Absence of Ni on the outer surface of Sr doped La2NiO4 single crystals†
Mónica
Burriel
*a,
Stuart
Wilkins
b,
John P.
Hill
b,
Miguel A.
Muñoz-Márquez
c,
Hidde H.
Brongersma
ad,
John A.
Kilner
ac,
Mary P.
Ryan
a and
Stephen J.
Skinner
*a
aDepartment of Materials, Imperial College London, Exhibition Road, London SW7 2AZ, UK. E-mail: m.burriel@imperial.ac.uk; s.skinner@imperial.ac.uk; Fax: +44 (0)20 7594 6757; Tel: +44 (0)20 7594 6782
bBrookhaven National Laboratory, Upton, NY 11973, USA
cCIC energiGUNE, Albert Einstein 48, 01510 – Miñano, Álava, Spain
dEindhoven University of Technology, Eindhoven, The Netherlands
First published on 7th November 2013
Abstract
A combination of surface sensitive techniques was used to determine the surface structure and chemistry of La2−xSrxNiO4+δ. These measurements unequivocally showed that Ni is not present in the outermost atomic layer, suggesting that the accepted model with the B-site cations exposed to the environment is incorrect.
Broader contextSolid oxide fuel cells (SOFC) offer the potential for efficient and clean energy generation using flexible fuel sources. In order to achieve market penetration of this technology operating temperatures, and hence cost, have to be reduced. A key component of the cell is the air electrode (cathode) where oxygen reduction occurs. This process depends strongly on surface atomic structure and chemistry which determines catalytic activity. In layered cathode materials, such as the La2−xSrxNiO4 compositions that are the focus of this work, it is assumed that the transition metal terminates the surface and hence controls activity. Using a combination of novel techniques which directly probe the outermost atomic layer we demonstrate that the surface is in fact (La/Sr) terminated. In light of this finding the mechanism of oxygen reduction on such surfaces should be reconsidered, with a view to better design of SOFC cathode materials. |
Solid oxide fuel cells (SOFC) offer a low carbon electrochemical route to energy conversion with high efficiency and low emissions, an attractive alternative to combustion of hydrocarbon fuels. Nickelate compounds with the K2NiF4-type structure are promising mixed conducting cathodes for intermediate temperature solid oxide fuel cells (IT-SOFC).1–4 Their fast oxygen transport properties are related to their ability to accommodate hyperstoichiometric oxygen in interstitial positions in the rocksalt layers of the structure.1 Recent molecular dynamics studies of La2NiO4+δ by Chroneos et al.5 have shown that the oxygen migration process takes place by an interstitialcy mechanism involving the migrating interstitial oxygen (Oi) and a neighbouring apical oxygen (Oa). In addition, there is evidence of the orientational dependence of oxygen surface exchange in La2NiO4+δ thin films.6 Substitution of La with the larger Sr atom can increase the stability of the La2NiO4+δ structure7 and also enhances the electronic conductivity with the shortening of the Ni–O1 (equatorial O) bond length.8 Additionally, upon chemical doping with Sr, oxidation of Ni2+ to Ni3+ is expected together with a decrease in the cell volume, which reveals the minor tendency to accommodate excess of oxygen.
As highlighted by Read et al.9 understanding of the surface chemistry of oxides is essential for a wide variety of industrial applications, from catalytic oxidation of NH3 to the oxygen reduction and incorporation of species in fuel cell electrodes. Even though the surface properties of these materials play a critical role in the ultimate device efficiency, as the surface oxygen exchange is often the rate-limiting step, the structure and chemistry of the surfaces have never been thoroughly investigated. In the case of lanthanum manganite (La1−xSrxMnO3, LSM), the state-of-the art cathode material, a significant effort is presently being directed towards the understanding of the atomic-scale processes governing the cathode performance.10–12 In this work we have focused on understanding the structure and related properties of the lanthanum nickelate oxides by combining three complementary and specialized techniques: X-ray scattering through crystal truncation rod (CTR) experiments, Low Energy Ion Scattering (LEIS) and angle-resolved X-ray photoelectron spectroscopy (AR-XPS) and have obtained information on the outermost surface layer and near-surface region of La2−xSrxNiO4+δ single crystals.
To date the outermost surface structure and surface chemistry of K2NiF4-based nickelates (with the A2BO4 structure) has only been explored using a theoretical simulation approach. Read et al.9,13 evaluated the surface energy and crystal morphology of low index surfaces of La2−xSrxNiO4+δ finding that by the incorporation of Sr into La2NiO4+δ electron holes are created. The Ni3+ species are postulated as being active sites responsible for the catalytic activity of Ni-containing perovskite-related oxides. Read also concludes that the (111) and (001) surfaces are terminated by Ni+2-O species, that the (110) surface is terminated by all three ion species, and the (100) surface is O2− terminated. Although the surface energies are similar, this (100) surface is suggested as the highest stability surface for the undoped La2NiO4+δ phase, in which the NiO6 octahedra on the outermost plane are distorted from the bulk symmetry. The higher activity of the transition metal cations, in comparison to that of the alkaline-earth metals or lanthanide metals, in A2BO4 oxides has also been predicted from density functional theory (DFT) simulations for the LaSrCoO4 (010) surface. In this case the O2 reduction was calculated to be more energetically favourable on Co sites (B-sites) than on La/Sr sites (A-sites), on the basis of the difference in adsorption energies.14 Furthermore, DFT simulations on single perovskite LaMnO3 (001) surfaces predict the cubic MnO2 surface (B-site) with adsorbed O atoms to be the most stable one15 and ab initio studies on several perovskite SOFC cathode materials (LaBO3 (001) with B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Mn, Fe, Co and Ni) suggest the LaO (A-site) termination to be catalytically inactive due to its high O vacancy formation and strong O adsorption energies.16
Mn, Fe, Co and Ni) suggest the LaO (A-site) termination to be catalytically inactive due to its high O vacancy formation and strong O adsorption energies.16
Typically surfaces are difficult to analyze due to their sensitivity to carbonation, and the issue of segregation of species to outermost layers. Traditionally XPS has been used to determine the surface chemistry of species, but the technique is inherently limited to the near-surface, as the chemical information provided is always an average over a distinct volume. Conventional XPS experiments at normal take-off angles for the detected photoelectrons and using laboratory X-ray sources provide signals integrated over the first 5–10 nm; a distance defined by the photoelectron inelastic mean free path (IMFP, λ) which depends on the photoelectron kinetic energy and consequently on the binding energy to a specific element. Because 95% of the photoelectrons will escape in 3λ, if the take-off angle is grazing to the surface the detection depth can be reduced down to sub-nanometric distances depending on the experimental set-up. To experimentally determine and fully elucidate the surface structure and chemistry of La2−xSrxNiO4+δ we have used a combination of surface sensitive techniques: CTR experiments to probe the surface structure and LEIS measurements to probe the surface chemistry. Complementary near-surface (0.5–7 nm) and chemical redox information on the same samples was provided by using angle-resolved-XPS.
Single crystals of La1.67Sr0.33NiO4+δ composition were grown by the floating-zone technique (described in detail elsewhere17). A large La1.67Sr0.33NiO4+δ single crystal was cleaved along the (001) and (110) crystallographic planes into several smaller crystals. One of the single crystal samples was used for the CTR measurements. The functional form of the CTRs is indicative of the precise form of the surface termination and is extremely sensitive to the atomic arrangement of the uppermost layers. Two further samples were used for LEIS measurements. LEIS is a surface analysis tool based on the scattering of low energy ions that provides elemental characterisation of the outermost surface layer of atoms in a solid material. Most importantly it yields quantitative, matrix independent, elemental characterisation of this layer and near surface depth profiles.18 The advent of LEIS now offers the opportunity of correlating surface exchange kinetics with chemical processes at materials atomic surfaces, giving unprecedented levels of information on electrochemical systems with isotopic discrimination (e.g.18O/16O ratios).19,20
LEIS experiments were performed on 2 single crystals: one “as-cleaved” and one “heat treated” (72 hours in air at 450 °C). For each of the samples two cleaved surfaces were measured: (001) and (110) which had been previously treated with atomic oxygen during 32 min to remove hydrocarbon contaminants from the surface. Each surface was measured using two different noble gas ions: He+/3 keV and 22Ne+/5 keV (further experimental details included in ESI†). Fig. 1 shows the 22Ne+ LEIS energy spectrum obtained for the (001) face of the La1.67Sr0.33NiO4+δ single crystal after heat treatment. The original data are plotted together with the extracted background (originated from sputtered ions and fit to an exponential function) and with the background corrected spectrum. The spectrum shows the peaks corresponding to the elemental composition of the outermost surface layer. The main peaks can be clearly attributed to single scattering of the Sr and La elements. Critically we find that there is no peak at the expected energy position for Ni indicating its absence from the surface monolayer. In addition to the main Gaussian peaks the contribution of the in-depth distribution of La appears at the lower energy side of the corresponding peak suggesting a high near-surface concentration of this species. Similar conclusions are drawn from the corresponding data recorded for the (110) heat-treated surface (ESI, Fig. S1†) and for the (110) and (001) faces of the as-cleaved sample. All the data are consistent with the presence of La and Sr, and the absence of Ni, at the surface. Preference of the A-site cations at the surface of had been previously measured by LEIS for polycrystalline BaZrO3 (ref. 21) and Sm1−xSrxCoO3−δ (ref. 22) perovskites. To obtain the sensitivity and detection limit of each element (Ni, La and Sr) high-purity NiO, SrO and La2O3 powders were used as reference materials (LEIS energy spectra and calculations included in ESI, Fig. S2†). Ni has the lowest sensitivity, with its detection limit calculated to be 10% of the surface fraction. In the He+ spectra, in addition to the La and Sr peaks, peaks corresponding to lighter elements such as lattice O, together with some surface contamination peaks (Na, K and F), were observed.
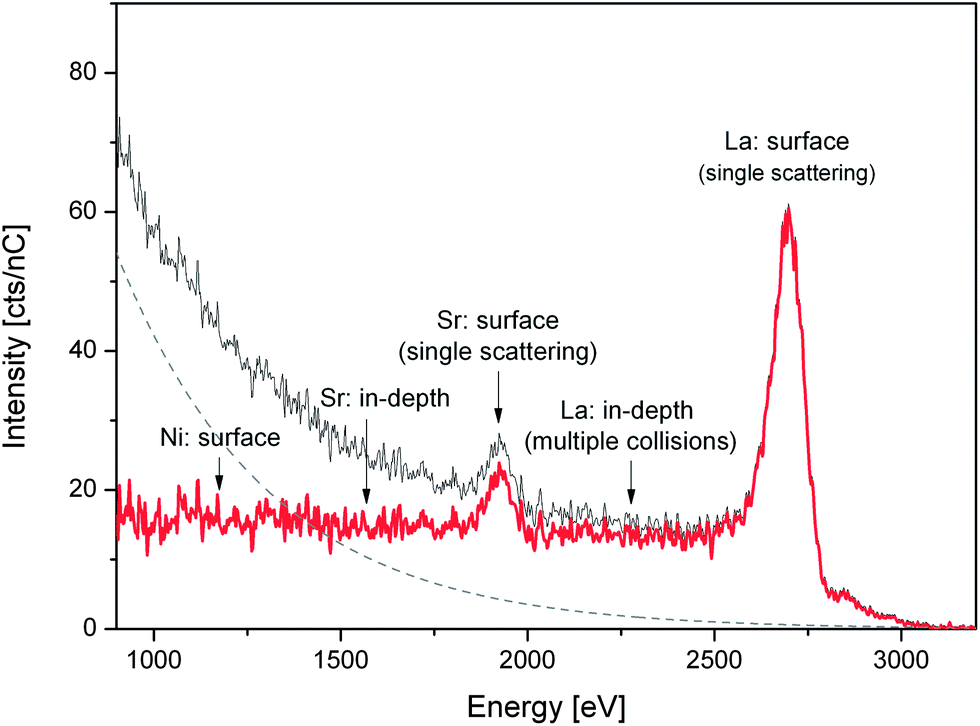 | ||
| Fig. 1 22Ne LEIS energy spectrum for the (001) face of the heat-treated La1.67Sr0.33NiO4+δ single crystal. Original data (grey line), background (dashed line) and obtained corrected spectrum (red line). | ||
X-ray scattering measurements were carried out at beamline X22C at the National Synchrotron Light Source (Brookhaven National Laboratory) with 12 keV X-rays using a six-circle diffractometer for surface scattering experiments. The experiments were carried out in an α = β mode, where α denotes the incidence angle, that is, the angle between the incident X-ray and the crystal surface, and β denotes the exit angle. The surface normal was parallel to the crystallographic c-axis. The CTR scattering intensity distributions along the (00L), (−20L) and (−11L) directions were measured in an air atmosphere at room temperature and 450 °C, to investigate more closely the system under near-operating fuel cell conditions. A Lorentzian squared fit was applied to each scan and optimised by the least square method. Footprint, Lorentz factor and Debye–Waller corrections were applied before comparing experimental and modelled truncation rod data.
The crystal truncation rod data for the (001) surface along the (00L) direction are shown in Fig. 2, along with the fits to the data for the two possible ‘perfect’ terminations: (La,Sr)O and NiO2. The data demonstrate that the surface is clearly (La,Sr)O terminated, and not NiO2 terminated, in good agreement with the LEIS results. Fig. 3a is a representation of the (La,Sr)2NiO4 structure, consisting of a stacking sequence of (La,Sr)2O2 bilayers and NiO2 monolayers along the c-axis. A 3D view of the crystal highlighting the two possible ideal terminations for the (001) face (top surface) is represented in Fig. 3c: NiO2 termination at the left and (La,Sr)O termination at the right side. Similar results (i.e., best fit for the (La,Sr)O termination) were obtained both at high temperature and room temperature for the (00L) direction, and for the two other measured CTR directions, namely (−20L) and (−11L) (ESI, Fig. S3†). On closer examination some deviation of the fit to the perfect (La,Sr)O surface termination is observed. This can be attributed to a combination of several surface effects, such as the variation in surface stoichiometry (Sr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :La and (Sr + La):Ni ratios) and to strain relaxation in the surface region. Attempts at introducing compositional and strain variations within the CTR model structure evidence a complex combination of chemical differences, rumpling and relaxation of the interplanar spacing in the surface region. Therefore a simple isotropic strain relaxation model cannot be applied.
:La and (Sr + La):Ni ratios) and to strain relaxation in the surface region. Attempts at introducing compositional and strain variations within the CTR model structure evidence a complex combination of chemical differences, rumpling and relaxation of the interplanar spacing in the surface region. Therefore a simple isotropic strain relaxation model cannot be applied.
 | ||
| Fig. 2 Crystal truncation rod scattering as measured along the 00L direction in air at 450 °C. Red dots indicate the raw data and solid lines the model CTR patterns for NiO2 (blue) or (La,Sr)O (green) terminations. | ||
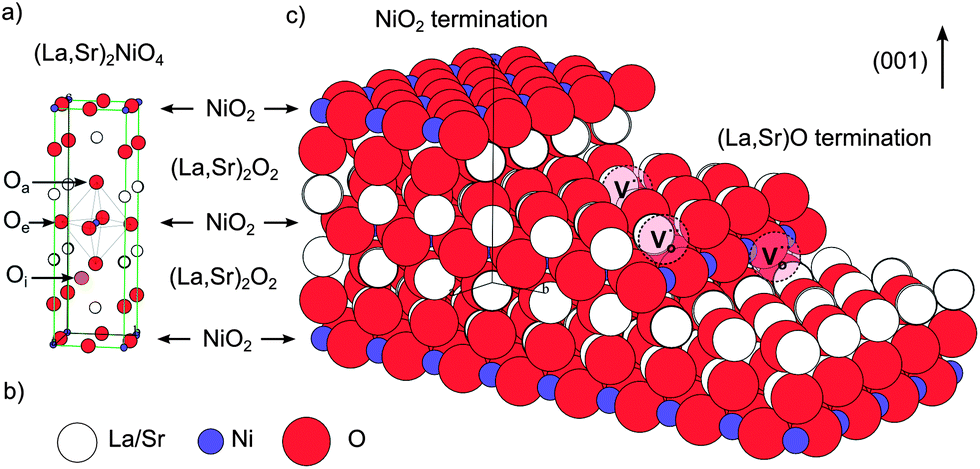 | ||
Fig. 3 (a) Representation of the (La,Sr)2NiO4 structure, consisting of a stacking sequence of (La,Sr)2O2 bilayers and NiO2 monolayers along the c-axis. The position of an oxygen interstitial in the (La,Sr)2O2 layer of the structure has been marked (orange atom) together with the positions of the apical (Oa) and equatorial oxygen (Oe) atoms. (b) Colour code for the Ni (purple), La/Sr (white) and O (red) atoms. (c) 3D view of the crystal highlighting the two possible ideal terminations for the (001) face (top surface), a step edge and possible oxygen vacancy positions (marked as 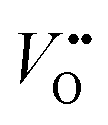 ). ). | ||
To complement these results and to provide information on the near-surface composition, angle-resolved XPS measurements were also performed on the (001) faces for both the “as-cleaved” and the “heat treated” single crystals to obtain a compositional depth-profile. La 3d and 4d, Ni 2p and 3p, Sr 3d, O 1s and C 1s photoelectron peaks were recorded in with a monochromatic AlKα source using a pass energy of 30 eV (further experimental details included in ESI†). Fig. S4 (ESI†) shows that in both crystals samples (as-cleaved and annealed for 72 hours in air at 450 °C) a mixture of Ni3+ and Ni2+ is present and the amount of Ni decreases as the surface is approached, in good agreement with LEIS and CTR results. Additionally, in the as-cleaved sample, the amount of Ni3+ is greater than in the annealed, especially when comparing the 4 nm surface regions. For a proper determination of the Ni3+/Ni2+ ratio, a careful study of the Ni 3p line has been performed for both samples (Fig. 4). The Ni 3p line shows coexistence of Ni3+ and Ni2+ that appear at approximately 71 and 67 eV respectively, which is consistent with the reported literature results.23 After the heat treatment both components are reduced and, as expected, Ni3+ noticeably decreases its contribution from being 34 to 17% of the total Ni component (for a depth of 3.5 nm). The average oxidation state varies therefore from +2.34 in the as-cleaved to +2.17 in the annealed crystal, following a similar behavior to that observed for La2NiO4+δ powder by X-ray absorption spectroscopy of the near-edge region (XANES).24Table 1 shows the thickness-dependent A:B site ratios, i.e. (Sr + La):Ni ratios, calculated from the fitted XPS spectra taken on the (001) faces at different angles. The peak areas used for this calculation correspond to the Ni 3p, La 4d and Sr 3d photoelectron peaks since their kinetic energy is very similar (i.e. 1418, 1380 and 1354 eV respectively) and therefore, the probed depth will be closely the same. For both samples the surface clearly shows an A-site enrichment, which decreases in depth towards the bulk theoretical A:B site ratio of 2. For the as-cleaved crystal the expected theoretical ratio is nearly reached for a depth of 7 nm, while as for the heat treated sample an A-site enrichment 2.25 times larger than the theoretical one (A:B ratio = 4.5) is measured at the same depth. However, it has to be taken into account that the depth profiling performed with XPS is a top-down analysis; this means that the outermost surface layer contribution is always present in the recorded data. By increasing the take-off angle the collected photoelectrons will be closer to the surface, therefore shallower regions close to the surface will be analysed. It should be pointed out that the concentration ratios calculated at each depth correspond to the average composition of an analysed volume from the surface to that depth. This means, that if the average A:B ratio at 7 nm is 2.32, and the external surface is A-site enriched, the exact composition at that depth would be smaller than 2.32.
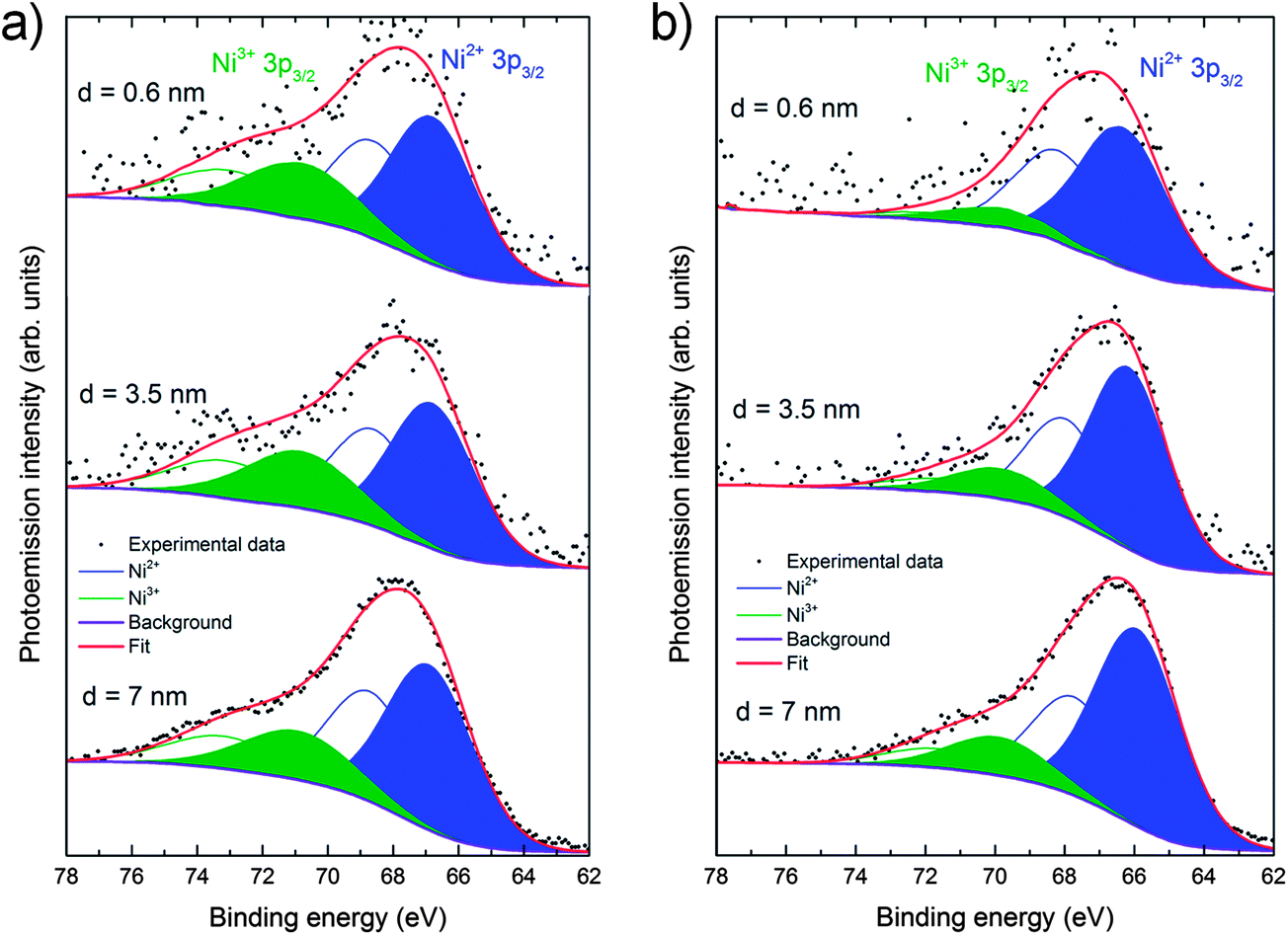 | ||
| Fig. 4 Angle-resolved experimental and deconvoluted Ni 3p XPS data for the (001) face of the La1.67Sr0.33NiO4+δ single crystal (a) before and (b) after heat treatment. The spectra correspond to three different surface regions 0.6, 3.5 and 7 nm. The contributions from Ni2+ and Ni3+ are marked in blue and green respectively. The full peaks correspond to the 3p3/2 lines and the empty curves to the 3p1/2 lines. The envelope of both components is shown in red. | ||
:Ni ratio calculated from the fitted XPS spectra taken on the (001) faces at different angles
| Depth (nm) | (La + Sr)/Ni | |
|---|---|---|
| As cleaved single crystal | Heat treated single crystal | |
| 0.6 | 5.3 ± 1.2 | 6.7 ± 1.9 |
| 1.8 | 8.5 ± 1.6 | 5.5 ± 1.1 |
| 3.5 | 6.5 ± 1.0 | 5.2 ± 0.9 |
| 4.8 | 3.9 ± 0.5 | 4.3 ± 0.7 |
| 5.9 | 3.4 ± 0.4 | 4.6 ± 0.6 |
| 6.6 | 3.1 ± 0.4 | 4.7 ± 0.5 |
| 7.0 | 2.3 ± 0.2 | 4.5 ± 0.4 |
| Bulk (theoretical) | 2.0 | 2.0 |
These complementary techniques show that Ni does not appear at the outermost crystallographic surface. This is counter to the hypothesis that Ni3+ species at the surface facilitates the catalytic activity of these materials. In Kröger–Vink notation, the overall exchange reaction at the gas/film interface can be written as:
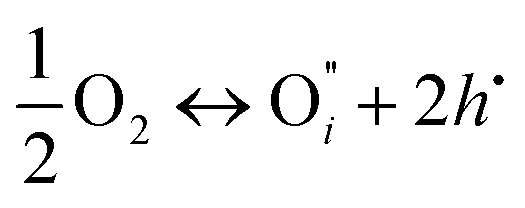 | (1) |
if the oxygen is incorporated into the crystal through the formation of oxygen interstitials or
 | (2) |
if the oxygen is incorporated into oxygen lattice vacancies. In both cases the surface exchange mechanism will involve a number of elementary reactions including O2 transport from the gas phase to the surface, adsorption on the surface, dissociation and charge transfer, and finally, incorporation into the surface layer. Although many different mechanisms are possible, they all include a charge transfer step involving the formation of electron holes either through the oxidation of B site ions (nickel holes) or by the formation of O− small polaron species. This possibility would involve the formation of oxygen holes on the surface, which could occur via different compensation mechanisms involving O− species, such as:
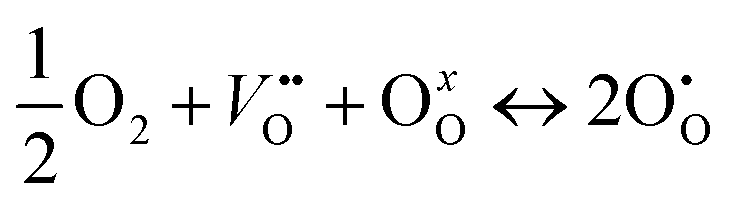 | (3) |
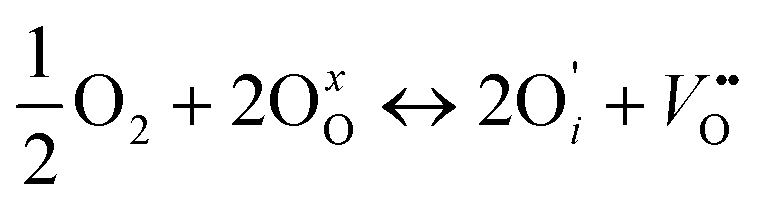 | (4) |
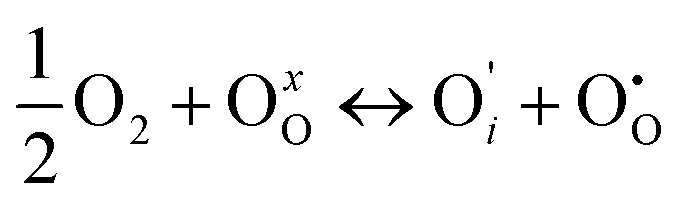 | (5) |
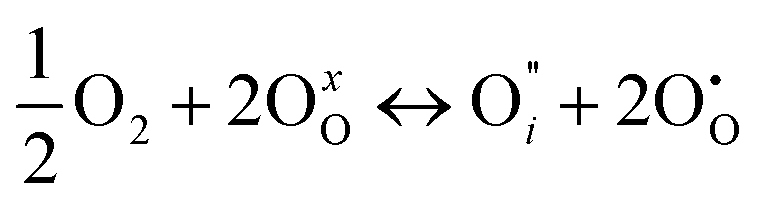 | (6) |
In the case of formation of Ni holes, as the Ni species are not directly accessible at the outermost surface, it is necessary to consider other possibilities, such as surface point defects and step edges. In Fig. 3b these 2 possibilities are shown: oxygen could be incorporated through vacant apical surface sites in the (La,Sr)O layer and/or through vacant equatorial sites at step edges in direct contact with Ni species. The existence and significance of the surface oxygen vacancies is in good agreement with the thermodynamic calculations performed by Read et al.,9 who found that the formation of oxygen vacancies has a lower energy at the surface than in the bulk and would also explain the high activation energies of the surface exchange (kinetic process) for Sr-doped La2NiO4+δ measured by Li et al.25 These authors relate it to oxygen vacancies being only minority defects in the nickelates, and being more energetically favourable on equatorial sites than on apical sites,26 as opposed to the case of perovskite materials, in which oxygen vacancies are the majority defects and which possess considerably lower activation energies. The authors suggested that both for undoped and Sr-doped La2NiO4+δ the rate limiting step for the surface exchange would involve oxygen vacancies and would thus be limited by the availability of oxygen vacancies on the surface. It should be pointed out that Fig. 3c constitutes an ideal representation of La2−xSrxNiO4+δ surface and that the real equilibrium surface structure is expected to be modified from the bulk one by rumpling and/or relaxation of the interplanar spacing in the surface region, as evidenced by the CTR results. Moreover A-site enrichment (several nm in depth) has been measured by XPS, which means that either the stacking sequence of (La,Sr)2O2 and NiO2 layers varies in the close-surface region, or a different A-site rich surface phase is formed.
As expected, for the sample that was not annealed a mixture of Ni2+ and Ni3+ is measured by XPS, as by the incorporation of Sr into La2NiO4+δ electron holes are created for charge compensation. It is also observed that the ratio between Ni2+ and Ni3+ species does not vary in depth (within the experimental error), which implies that the differences in La/Sr composition could be compensated by differences in the oxygen content (interstitials and/or vacancy concentration) in the top surface region. When the sample is annealed, oxygen will partially desorb to reach the new equilibrium state accompanied by the reduction of Ni3+ to Ni2+ to maintain electroneutrality. The XPS Ni 3p spectra show a larger Ni2+ component for the annealed sample, which clearly evidences a reduction in the oxidation state of the Ni species in the near surface region.
Conclusions
In this study we have clearly shown that for La1.67Sr0.33NiO4+δ, a promising mixed conducting cathode, Ni is not present on the outermost atomic layer of the low-index (110) and (001) faces of a single crystal. This significant discovery suggests that the accepted model with the B-site cations exposed to the environment is incorrect and other possibilities which explain the catalytic activity of the surface have been proposed. The LEIS results have been complemented with the chemical redox information provided by AR-XPS and correlated to the crystallographic data of the crystal surface provided by CTR measurements. All of these methods are in good agreement. Hence, in light of these compelling new data, it is essential that the simulation of these surfaces is revisited. More generally it is clear that the unique monolayer sensitivity provided by LEIS, complemented by other state-of-the-art surface techniques, can provide a complete picture of the surface and near-surface chemistry of this important class of materials. It is anticipated that the new surface information provided by these techniques will contribute significantly to the understanding of the surface processes in mixed conducting materials, with great importance in the catalysis and solid state ionics fields.Acknowledgements
This research was supported by a Marie Curie Intra European Fellowship within the seventh European Community Framework Programme (PIEF-GA-2009-252711) and by KAUST (King Abdullah University of Science and Technology) Academic Excellence Alliance (for M.B). Use of the National Synchrotron Light Source, and work carried out at Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract no. DE-AC02-98CH10886. The authors also thank D. Prabhakaran (University of Oxford) for providing single crystals and M. Fartmann (TASCON GmbH), T. Grehl, P. Brüner (ION-TOF GmbH), S. Fearn and H. Téllez (Imperial College) for LEIS measurements and support with LEIS data analysis.Notes and references
- C. N. Munnings, S. J. Skinner, G. Amow, P. S. Whitfield and I. J. Davidson, Solid State Ionics, 2005, 176, 1895–1901 CrossRef CAS PubMed.
- E. Boehm, J. M. Bassat, P. Dordor, F. Mauvy, J. C. Grenier and P. Stevens, Solid State Ionics, 2005, 176, 2717–2725 CrossRef CAS PubMed.
- E. Boehm, J. M. Bassat, M. C. Steil, P. Dordor, F. Mauvy and J. C. Grenier, Solid State Sci., 2003, 5, 973–981 CrossRef CAS.
- S. J. Skinner and J. A. Kilner, Solid State Ionics, 2000, 135, 709–712 CrossRef CAS.
- A. Chroneos, D. Parfitt, J. A. Kilner and R. W. Grimes, J. Mater. Chem., 2010, 20, 266–270 RSC.
- M. Burriel, G. Garcia, J. Santiso, J. A. Kilner, J. C. C. Richard and S. J. Skinner, J. Mater. Chem., 2008, 18, 416–422 RSC.
- X. Granados, J. Fontcuberta, M. Valletregi, M. J. Sayagues and J. M. Gonzalezcalbet, J. Solid State Chem., 1993, 102, 455–464 CrossRef CAS.
- A. Aguadero, M. J. Escudero, M. Perez, J. A. Alonso, V. Pomjakushin and L. Daza, Dalton Trans., 2006, 4377–4383 RSC.
- M. S. D. Read, M. S. Islam, G. W. Watson and F. E. Hancock, J. Mater. Chem., 2001, 11, 2597–2602 RSC.
- T. T. Fister, D. D. Fong, J. A. Eastman, P. M. Baldo, M. J. Highland, P. H. Fuoss, K. R. Balasubramaniam, J. C. Meador and P. A. Salvador, Appl. Phys. Lett., 2008, 93, 151904 CrossRef.
- W. A. Harrison, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155437 CrossRef.
- H.-T. Chen, P. Raghunath and M. C. Lin, Langmuir, 2011, 27, 6787–6793 CrossRef CAS PubMed.
- M. S. D. Read, M. S. Islam, F. King and F. E. Hancock, J. Phys. Chem. B, 1999, 103, 1558–1562 CrossRef CAS.
- J. Zhou, G. Chen, K. Wu, Y. Cheng, B. Peng, J. Guo and Y. Jiang, Appl. Surf. Sci., 2012, 258, 3133–3138 CrossRef CAS PubMed.
- E. A. Kotomin, Y. A. Mastrikov, E. Heifetsa and J. Maier, Phys. Chem. Chem. Phys., 2008, 10, 4644–4649 RSC.
- Y.-L. Lee, J. Kleis, J. Rossmeisl and D. Morgan, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224101 CrossRef.
- D. Prabhakaran, P. Isla and A. T. Boothroyd, J. Cryst. Growth, 2002, 237, 815–819 CrossRef.
- H. H. Brongersma, M. Draxler, M. de Ridder and P. Bauer, Surf. Sci. Rep., 2007, 62, 63–109 CrossRef CAS PubMed.
- J. A. Kilner, S. J. Skinner and H. H. Brongersma, J. Solid State Electrochem., 2011, 15, 861–876 CrossRef CAS PubMed.
- H. Tellez, R. J. Chater, S. Fearn, E. Symianakis, H. H. Brongersma and J. A. Kilner, Appl. Phys. Lett., 2012, 101, 151602 CrossRef.
- J. Rosink, J. P. Jacobs and H. H. Brongersma, in Surfaces, Vacuum, and Their Applications, ed. I. Hernandez Calderon and R. Asomoza, 1996, pp. 44–51 Search PubMed.
- I. C. Fullarton, J. P. Jacobs, H. E. van Benthem, J. A. Kilner, H. H. Brongersma, P. J. Scanlon and B. C. H. Steele, Ionics, 1995, 1, 51–58 CrossRef CAS.
- J. F. Marco, J. R. Gancedo, J. Ortiz and J. L. Gautier, Appl. Surf. Sci., 2004, 227, 175–186 CrossRef CAS PubMed.
- R. J. Woolley, B. N. Illy, M. P. Ryan and S. J. Skinner, J. Mater. Chem., 2011, 21, 18592–18596 RSC.
- Z. Li, R. Haugsrud and T. Norby, Solid State Ionics, 2011, 184, 42–46 CrossRef CAS PubMed.
- L. Minervini, R. W. Grimes, J. A. Kilner and K. E. Sickafus, J. Mater. Chem., 2000, 10, 2349–2354 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ee41622d |
| This journal is © The Royal Society of Chemistry 2014 |