homogeneous form. However, they, too, did not see any sustained water oxidation.
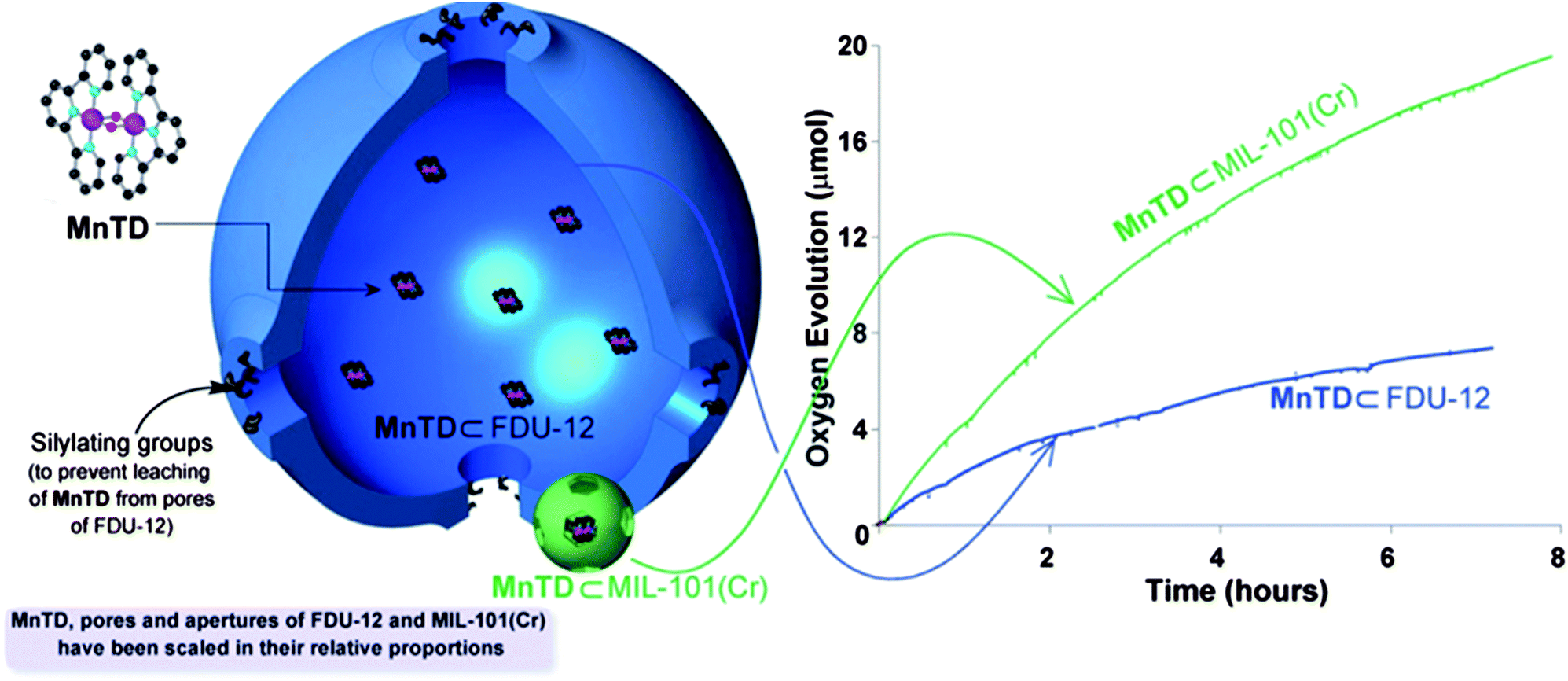 | ||
| Fig. 4 Presence of multiple molecules of MnTD in a pore does not result in the high rate and sustenance in water oxidation. Cartoon representation of individual pores MnTD⊂MIL-101(Cr) (green) and MnTD⊂FDU-12 (blue). The pores, apertures of materials and metal complex of MnTD are scaled according to their relative dimensions. FDU-12, a mesoporous silica, has pores of diameter 186 Å, and thus encages multiple molecules of MnTD, unlike MIL-101(Cr) (pore diameter ≤30 Å). Right-hand side: O2 evolution with 4 mg MnTD⊂MIL-101(Cr) and 4 mg of MnTD⊂FDU-12 in 6 mL pH = 1 solution of 75 mg CAN (133 μmol). Previous quantification: MnTD⊂MIL-101(Cr) has one catalyst molecule per pore of MIL-101(Cr) on average.1 | ||
While we initially expected that MnTD⊂FDU-12 would not show any catalytic activity, it looks like heterogenization in cages, in general, has a positive effect on the catalytic activity of the catalyst. This is somewhat consistent with the previous observation with MnTD adsorbed onto Kaolin clay, that gave a TON ∼2 with CAN.27 However, sustained catalysis, our major goal, requires cage-isolation of each catalyst molecule.
2.5. Rate-dependence on oxidant concentration
We performed preliminary study of the kinetics of the catalysis with respect to CAN. While it is true that the TOF increases with increase in CAN concentration, no clear relation between the amount of CAN and catalytic rate was found (Fig. 5). This could be the consequence of several factors: (a) diffusion rate of CAN into the pores of the MOF follows a complex pattern; (b) as CAN gets reduced, insoluble products of CAN reduction start clogging the pores, further complicating the process of interaction of CAN with MnTD. We will continue our efforts to further investigate both the kinetics and the mechanism of water oxidation with this Mn-based catalyst with CAN. However, practical applications of water oxidation will necessitate a chemical oxidant-free system, a direction we are pursuing fervently.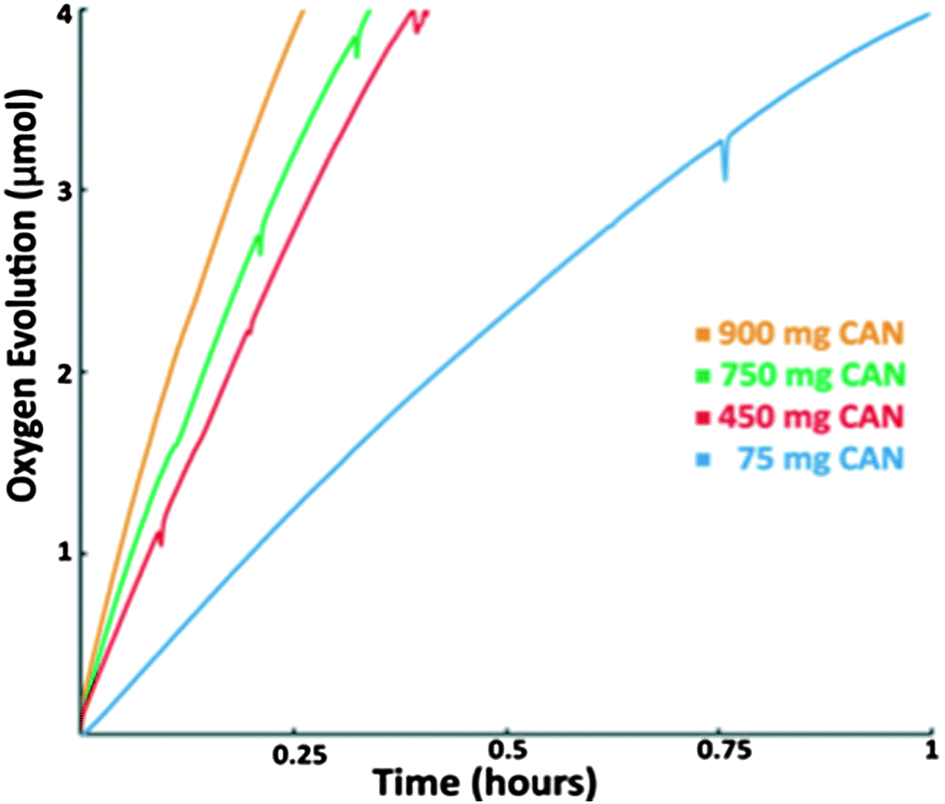 | ||
| Fig. 5 Oxygen evolution plots with 4 mg of MnTD⊂MIL-101(Cr) (contains ∼0.4 μmol of MnTD) in 6 mL pH = 1 solution (with HNO3) of various amounts of CAN: 900 mg (1.6 mmol), 750 mg (1.33 mmol), 450 mg (0.8 mmol) and 75 mg (0.13 mmol). Although increase in CAN increases the rate of the O2 evolution, there is no systematic correlation between the two. | ||
3. Conclusions
In summary, we report the first example of a Mn-based catalyst performing water oxidation at a high rate (TOF (mol O2/mol MnTD) = 40 h−1) with CAN as an external oxidant. MnTD⊂MIL-101(Cr) maintains its water oxidation activity for hours, and even after a week, in the harsh conditions required for water oxidation with CAN (pH = 1). We show that while mere encaging a reactive catalyst in pores of a porous material tend to show improved activity of the catalyst, the isolation of single molecules of catalyst in small pores (one molecule of catalyst per pore) is necessary for sustained activity: an important criterion for CO2 sequestration and water splitting catalyses. Judging (a) the mechanistic similarity between water oxidation with CAN and photoelectrochemical water oxidation, (b) the high and sustained rate in water oxidation with this biomimetic catalyst-construct, and (c) the potential tunability of this catalyst-construct, we feel that the current system, especially the architectural philosophy behind MnTD⊂MIL-101(Cr) could play an important role in the future development of robust and cheap catalysts for clean energy.Acknowledgements
This work has been supported with start-up funding from Utah State University. We thank A. J. Stewart (undergraduate student) for the synthesis of FDU-12, Moumita Bhattacharya (graduate student) for help with product identification of stilbene epoxidation, Stacy Anderson (Berreau Group, USU) for help with GC-MS detection of evolved gases.References
- B. Nepal and S. Das, Angew. Chem., Int. Ed., 2013, 52, 7224–7227 CrossRef CAS PubMed.
- S. Das, D. E. Johnston and S. Das, CrystEngComm, 2012, 14, 6136–6139 RSC.
- J. Limburg, J. S. Vrettos, H. Y. Chen, J. C. de Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423–430 CrossRef CAS PubMed.
- R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2008, 47, 1815–1823 CrossRef CAS PubMed.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444–455 CrossRef CAS PubMed.
- S. Das, G. W. Brudvig and R. H. Crabtree, Inorg. Chim. Acta, 2009, 362, 1229–1233 CrossRef CAS PubMed.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS.
- R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2007, 46, 2193–2203 CrossRef CAS PubMed.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247–2252 RSC.
- A. Mills, Chem. Soc. Rev., 1989, 18, 285–316 RSC.
- J. L. Fillol, Z. Codola, I. Garcia-Bosch, L. Gomez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS PubMed.
- M. Wiechen, H. M. Berends and P. Kurz, Dalton Trans., 2012, 41, 21–31 RSC.
- B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451–1467 CrossRef CAS PubMed.
- R. Tagore, H. Y. Chen, H. Zhang, R. H. Crabtree and G. W. Brudvig, Inorg. Chim. Acta, 2007, 360, 2983–2989 CrossRef CAS PubMed.
- H. Y. Chen, J. W. Faller, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2004, 126, 7345–7349 CrossRef CAS PubMed.
- C. Baffert, S. Romain, A. Richardot, J. C. Lepretre, B. Lefebvre, A. Deronzier and M. N. Collomb, J. Am. Chem. Soc., 2005, 127, 13694–13704 CrossRef CAS PubMed.
- E. A. Karlsson, B. L. Lee, T. Akermark, E. V. Johnston, M. D. Karkas, J. L. Sun, O. Hansson, J. E. Backvall and B. Akermark, Angew. Chem., Int. Ed., 2011, 50, 11715–11718 CrossRef CAS PubMed.
- S. A. Hayes, P. Yu, T. J. O'Keefe, M. J. O'Keefe and J. O. Stoffer, J. Electrochem. Soc., 2002, 149, C623–C630 CrossRef CAS PubMed.
- S. Das, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2008, 130, 1628–1637 CrossRef CAS PubMed.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS PubMed.
- S. Kundu, M. Schwalbe and K. Ray, BioInorganic Reaction Mechanisms, 2012, 8, 41–57 CrossRef.
- S. Kundu, E. Matito, S. Walleck, F. F. Pfaff, F. Heims, B. Rabay, J. M. Luis, A. Company, B. Braun, T. Glaser and K. Ray, Chem.–Eur. J., 2012, 18, 2787–2791 CrossRef CAS PubMed.
- S. Romain, F. Bozoglian, X. Sala and A. Llobet, J. Am. Chem. Soc., 2009, 131, 2768–2769 CrossRef CAS PubMed.
- B. Li, S. Y. Bai, X. F. Wang, M. M. Zhong, Q. H. Yang and C. Li, Angew. Chem., Int. Ed., 2012, 51, 11517–11521 CrossRef CAS PubMed.
- B. Li, F. Li, S. Y. Bai, Z. J. Wang, L. C. Sun, Q. H. Yang and C. Li, Energy Environ. Sci., 2012, 5, 8229–8233 CAS.
- M. Yagi and K. Narita, J. Am. Chem. Soc., 2004, 126, 8084–8085 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; synthetic procedures; catalytic conditions; FT-IR spectra. See DOI: 10.1039/c3ee43040e |
| This journal is © The Royal Society of Chemistry 2014 |