Lithium ion battery applications of molybdenum disulfide (MoS2) nanocomposites†
Tyler
Stephenson
*ab,
Zhi
Li
ab,
Brian
Olsen
ab and
David
Mitlin
*ab
aDepartment of Chemical an d Materials Engineering, University of Alberta, Edmonton, Alberta, Canada T6G 2G6. E-mail: tjs10@ualberta.ca; dmitlin@ualberta.ca; Tel: +780-554-8420 Tel: +780-492-1542
bNational Institute for Nanotechnology, National Research Council of Canada, 11421 Saskatchewan Drive, Edmonton, Alberta, Canada T6G, 2M9
First published on 17th October 2013
Abstract
This is the first targeted review of the synthesis – microstructure – electrochemical performance relations of MoS2 – based anodes and cathodes for secondary lithium ion batteries (LIBs). Molybdenum disulfide is a highly promising material for LIBs that compensates for its intermediate insertion voltage (∼2 V vs. Li/Li+) with a high reversible capacity (up to 1290 mA h g−1) and an excellent rate capability (e.g. 554 mA h g−1 after 20 cycles at 50 C). Several themes emerge when surveying the scientific literature on the subject: first, we argue that there is excellent data to show that truly nanoscale structures, which often contain a nanodispersed carbon phase, consistently possess superior charge storage capacity and cycling performance. We provide several hypotheses regarding why the measured capacities in such architectures are well above the theoretical predictions of the known MoS2 intercalation and conversion reactions. Second, we highlight the growing microstructural and electrochemical evidence that the layered MoS2 structure does not survive past the initial lithiation cycle, and that subsequently the electrochemically active material is actually elemental sulfur. Third, we show that certain synthesis techniques are consistently demonstrated to be the most promising for battery applications, and describe these in detail. Fourth, we present our selection of synthesis methods that we believe to have a high potential for creating improved MoS2 LIB electrodes, but are yet to be tried.
 Tyler Stephenson | Tyler Stephenson is currently finishing his PhD in Materials Engineering at the University of Alberta. His strong interest in electrochemistry and energy storage is an asset for his lithium ion battery research. His other projects focus around corrosion and fouling for the oil and gas industry, nuclear industry, and on the remediation of tailings from the Alberta oil sands. In 2010, Tyler completed his BSc in Materials Engineering, with a specialization in nano and functional materials, at the University of Alberta. |
 Zhi Li | Zhi Li received his bachelor and master degree at the Shanghai Jiaotong University, China. Thereafter he joined the department of Chemistry and Geochemistry, Colorado School of Mines, USA, and received his PhD in 2009. In 2010 he joined the University of Alberta and National Institute for Nanotechnology in Canada as a post-doctoral research fellow. His research interests are in the field of materials for electrochemical energy storage and catalysis. |
Brian Olsen received his BSc in Engineering Physics and MSc in Materials Engineering from the University of Alberta. He is currently working at the University of Alberta as a research engineer specializing in energy storage, microscopy, and data analysis. |
 David Mitlin | David Mitlin is a Professor in the Department of Chemical and Materials Engineering at the University of Alberta and a Principal Research Officer at the National Institute for Nanotechnology NRC. Dr Mitlin has published over 100 peer-reviewed journal articles, holds two U.S. patents, is an Editor for Journal of Materials Science, and serves on the Board of Review for Metallurgical and Materials Transactions. Prior to joining the University of Alberta in 2004, Dr Mitlin was at Los Alamos National Laboratory (USA), where he was awarded a Directors Funded Post Doctoral Fellowship. From 2000 to 2002, Dr Mitlin worked as an Integration Engineer at the IBM Semiconductor Research and Development Center, in Hopewell Junction NY. He received a Doctorate in Materials Science from U.C. Berkeley in 2000. |
Broader contextThe unique properties of molybdenum disulfide engender a versatility that has enabled its use in a wide range of scientific fields. The global prevalence of lithium ion battery (LIB) technology creates a strong driving force for the development of advanced electrode materials. This is the first targeted review on the synthesis – microstructure – electrochemical performance relationships of MoS2 electrodes for secondary LIBs. The use of MoS2 is discussed both in terms of its application for LIB anodes, and for cathodes – an emerging and highly promising area. A comparative assessment of MoS2 as an electrode material places it in a highly competitive position. While MoS2–lithium metal batteries were unsuccessfully commercialized in the 1980's, we will show that advancements in nanostructuring of the material, ionic liquid electrolytes, and nanocomposites have revitalized the research effort. We present a detailed and critical analysis regarding what is known about the lithiation/delithiation mechanism in MoS2, and highlight key areas requiring further study. |
1 Introduction
In this review we focus on synthesis – microstructure – electrochemical performance relationships for MoS2 – based electrodes for secondary lithium ion batteries (LIBs). The use of MoS2 is discussed both in terms of its application for LIB anodes (where most work has been done) and for cathodes (an emerging and highly promising area). We treat these aspects in detail, while avoiding discussions regarding other dichalcogenides. We present a detailed and critical review regarding what is known about the lithiation/delithiation mechanism in MoS2; a topic that is currently under significant debate in the energy storage community. The outline of this review is as follows: Section 2.0 presents other applications of MoS2 beyond rechargeable batteries. Section 3.1 outlines the long and rich history of MoS2 in the battery field, including its commercial applications in the 1980's by Moli Energy. Section 3.2 contrasts the scope and the aims of this article with those of several other recent reviews that adopt a broader context for the various energy and non-energy applications of dichalcogenides. Section 4.0 provides an overview of MoS2 in the LIB material landscape, contrasting its electrochemical performance to that of other promising electrode materials. Section 5.0 details the structure and bonding of MoS2. In Section 6.0 we discuss the lithiation mechanisms of MoS2, which are in fact highly dependent on the cut-off voltage at discharge. Section 7.0 presents a detailed discussion on MoS2 nanocomposites for LIBs. Here we consider the interrelation between the various synthesis approaches that have been employed for creating nanostructured MoS2. In addition we examine the resultant microstructures that often contain a secondary nanodispersed carbon phase, and the several criteria used to judge electrochemical cycling performance (e.g. maximum reversible capacity, cycle 1 and steady-state cycling coulombic efficiency, cycling capacity retention, and rate capacity retention). Finally in Section 8.0, we present an overview of what we believe are the additional promising approaches for achieving MoS2 – based nanomaterials for lithium storage, but which have not been investigated to date. Section 9.0 contains the concluding remarks.2 Applications of MoS2
Molybdenum disulfide exhibits a remarkably diverse range of unique properties, many of which are effectively summarized by the reviews of Strano et al.1 and Zhang et al.2 Due to these properties MoS2 is currently the focus of research groups across a broad range of disciplines. In the field of tribology, MoS2 is often referred to as a super lubricant. The material has long been recognized for its exceptional lubricity, owing to the weak van der Waals bonds between S–Mo–S layers, and to the relative inertness of the sulfur basal planes.3 It has been a favored dry lubricant for aerospace applications due to its excellent lubricity in vacuo and under high load.4,5 MoS2 is also a key additive in many types of commercial lubricating fluids and greases. More recently, a fascinating negative compressibility effect for MoS2 under conditions of dynamic shear has revealed a new property of the interlayer glide mechanism.6 MoS2 is also making an impact in the field of chemical sensing. Owing to the variance in reactivity between basal and edge sites, functionalized MoS2 nanosheets have been used to immobilize DNA strands and immunoglobins for biosensing applications.7,8 Additionally, flexible MoS2 based thin film transistors have successfully been used to detect minute changes in nitrogen oxide (NO),9 and nitrogen dioxide (NO2)10 concentrations under ambient conditions. This type of sensor is highly sought after in the field of toxic gas detection.The tunable direct band gap and highly deformable nature of a monolayer of MoS2 has led many researchers to consider it as a viable material for photovoltaic applications.11–15 In addition to this, the use of MoS2 in catalysis is widespread due again to its band gap (which couples well with the solar spectrum), as well as the catalytic activity of its edge sites.16,17 As a photocatalyst for the oxidation of environmental contaminants, MoS2 is frequently coupled with an additional semiconducting phase (such as TiO2) as a nanocomposite with great positive effect.18,19 Its catalytic effects for hydrodesulfurization in oil refining are well known.20 MoS2 has also been shown as a synergistic catalytic support for gold nanoparticles21 as well as for a nickel–iron alloy used in the electrooxidation of hydrazine, an important fuel.22 Furthermore, many groups are exploring the catalytic effect of MoS2 for hydrogen evolution.23–28 Finally, methods are being elucidated to employ MoS2 nanocomposites as flexible and rewritable memory diodes.29 While this article will focus on the application of MoS2 to the field of energy storage in lithium ion batteries, the material remains exquisitely versatile.
3 History
3.1 Background and Motivation
Efficient energy storage is a long-standing technological and scientific problem that has global implications for all of humanity. The requirements of progressively smaller scale and larger capacity for a wide range of portable, automotive and stationary systems continue to be strong driving forces for the development of advanced lithium ion batteries. Though lithium has been incorporated into battery systems since the late 1950's,30–32 commercial secondary lithium ion batteries remain a challenge for many applications requiring capacity retention over thousands of cycles and progressively higher energy storage densities. Early work revealed the propensity of mixed valence state transition metal dichalcogenides to intercalate alkali metal ions.33–35 These studies led to various commercial primary lithium battery systems in the 1970's. Of these, lithium sulfur dioxide (Li/SO2), lithium thionylchloride (Li/SOCl2), and lithium sulfurylchloride (Li/SO2Cl2) were influential. In addition, the viability of molybdenum and other disulfides was also explored.36–43Given the emergence of nanostructured materials, MoS2 is once again becoming the subject of significant attention as a battery anode material.44,45 The material is quite promising as a negative electrode, since its capacity can be three and a half times that of commercial graphite anodes (372 mA h g−1). For example, reversible values as high as 1290 mA hg−1 have been reported for nanostructured MoS2–graphene composite electrodes.46 Moreover, compared to other emerging negative electrode materials, such as silicon or germanium, MoS2 generally displays much better rate capability and lower rates of cycling induced degradation. While silicon anodes possess initial capacities around 3500 mA h g−1 when tested at low rates such as 0.1 or 0.2 C, they retain minimal capacity at rates of 10 C and higher, e.g.47 Conversely, as will be discussed later, properly designed MoS2 electrodes are capable of being cycled at high current densities, retaining a capacity of 554 mA h g−1 after 20 cycles at a rate of 50 C.48 From a practical battery design perspective, MoS2 electrodes are also quite attractive since they possess significantly less volumetric expansion upon lithiation compared to some other conversion materials. For example, while silicon expands 280% upon full lithiation (Li15Si4), the conversion reaction of MoS2 to Li2S and molybdenum yields “only” 103% expansion.
A key disadvantage of employing MoS2 based anodes is their intermediate lithiation voltages (1.1–2.0 V vs. Li/Li+, depending on the degree of lithiation), which substantially narrows the voltage window and hence the net energy density of a full cell LIB. While one can argue that a higher potential versus lithium makes MoS2 safer than graphite due to a reduction in the driving force for lithium dendrite formation, there is always a trade-off.49 Moreover, recent studies by Cui et al.50 and Xie et al.51 have demonstrated that by employing heteronanostructures that can accommodate the volume expansion, the cyclability of silicon may be substantially improved. As is going to be documented in this review, nanostructured MoS2-based anodes are also highly stable during cycling. However a polysulfide shuttling problem, well known for Li–S batteries, may cause premature electrode failure via electrochemical degradation of the active material.
In addition, the intermediate voltage profile versus Li/Li+ has led researchers to consider MoS2 as a positive electrode, with the system often being pre-lithiated prior to device assembly. The application of MoS2 as a cathode was patented in 1980 (ref. 52) and has since been explored by others.53–57 Similar to the case of using MoS2 as an anode, the intermediate voltage of MoS2 is generally viewed as a disadvantage for its use as a cathode. The cathodic voltage of LiMoS2 is lower than other commercial cathode materials such as lithium cobalt oxide (LiCoO2) and related four and five component oxides (2.5–4.5 V vs. Li/Li+).58–61 However in some applications this may be compensated by the higher charge storage capacity of LiMoS2 to yield energy densities on par or even higher than LiCoO2 and related materials (for example 1.5 V × 1000 mA h g−1vs. 3.5 V × 150 mA h g−1).62,63 Secondary lithium metal batteries using MoS2 as a cathode and lithium as an anode were commercialized in the early 1980's by Moli Energy. However these batteries were prone to the growth of lithium dendrites from the anode which resulted in poor cycle life and safety concerns due to short circuiting.
Archer et al.64 have recently shown that this effect can be mitigated by the use of carefully selected ionic liquid electrolytes. These researchers constructed a half cell of MoS2 and lithium, using an electrolyte consisting of a blend of silica nanoparticles with 1-methyl-3-propylimidazolium bis(trifluoromethanesulfone) and propylene carbonate (SiO2-IL-TFSI/PC). The cell retained a reversible capacity of 750 mA h g−1 after 15 cycles. Moreover, the use of their hybrid electrolyte prolonged short circuit times by an order of magnitude compared to a “standard” electrolyte of ethylene carbonate and dimethyl carbonate. This achievement should provide a research path forward to optimize pre-lithiated MoS2-based microstructures for positive electrode applications. Not only would the revival of the lithium–metal battery solve electrode compatibility issues, it would enable the use of a wider range of high capacity cathodic materials. The emergence of nanostructured materials has led to a performance enhancement of a number of traditional lithium ion battery materials. As a result, molybdenum disulfide is presently being re-explored as an advanced lithium ion battery material and will hence be the focus of this article.
3.2 Related reviews
Recently there have been several high quality review articles on the synthesis and structure of a variety of sulfides which include their general application in a multitude of functional and energy storage fields. The relatively broad and excellent review by Chen et al.65 discussed a wide range of sulfides and chalcogenides, including MoS2. This review gave a comprehensive description of chalcogenide properties including optical, magnetic, electrical, field-emission, photoelectric, thermoelectric, and photocatalytic activity. In addition to the energy storage applications of these materials (lithium ion batteries), the authors discussed a wide range of others including fuel cells, solar cells, and electrical nanogenerators.The comprehensive article by Yu et al.66 provides another exceptional discussion on a wide variety of nanostructured metal chalcogenides, including sulfur, selenium and tellurium compounds, for energy conversion and storage. The thrust of their work is to summarize and critically compare winning synthesis and modification strategies across a range of energy related applications, making the review highly pertinent across those fields. Emphasis is placed on methods for creating a wide array of nanomaterials, including discussions of an array of liquid-phase synthesis methodologies and strategies for modification of metal chalcogenide nanomaterials. A diverse range of synthesis methods are covered including liquid exfoliation, hot-injection, mixed solvent, microwave, Kirkendall-effect-induced and photochemical. Modification of metal chalcogenide nanomaterials with carbon, noble metals, metal oxides and with other metal chalcogenides is also discussed in detail. The applications covered include fuel cells, water splitting, supercapacitors and solar cells, in addition to lithium ion batteries.
The highly relevant review by Zhang et al.67 focuses on metal dichalcogenide (mostly MoS2) nanosheets, and covers a broad array of synthesis methods, properties and applications. The section on preparation methods emphasizes the optimum approaches for yielding such morphologies as 2D graphene-like single and multi-layers. Moreover this particular article offers a uniquely in-depth discussion of device and sensor applications of MoS2. The authors begin with the synthesis approach that really began the “graphene revolution” i.e. mechanical cleavage, and demonstrate how it has been applied to a variety of materials such as sulfides, nitrides, selenides, and oxides. The review then covers synthesis by electrochemical lithium intercalation, exfoliation, and sonication in various solvents as well as CVD growth. The authors provide a critical review of MoS2 crystal structure (structures 2H and 1T are emphasized), mechanical properties, electronic structure and optical properties. In the Applications section the review provides a detailed discussion regarding the use of MoS2 nanosheets for electronic devices, optoelectronic devices, sensing platforms, and energy storage devices that includes both electrochemical supercapacitors and lithium ion batteries.
Researchers focused a state-of-the-art review on several highly technologically promising two-dimensional layered nanomaterials: molybdenum trioxide (MoO3), disulphide (MoS2), diselenide (MoSe2) and ditelluride (MoTe2).68 Their manuscript provides an accurate overview of the crystal structure and bonding of the oxide and of each dichalcogenide, and explains and contrasts their electronic band structure, electrical, optical, mechanical, thermal and magnetic properties. Synthesis methods for layered crystals including vapor phase deposition (PVD and CVD methods), liquid phase deposition and solid state reactions are discussed. Moreover this review offers a detailed section discussing methods for layer exfoliation and identification. Approaches such as mechanical exfoliation, liquid exfoliation, laser thinning, as well as AFM and optical methods for thickness and layer number identification are examined. The authors span numerous application fields by discussing uses of these materials as lubricants, in electrochromic systems, in electronic devices, in battery electrodes, as catalysts, in optical devices, in sensors and in superconductors.
The key aspect differentiating this review from others is that while being quite comprehensive, we focus almost entirely on synthesis – microstructure – electrochemical performance relationships of MoS2 – based electrodes. These are discussed in terms of their application to anodes and cathodes in LIBs. We treat these aspects in substantial detail, and keep our focus relegated to MoS2. We limit our discussion of the synthesis methods to those approaches that have either been demonstrated to be optimum for LIBs or to those that in our opinion have much promise. Moreover, a substantial portion of this manuscript contains a critical discussion regarding the ambiguity in the battery literature concerning the actual lithiation sequence of MoS2, with and without the addition of nanostructured carbons.
4 MoS2 in the LIB material landscape
While bulk MoS2 offers little in the way of exciting electrochemical properties for lithium storage, its nanostructured counterparts are the focus of much attention. The nanostructuring of materials for lithium ion batteries embodies a number of well-known advantages and disadvantages.44,69–71 The trend that nanostructured analogues routinely outperform their bulk equivalents in terms of capacity and cycle life has been demonstrated in many other emerging negative electrode materials. As such, there exists a wide range of nanostructured materials that provide a highly competitive landscape in terms of electrochemical performance in LIBs. Compared to these, MoS2 stacks up well in terms of experimental reversible specific capacity, and values as high as 1290 mA h g−1 have been reported for nanostructured MoS2–graphene composite electrodes.46 While there are a few materials with higher theoretical capacity, such as certain nanostructured carbons, silicon,72 tin,73,74 and tin dioxide (SnO2),75,76 MoS2 is advantageous in terms of rate capability and capacity retention, as well as cost (e.g. sub-micron MoS2 powder retails for dollars per kg).Nanostructured carbons show significantly higher lithium storage capacity than bulk graphite, especially at high current densities.77,78 Single-walled carbon nanotubes exhibit a range of capacities between 400 and 460 mA h g−1, while multi-walled carbon nanotubes have a capacity of 340 mA h g−1, similar to graphite (372 mA h g−1).79–81 Reversible capacities from 540–780 mA h g−1 have achieved for graphene, which can be further enhanced by forming mixtures with other carbon allotropes such as carbon nanotubes and fullerenes.82–84 Furthermore, an impressive 800 mA h g−1 specific capacity was reported for oxidized graphene nanoribbons.85 More recently, researchers often employ graphene as a nanocomposite additive with great positive effect.86 However, the low packing density of carbons, especially for the nanostructured variety limits their volumetric energy density, one of the most important parameters for portable applications.77,87 Also, the high surface area often leads to the excessive formation of solid electrolyte interphase (SEI) which results in large irreversible capacities and capacity fading.77,88 In addition, most varieties of nanostructured carbons, such as graphene and carbon nanotubes, remain far too expensive for commercial electrode applications.
Among the other emerging anode materials, nanostructured metal oxides remain attractive in terms of capacity, though they generally fall short in their rate capability, significant overpotential, and capacity retention. For example, SnO2 can exhibit a large specific capacity (∼800 mA h g−1) when coupled with carbon.89 However, similar to what is found for most conversion oxides, poor cycling performance has impeded tin dioxide's usefulness. Cobalt oxide (Co3O4) is also promising owing to its large theoretical specific capacity (890 mA h g−1).90 However it too demonstrates poor cycling stability. There are some general exceptions to the rule that oxides cycle poorly, and have poor rate performance. Some notable cases are the insertion electrodes based on TiO2 nanostructures.91–94 However, the capacity of TiO2 (250 mA h g−1) is less than that of graphite. Molybdenum dioxide (MoO2) graphene nanocomposites (there is some debate concerning whether these are conversion or insertion electrodes) have been reported to retain 675 mA h g−1 after 100 cycles.95 Recently, an MoO2 nanocomposite with multiwalled carbon nanotubes was reported to have a reversible capacity of 1144 mA h g−1 after 200 cycles.96 MoO2 has also been successfully employed as a single component anode demonstrating a simple, low cost fabrication process.97 Other materials, such as nickel oxide (NiO) also exhibit enhanced performance (1031 mA h g−1 after 40 cycles) when formed as a nanocomposite with graphene.98–100
Other materials which exhibit high capacities for lithium storage include silicon and sulfur compounds. Silicon nanoparticles in a composite with graphene, as well as aluminum coated silicon nanowires have been shown to exhibit large reversible capacities of 1866 mA h g−1 (ref. 101) and 3300 mA h g−1 (ref. 102) respectively. However the majority of silicon nanostructures are prone to rapid capacity degradation due to volumetric expansion upon lithium intercalation which pulverizes the electrode.103–106 The samples in ref. 102 suffered a 25% capacity degradation after 50 cycles. Tin disulfide (SnS2) is also being considered as a replacement for the commercial graphite anode. SnS2 nanoplates were shown to retain a capacity of 583 mA h g−1 after 30 cycles but did not survive for longer durations.107,108 Elemental sulfur has long been recognized for its large theoretical specific capacity of 1675 mA h g−1.109 Unfortunately, lithium–sulfur batteries suffer from poor rate capability (due to poor electrical conductivity of sulfur) and dissolution of lithium–sulfur compounds. Promising efforts are underway to stabilize these compounds (usually with a carbon phase) and capacities as high as 455 mA h g−1 after 50 cycles at higher current densities have been reported.110 These materials, as well as others are effectively summarized in recent reviews.111–121
Table 1 provides a summary of the electrochemical performance of various LIB electrode materials. From the comparison, we can see that MoS2 is a highly competitive LIB material in terms of charge capacity, rate capability and cycle life. The main disadvantage of MoS2 is the intermediate voltage of 2.0 V that prevents it from coupling well with other materials in a full cell. Reported voltages vs. Li/Li+ are experimentally measured values.
| Anode materials | Theoretical specific capacity (mA h g−1) | Voltage vs. Li/Li+ | First discharge capacity (mA h g−1) | First charge capacity (mA h g−1) | Reversible capacity after (W) cycles (mA h g−1) | Coulombic efficiency after (X) cycles (%) | Current density | Reversible capacity (mA h g−1) after (Y) cycles at (Z) current density | Reference |
|---|---|---|---|---|---|---|---|---|---|
| a * – indicates a value estimated from a published graph. | |||||||||
| MoS 2 | 669–1675 | 2.0 | 1062 | 917 | 907 (50) | 98* (50) | 1 C | 554 (20) (50 C) | 48 |
| MoS2–GNS | 669–1675 | 2.0 | 1300 | 2200 | 1290 (50) | 99.2 (50) | 100 mA g−1 | 1050 (5) (1000 mA g−1) | 46 |
| MoO2–MWCNT | 840 | 1.6 | 2270 | 1243 | 1144 (200) | 99 (200) | 100 mA g−1 | 408 (5) (1 A g−1) | 96 |
| TiO2 | 335 | 1.5 | 334 | 245 | 243 (30) | 98.7 (30) | 66 mA g−1 | 6 (10) (6.67 A g−1) | 91 |
| Co3O4 | 890 | 1.2 | 1285 | 1108 | 1004 (50) | 98 (50) | 50 mA g−1 | 790 (5) (1 A g−1) | 90 |
| Sn–C | 994 | 0.6 | 490* | 350* | 510* (200) | 99* (200) | 0.8 C | 200 (10) (5 C) | 74 |
| SnO2–GNS | 790 | 0.6 | 1875* | 1120* | 872 (200) | 99.5 (200) | 100 mA g−1 | 519 (10) (2 A g−1) | 75 and 76 |
| SiNW–Al | 4200 | 0.5 | 3347 | 3105 | 1300 (100) | 98.8 (100) | 0.1 C | 1300 (100) (0.1 C) | 102 |
| NiO–GNS | 718 | 0.5 | 1600* | 1056 | 1031 (40) | 98 (40) | 0.1 C | 460* (5) (5 C) | 98 and 99 |
| Graphene | 372–1116 | 0.5 | 945 | 650 | 460 (100) | 99* (100) | 1 C | 460 (100) (1 C) | 82–84 |
| Graphite | 372 | 0.3 | 320* | 320* | 240 (20) | 99* (1) | 50 mA g−1 | 240 (20) (50 mA g−1) | 84 |
| Li | 3600 | 0.0 | — | — | — | — | — | — | 111 |
| Cathode materials | |||||||||
| Li Ni0.5Mn1.5O4 | 331 | 4.6 | 311 | 367 | 294 (80) | 99* (80) | 0.3 C | 245* (30) (7 C) | 58 |
| LiCoO2 | 272 | 4.5 | 190 | 153 | 110* (14) | — | 47 mA g−1 | 110* (14) (47 mA g−1) | 62 and 63 |
| Sulfur | 1675 | 2.0 | 960 | 830* | 650 (40) | 95 (30) | 0.2 C | 350 (45) (1 C) | 109 |
5 Structure and bonding of MoS2
A considerable amount of work has been done to characterize the electronic, optical and physical properties of molybdenum disulfide.122–145 It is known to be a polytypic material,125,127,130 and along with a myriad of higher order sulfides,125 three main structural polytypes of the material have been identified: 2H–MoS2, 3R–MoS2, and 1T–MoS2. Fig. 1 shows idealized molecular models of these structures, as well as their lithiated counterparts. Fig. 1a and b show the neat and the lithiated 2H–MoS2 structures, with a 5% lattice expansion in the c-direction and a-direction due to lithium intercalation. Fig. 1c shows the lithiated 1T–MoS2, with the lithium ions occupying octahedral interstices. Fig. 1d shows 3R–MoS2 while Fig. 1e shows Li2S, which will be pertinent when discussing the lithiation-cycled structures. Of these, the 2H and 3R polytypes have been found to be naturally occurring (the 2H is found in much higher quantities) and thus their structures are very well characterized.124 The 1T polytype is a synthetic material, and as a result, there is some disagreement in open literature regarding its structure. The 2H and 3R polytypes exhibit stacking sequences of ABA, and ABC respectively, with the molybdenum cations having trigonal prismatic coordination.123,142Fig. 1a shows the stable 2H structure (space group P63/mmc), with lattice parameters of a = 3.16 Å, c = 12.29 Å and Wyckoff positions of molybdenum and sulfur atoms being: 2 Mo at ±(1/3,2/3,1/4) and 4 S at ±(1/3,2/3,z and 1/3,2/3,1/2 − z) with z = 0.621.122,130Fig. 1d shows the 3R structure which is of the R3m space group, with lattice parameters of a = 3.16 Å and c = 18.37 Å.131 Here, Wyckoff positions of molybdenum and sulfur were reported as Mo at (2/3,1/3,0), and S at (0,0, ±z) with z = (2/3 × 0.127).131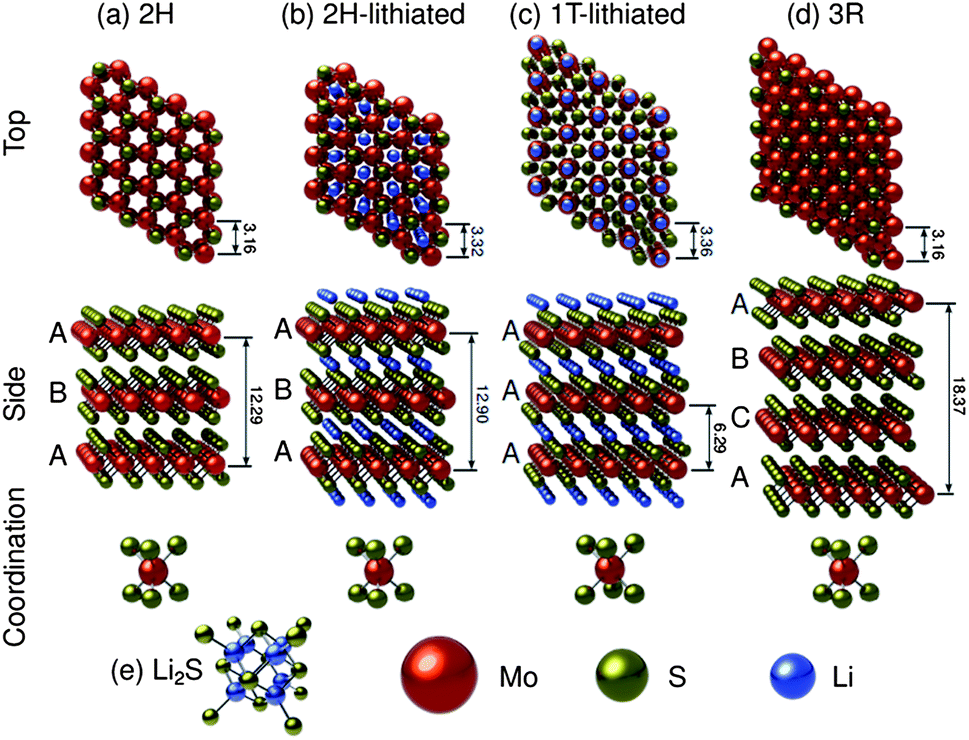 | ||
| Fig. 1 Molecular models of (a) 2H–MoS2. (b) Lithiated 2H–MoS2 showing a 5% lattice expansion in the c-direction and a-direction due to intercalation. (c) Lithiated 1T–MoS2 showing lithium ions occupying octahedral interstices. (d) 3R–MoS2 (e) Li2S (domains of this phase would be interspersed with molybdenum nanoparticles). Dimensions are shown in Angstroms. | ||
While theory predicts the trigonal (1T) polytype of MoS2, its unstable nature has made it difficult to characterize.126,127,130 In literature pertaining to lithium ion batteries, the 1T polytype is only observed after lithiation. We employed the crystallographic data from the study by Py and Haering to construct Fig. 1c.146 The lattice parameters for the 1T polytype are reported as a = 3.36 Å and c = 6.29 Å.146 The model in Fig. 1c was constructed using the space group P1, a = b = 3.36 Å, c = 6.29 Å, α = β = 90°, γ = 120°, with Wyckoff positions of Li at (0,0,1/2), Mo at (0,0,0) and S at (1/3,2/3,3/4) and (2/3,1/3,1/4).
As a layered transition metal dichalcogenide the electrical, optical, and physical properties of MoS2 are extremely anisotropic.123 Electrical and thermal conductivities are orders of magnitude smaller in the direction perpendicular to the basal plane, and thermal expansion is an order of magnitude greater.123 The layered hexagonal crystal structure is formed by strong Mo–S covalent bonds in the layers, and weak van der Waals forces between S–Mo–S layers.122,129 The van der Waals gap has been measured at approximately 3.49 Å via XRD.123,130,143 Within the S–Mo–S layers, the intermediate difference in electronegativities between sulfur and molybdenum lead to covalent bonds that are partially polarized. The molybdenum cations give up (primarily d-band) valence electrons to the sulfur anions and are left in an oxidation state of (4+) while the oxidation state of the sulfur anions becomes (2−).123 However, within the layer, each molybdenum atom is coordinated with six sulfur atoms while each sulfur atom becomes coordinated with three molybdenum atoms to give the hexagonal unit cell of 2H–MoS2, as shown in Fig. 1a. Looking in the direction perpendicular to the basal plane, the molybdenum and sulfur atoms are arranged in hexagonal sheets.123
The trigonal prismatic coordination of the molybdenum atoms gives rise to six equivalent cylindrical bond functions. These are a combination of the 4d, 5s, and 5p orbitals.129,133,134,143 This type of orbital combination has been described as d4sp hybridization by the work of Pauling147 and Hultgren.148 On the molybdenum atom, four valence electrons primarily from the 4d orbital are responsible for the bonding to the sulfur atoms, and the remaining two valence electrons of molybdenum reside in non-bonding orbitals. Each sulfur atom achieves coordination to three molybdenum atoms via hybridization of 3p and 3d orbitals. The van der Waals bonding between S–Mo–S sandwiches is a result of the interaction between saturated sulfur 3s subshells, which extend perpendicular to the basal plane.123,129 The weak inter-layer van der Waals bonding allows for expansion of the bulk structure in the c-direction upon intercalation.
6 Lithiation mechanism of MoS2
6.1 Lithiation of MoS2 from 3.0 to 1.1 V
Intercalation of lithium into MoS2 is known to occur within the voltage range of 3–0 V with a significant change in lithiation mechanism occurring below approximately 1.1 V versus Li/Li+. In the range of 3 through 1.1 V, lithium ion insertion is fully reversible which is shown as reaction (1) and idealized in Fig. 1b. This sequence, which is well agreed upon in literature, is commonly observed up to the voltage plateau occurring at approximately 1.1 V during initial discharge. Here, x is in the range of 0 ≤ x ≤ 1.| MoS2 + xLi+ + xe− → LixMoS2 (∼1.1 V vs. Li/Li+) | (1) |
The theoretical specific charge capacity of this reaction is 167 mA h g−1, corresponding to the intercalation of one lithium ion per molybdenum atom.146 At voltages below this plateau there appears to be one or several disproportionation reactions as well as the presence of intermediate metastable sulfide species. These reactions have been suspected since early work on Li–MoS2 was conducted,36 and were partially clarified in the late 1980's when Selwyn et al.149 published seminal work on the decomposition of molybdenum and tungsten dichalcogenides during lithiation. The exact potential onset as well as the nature of these reactions have only recently been elucidated, and are still not fully understood. These will be discussed in the next section of the review.
Since then, lithium intercalation into molybdenum disulfide has been studied in detail by various groups.53,142,150–154 More recent work has also been instrumental in the understanding of the complex mechanisms involved in lithium intercalation into molybdenum disulfide host lattices.155–159 Various methods were employed to study the insertion of lithium into molybdenum disulfide. These include physical vapor deposition (PVD) of lithium onto cleaved MoS2 in high vacuum,150 liquid phase intercalation by immersion of MoS2 in n-butyl lithium (C4H9Li),40,42,156,157 and intercalation via electrochemical methods.36–43,55,151,158 Intercalated samples were characterized via X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), nuclear magnetic resonance (NMR), transmission electron microscopy (TEM), and scanning electron microscopy (SEM).
Below approximately x = 1, intercalation of lithium into molybdenum disulfide is commonly described as an ion/electron transfer topotactic reaction producing a metallic paramagnetic product.36,38,155 For lithium atoms, the occupied valence electron states have a higher energy than the unoccupied conduction band states of the molybdenum cations in MoS2 (primarily the 4d bands), and so electron transfer results upon intercalation.155 The weak van der Waals forces that hold the S–Mo–S layers together in 2H–MoS2 allow for the insertion of lithium between them. As intercalation proceeds, lithium diffuses between the MoS2 planes and occupies vacant octahedral interstices in the hexagonal crystal lattice as shown in Fig. 1b. It has been suggested that the diffusion of lithium ions involves tetragonal interstices in the van der Waals gap, alluding to the fact that these ions are fast diffusers.36,39,43,153,160 The occupation of tetragonal interstices is however, less likely, since the energy to occupy a tetrahedral site is larger. Alkali ion conductivity in transition metal sulfides is due to a number of factors.43 First, there is a high concentration of potential charge carriers. Second, there is a high concentration of vacancies and interstitial sites, and third, the activation energy for ionic hopping to adjacent sites is relatively low (on the order of the formation enthalpy for the ternary compound LixMX2).43 Diffusion of lithium ions takes place primarily in two-dimensional intercalation planes in the van der Waals gap.41 For Li+ ions in MoS2 this occurs primarily between octahedral interstices parallel to the basal plane.161
Below approximately x = 0.1, intercalation proceeds into the 2H–MoS2 lattice with little disruption. The host lattice supplies intercalation sites and redox centers, but otherwise assumes a passive role in the reaction. As the lithium concentration increases in the 2H–MoS2 lattice, the addition of 1 or more electrons to the host phase, creating [MoS2]−, leads to the formation of a distorted octahedral environment for the metal cations, which experience substantial alterations in electronic band structure and electrochemical potentials.38–40,142 Researchers have commonly observed a lithium superlattice forming in the van der Waals gap as the lithium concentration rises in the range of approximately 0.1 ≤ x ≤ 1.157,160–164 This superlattice, described as 2ao × 2ao, is an ordering of lithium ions in the van der Waals gap, which occupy octahedral and then tetrahedral interstices above the saturation limit. This effects a phase change in which the coordination of the molybdenum atoms shifts from trigonal prismatic in the 2H phase, to octahedral in the 1T phase. This phase change is associated with the voltage plateau that is commonly observed around 1.1 V vs. Li/Li+ in galvanostatic cycling. The mechanism of this phase change, as explained by the work of Py and Haering,146 is described as a glide process between the molybdenum and sulfur planes. A similar process has been observed in the lithiation of graphitic carbons.165 Mulhern166 has gone on to demonstrate that the resulting intercalated phase of MoS2 is highly distorted, and undergoes appreciable lattice expansion. Intercalation-induced lattice strain in the basal plane causes the formation of dislocations, which fragment the lattice and may create diffusion pathways for lithium ions, allowing them to penetrate further into the host material.160 Furthermore, this fragmentation may lead in part to the disproportionation reaction associated with the formation of lithium sulfide (Li2S) and molybdenum metal particles that will be outlined in the next section.
An examination of 2H–MoS2 octahedral site radius and lithium ionic radius indicates that the two are approximately 0.7 Å.167 Here we consider that the van der Waals gap in 2H–MoS2 is approximately 3.49 Å and the sulfur atom covalent radius is 1.04 Å.167 Therefore, it can be inferred that lithium intercalation should proceed into molybdenum disulfide without any appreciable change in host lattice parameters. However experimentally we know this is not the case. XRD studies of intercalated MoS2 indicate that there is an approximate 4–6% lengthening of the c-axis and a-axis in the hexagonal unit cell upon intercalation with lithium ions as shown in Fig. 1b and c.36,38,146,160 While it is true that a portion of this expansion in the c-direction is attributed to the co-intercalation of solvent molecules (most of the intercalation studies are done using n-butyl lithium solutions in hexane), there is still appreciable expansion that results from lithium ion insertion.
Nuclear magnetic resonance studies have shown that lithium atoms can vary in size based on their ionic character (having a fully ionized radius of approximately 0.7 Å and a neutral radius of approximately 1.4 Å).40 It is thought that the ionicity of the lithium decreases, as its concentration in the host lattice rises.38,40 Therefore, the ionicity will have an effect on the lattice strain being exerted by the lithium ions. The first lithium atoms to intercalate each donate an electron to the empty 4d band of the molybdenum cations and are thus stored in a completely ionized state. However, it has been shown that as the lithium ion concentration increases, their ionicity decreases, and the ions themselves are thought to undergo a slight increase in ionic radius. This brings about an expansion of the lattice in the c-direction.36–39,146 In MoS2, lattice expansion in the a-direction is partially attributed to alterations in the electronic band structure of the host as intercalation continues.150 It is known that the transition metal dichalcogenides have strong overlap–covalency interactions between the metal d-bands, and the chalcogen s–p bands.143 Therefore, as intercalation continues, the sulfur atoms in MoS2 experience a slight increase in atomic radius as the electron density in the host lattice rises.40,143 These affects, along with solvent co-intercalation, are the likely source of the observed increase in the lattice parameters of MoS2 during intercalation (Fig. 1b).
6.2 Lithiation of MoS2 from 1.1 to 0 V
More recently, an increasing number of authors are validating the existence of one, or a series of decomposition reactions when MoS2 cells are discharged below 1.1 V vs. Li/Li+.46,168–182 Furthermore, since these decomposition reactions all yield a lithium sulfide product, an examination of some of the more prevalent lithium sulfur (Li–S) battery literature provides us with some clarification of the reaction mechanisms. For this system the formation of polysulfides and the sulfide shuttling effect are well characterized.183–193 For the more “bulk-like” structures of MoS2, significant voltage plateaus are observed upon the first discharge at approximately 1.1 and 0.6 V vs. Li/Li+, although the exact values vary by study.46,168–182For the case of truly nanoscale structures the five issues that arise when interpreting or even comparing the charging profiles are: first, the morphology of the nanostructured MoS2 electrodes deviates significantly from bulk, for example possessing much larger interlayer spacing and surface area to volume ratio, and/or a much higher defect density.168,180,181,194–198 For the case of interlayer spacing, it has been shown that MoS2 nanostructures with a wider spacing resulted in enhancements in electrochemical performance in terms of the initial lithiation kinetics and the charge storage capacity.168,177,180–182 Here, the effect is attributed to the increased volume associated with the layer expansion leading to faster ionic diffusion and better material utilization during the initial lithiation. The effect of enhanced lithium storage in a wider van der Waals gap has also been conclusively demonstrated with graphene.82,84,101,199 The increased surface area effect is expected to remain influential during the life of the material. However, effects due to the non-equilibrium spacing of the basal planes in MoS2 or high defect content will become unimportant after the structure irreversibly decomposes to molybdenum and Li2S.
Interestingly, an examination of literature pertaining to MoS2 nanostructures as hydrodesulfurization (HDS) catalysts leads to some useful information regarding point defects in the material.200–204 The structure of HDS catalysts must be well characterized, as it is usually defect sites on the crystal that lead to the bonding of the sulfur functional group (or sulfur-containing groups) in organic compounds. Using techniques such as scanning tunneling microscopy (STM), the morphology of these MoS2 nanostructures (such as nanosheets and fullerenes) have been meticulously characterized by these researchers. In the nanostructures, they see a much higher defect density in the form of vacancies (mainly sulfur edge vacancies) that lead to sub-coordinated molybdenum centers, and have concluded that they exhibit increased reactivity due to the presence of dangling bonds. It seems plausible that these same sites could act as adsorption sites for lithium ions, and may help to explain the elevated lithium storage capacities that are so often observed in the MoS2 nanostructures during the first cycle.157 Also, the presence or creation of point defects along the sulfur basal plane may serve as nucleation points for the formation of the lithium superlattice that has been observed to develop in the van der Waals gap.157,160,162,164 Researchers are therefore encouraged to attempt atomic force microscopy and STM studies on lithiated MoS2 samples to observe lithiation patterns, similar to what was done by Kalinin et al.205
Second, the structures often contain significant amounts of carbon-based phases, which are electrochemically active towards lithium.170,172,174,181,182 For example Archer et al.172 describe a system with 22 wt% carbon that had the best electrochemical performance. In many studies, the authors employ carbon nanostructures that possess charge storage capacities well in excess of graphite's 372 mA h g−1.77,78,82–85 Materials like multilayer “graphene”, highly graphitic nanoparticles, or carbon nanotubes bind lithium via adsorption, pore filling, and intercalation, routinely yielding capacities as high as 650 mA h g−1. Were the carbons to also contain substantial amounts of nitrogen heteroatoms, reversible capacities as high as 1780 mA h g−1 are possible.88,206 In the authors' opinion the coaptation of an active material with carbons goes a long way towards accounting for the tremendously enhanced capacity of the nanocomposites, since in general this contribution is neglected in the calculations.
Third, in nanoscale materials, the formation of an SEI layer (commonly described as a gel-like polymeric layer) can also have a significant contribution to the overall voltage profile.170 Such capacity contribution is of course detrimental, resulting in poor coulombic efficiency of the electrodes. Fourth, it is likely that similar to the lithium-sulfur battery, full charging–discharging for the Li–MoS2 system involves intermediate “molecular” polysulfides. For the Li–S system these reactions are well characterized,183–193 however their exact nature remains to be elucidated in the Li–MoS2 system.177–182 Fifth, it is therefore plausible that the surfaces of molybdenum nanoparticles present after full discharge have dangling bonds that will attract and immobilize polysulfides, and could serve as adsorption sites for lithium ions in subsequent cycles. As a corollary, sub-coordinated molybdenum centers have been shown to have a high affinity for sulfur-containing molecules.200 However molybdenum metal nanoparticles are generally X-ray amorphous and are thus difficult to track during the lithiation studies.181
The lower voltage plateau evident upon first discharge, occurring at approximately 0.6 V vs. Li/Li+, has been attributed to either the reversible conversion reaction of MoS2 to Li2S and metallic molybdenum through reactions (1) and then (2),46,168–176 or the irreversible decomposition with the same redox chemistry, followed by cycling between Li2S (Fig. 1e) and elemental sulfur (reaction (3)).177–182 The theoretical specific charge capacity of reaction (1) is 167 mA h g−1, while the theoretical capacity of MoS2 lithiating by reactions (1) and then by (2) (i.e. full discharge) is 669 mA h g−1.
| LixMoS2 + (4 − x)Li+ + (4 − x)e− → Mo + 2Li2S (∼0.6 V vs. Li/Li+) | (2) |
The reaction of elemental sulfur and lithium may be described by equation (3), which yields a theoretical capacity of 1675 mA h g−1 if the weight of the molybdenum is not taken into account.
| S + 2Li+ + 2e− → Li2S (∼2.2 V vs. Li/Li+) | (3) |
Though the majority of open literature favors the reversible conversion reaction sequence, recent detailed XRD studies indicated that upon delithiation, MoS2 was no longer detectable.177–181 This may be due to an amorphization of the electrode materials. However since both the 1.1 and the 0.6 V plateaus are observed only during the first lithiation, we believe that the irreversible decomposition of MoS2 is more likely. At room temperature solid-state sulfidation reactions are quite sluggish, so it is unlikely that MoS2 would re-form especially at higher charging rates. This conclusion is also based on a recent TEM study of post-cycled MoS2 electrodes (Fig. S8 in their ESI†).182 The authors conclusively detected metallic molybdenum in the delithiated state. One difficulty in characterizing the cycle 1 discharge plateaus for the MoS2 half-cell (vs. Li/Li+) is that the plateau at 0.6 V is difficult to distinguish from the one attributed to SEI formation. Unfortunately, the formation of an SEI layer occurs in most battery anodes during the first discharge cycle around 0.6 V. In fact it may be very difficult to separate the two processes for high surface area electrodes where substantial capacity is lost due to SEI formation, while the initial lithiation reaction irreversibly alters the lithium active phases. However extensive microstructural evidence, discussed in this manuscript, supports the formation of metallic molybdenum during discharge cycle 1. Moreover low surface area “bulk” MoS2, where the total irreversible capacity due to SEI should be relatively low, clearly demonstrates the 0.6 V plateau (as seen by the CV peak at ∼0.6 V in Fig. 2a).170
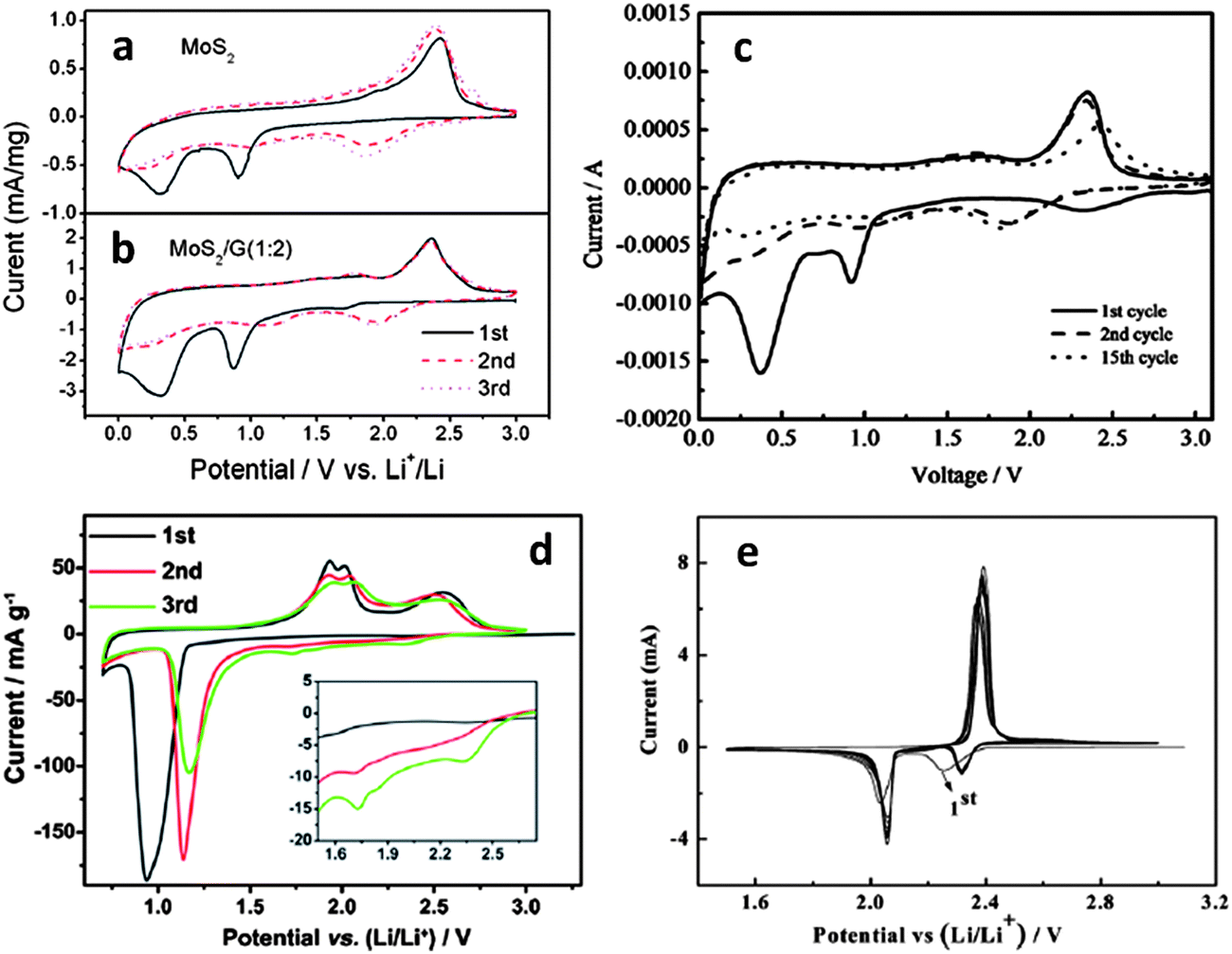 | ||
| Fig. 2 Cyclic voltammograms for (a) MoS2 powder.170 (b) MoS2–graphene nanocomposite (scan rate 0.5 mV s−1).170 (Copyright 2011 American Chemical Society) (c) MoS2–amorphous carbon nanocomposite (scan rate 0.2 mV s−1).172 (d) Commercial MoS2 powder in a smaller voltage window (scan rate 0.05 mV s−1).179 (Copyright 2012 Elsevier Ltd.). (e) Sulfur (scan rate 0.05 V).207 Adapted from ref. 170 (DOI: 10.1021/nn200659w), ref. 172 (DOI: 10.1039/c2jm32468g), ref. 179 (DOI: 10.1016/j.electacta.2012.07.020), and ref. 207 (DOI: 10.1039/c2jm15041g) with permission. | ||
The irreversibility of the MoS2 decomposition reaction upon initial discharge is further supported by the trends observed in cyclic voltammetry (CV). Fig. 2a (ref. 170) and 2d (ref. 179) highlight CV curves for commercial MoS2 powders in different voltage ranges (scan rates 0.5 and 0.05 mV s−1 respectively). Fig. 2b (ref. 170) shows a typical CV curve for an MoS2/graphene nanocomposite (scan rate 0.5 mV s−1). Fig. 2c (ref. 172) is a CV curve for an amorphous carbon–MoS2 nanocomposite (scan rate 0.2 mV s−1). Fig. 2e (ref. 207) is a CV curve for a typical lithium–sulfur redox couple, shown for comparison (scan rate 0.05 mV s−1). In Fig. 2e, the two cathodic peaks at ∼2.3 and ∼2.1 V are attributed to the stepwise reduction of sulfur to Li2S. The first step (∼2.3 V) involves the reduction of sulfur to intermediate lithium polysulfides (Li2Sn, 2 < n < 8) and the second step (∼2.1 V) is attributed to the reduction of higher order polysulfides to Li2S.109,118 The dominant oxidation peak at 2.4 V is recognized as the conversion of all lithium polysulfides to S82− accomplished by facile charge transfer kinetics.109 This CV plot also demonstrates the excellent reversibility of the lithium–sulfur redox couple.
Fig. 2a–c show similar trends in all cathodic and anodic sweeps. In the first cathodic sweep, peaks at approximately 1.0 V are observed on all plots and attributed to the formation of LixMoS2 and the resulting 2H to 1T phase transition.170,177 This peak is also observed in 2d, although here, the authors only discharge to 0.8 V in an attempt to investigate the reversibility of reaction (1) in this voltage range. The large cathodic peak at approximately 0.4 V (Fig. 2a–c) is attributed to the conversion reaction of MoS2 to Li2S and molybdenum (reaction (2)). The irreversibility of this reaction is supported by the disappearance of these peaks in subsequent reduction cycles. Instead, a dominant cathodic peak at approximately 2.0 V (consistent with 2e) is observed in Fig. 2a–c, while the peaks at 1.0 and 0.4 V previously discussed are greatly diminished in subsequent cycles. The dominant cathodic peak forming at ∼2.0 V is well known in lithium–sulfur battery systems and is attributed to the formation of Li2S.109,186,188,208 Upon recharging, Fig. 2a–c show two anodic peaks (one shallow peak at ∼1.7 V and a large peak at ∼2.4 V). The first shallow anodic peak is likely due to the delithiation of residual LixMoS2 which has not undergone conversion. The dominant anodic peak at ∼2.4 V is due to the conversion of Li2S to S82− consistent with Fig. 2d and e.110,187,208–210
The material in Fig. 2d (ref. 179) was discharged too deeply to demonstrate full reversibility of reaction (1). However by initiating it in a stepwise manner the MoS2 conversion reaction was successfully observed. This is similar to the results obtained by Py and Haering,146 where the formation of LixMoS2 was conclusively identified with the long voltage plateau at 1.1 V. In Fig. 2d, the first discharge to 0.8 V (ref. 179) allowed the conversion of MoS2 to Li2S and molybdenum metal, but not completely. The first anodic sweep reveals the delithiation of LixMoS2 (the doublet centered at 2.0 V) as well as a large broad peak at ∼2.5 V, which is attributed to the formation of sulfur (similar to Fig. 2e). With subsequent cycling the anodic doublet centered around 2.0 V and the dominant cathodic peak at around 1.0 V both get weaker. This indicates the consumption of the active materials initially present, i.e. LixMoS2 and MoS2. Such cycling induced degradation has also been observed in other studies172,178 and attests to the instability of the LixMoS2 compound, and its tendency to decompose at higher values of x. By the third cycle, two small cathodic peaks at 1.75 and 2.34 V appear (shown inset in Fig. 2d), which are likely due to the reduction of higher order lithium polysulfides.109 Based upon the trends observed in literature, the pertinent redox reaction after first discharge involves lithium and sulfur as the electro-active species (reaction (3)).
Fig. 3 presents recent XRD data that further supports the argument regarding a lithiation sequence that involves the irreversible formation of molybdenum.172,177Fig. 3a shows an XRD scan of an MoS2 electrode in the as-received state (bottom) and after it was discharged to 0.01 V (top).172 The as-received material is clearly 2H–MoS2, while the discharged material is Li2S and molybdenum metal. Fig. 3b shows an XRD pattern of a commercial MoS2 powder after the first discharge to 0.01 V.177 Again, there is strong evidence that Li2S and molybdenum are the dominant phases. Fig. 3c shows the Li2S + molybdenum composite (material analyzed in Fig. 3b) after it was recharged to 3.0 V. While there is substantial peak broadening due to partial amorphization and/or nanocrystallization, elemental sulfur and molybdenum metal are definitively present in the charged state. This also indicates that the material was not fully lithiated, consistent with it being a micro-scale powder rather than a nanocomposite. In situ XRD studies of MoS2 have been performed, though no study has done this across the entire voltage range of 0.01 to 3 V.146,179 The elucidation of the microstructural evolution in MoS2 based electrodes during cycling would benefit greatly from an in situ XRD study throughout the entire voltage range. XRD and FTIR have been used by others to track the lithiation of MoS2, with the results being in agreement with the trends discussed here.179,181
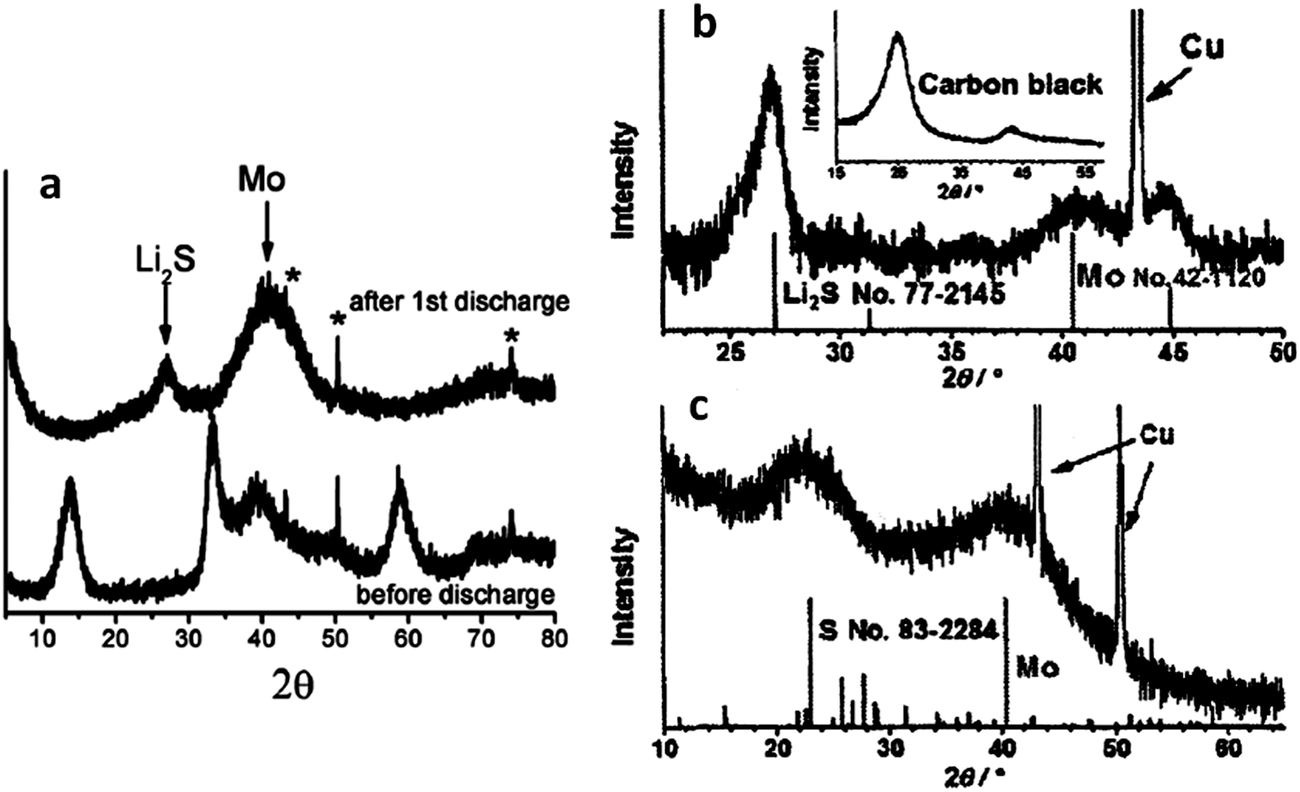 | ||
| Fig. 3 XRD scans of MoS2 electrode at various states of charge. (a) (Bottom) as received, and (top) after discharge to 0.01 V. Peaks marked by * are from the copper current collector.172 (b) After discharge to 0.01 V.177 (c) After recharge to 3.0 V.177 (Copyright 2012 Wiley-VHC Verlag GmbH &Co, KGaA, Weinheim) Adapted from ref. 172 (DOI: 10.1039/c2jm32468g), and ref. 177 (DOI: 10.1002/asia.200100796) with permission. | ||
It appears that after the first discharge, the molybdenum nanoparticles may have a multifunctional beneficial role: first, the particles serve to enhance the electrical conductivity of the Li2S matrix, which partially alleviates the poor electrical conductivity concern associated with both the sulfur and Li2S phases. Second, the nanoparticles may serve as pinning sites for soluble polysulfides, preventing their dissolution, and thus mitigating the shuttling effect that causes electrochemical degradation of lithium–sulfur batteries. While the first effect is quite reasonable and should be expected were the MoS2 to irreversibly decompose, the second effect is hypothetical and requires substantial experimental evidence before being considered a real benefit. Authors have argued that it may be the carbon that in fact pins the soluble polysulfide anions.186,187,191
The theoretical specific capacity of reaction (2) is 669 mA h g−1 while that of reaction (3) is 1675 mA h g−1. Therefore reaction (2) does not fully explain the enhanced specific capacities well in excess of 700 mA h g−1 that are commonly observed in the Li–MoS2 system.46,169–182 While reaction (3) is quite likely, it can only follow reaction (2), which means the mass of molybdenum must also be taken into account. Therefore in the authors' opinion, reactions (2) and (3) do not fully capture the complexity of the charging/discharging process in the MoS2-based system. We believe that there are three additional, and by no means mutually exclusive, contributions to the net charge storage: first, nanostructured molybdenum particles may also participate to some extent in the lithiation reaction, serving as physical adsorption sites for the Li+ ions. Bulk molybdenum is inactive towards lithiation, so any binding would have to be at or near the surface.211 Reports in literature are consistent in showing a much higher capacity for materials that are nanoscale rather than their bulk counterparts.170,174,180,181,185,212–214 Researchers have characterized these effects well for a variety of transition metal nanoparticles formed from oxide conversion electrodes.215–218 These authors comment on the various charge storage mechanisms in conversion electrodes and allude to enhancements from capacitive effects brought about by the high surface area metallic nanoparticles. Authors also point out that there may be a contribution to the reversible capacity from the polymeric SEI layer that forms around the metallic nanoparticles.215 However in our opinion such reactions would be either fully irreversible or very poorly reversible, and would only adversely affect the coulombic efficiency without boosting the reversible capacity. Another possibility is that more than two lithium ions react per sulfur atom in reaction (3). We believe that this is unlikely since no analogue has been reported for the well-characterized Li–S system.
The present authors believe that much of the capacity enhancement beyond 669 mA h g−1 is largely due to the presence of a nanostructured carbon phase whose contribution to the total electrode capacity is either underappreciated or perhaps not accounted for at all. As was discussed previously, many of the carbon allotropes interspersed with MoS2 possess quite a high lithium storage capability that well-exceeds that of carbon black and even that of graphite. There is also a likely synergy between the two nanodispersed phases that cannot be captured by a standard rule of mixtures calculation even when the capacity of each phase is obtained separately. The enhancement of lithium capacity by carbon phases is routinely demonstrated in MoS2 and Li–S literature, though the exact mechanism remains unclear.46,169,170,172,180–182,185,187,193
7 MoS2 nanocomposites in LIBs
With the development of nanostructured materials, there is a resurgence of synthesis research directed at creating MoS2-based nanocomposite structures for lithium ion battery applications. The effort is primarily directed at utilizing MoS2 as an anode material to be used against a pre-lithiated cathode. A particle size refinement of MoS2 down to the nanoscale greatly shortens the lithium ion diffusion distances, providing a substantial boost in the rate-dependent capacity retention as compared to the more “micro” MoS2 counterparts.44,46,159,172,174,180,182 In addition, it is consistently demonstrated that it is the hybrid MoS2–carbon systems, in particular the ones with nanoscale structure, that offer the optimum combination of energy density, cycling stability, and high rate capability.190,219–229There are several microstructural scenarios during the conversion reaction, where nanostructured carbon would enhance electrode performance. These include one or a combination of (a) carbon acting as a binder between the S/Li2S and the molybdenum nanoparticles; (b) carbon encapsulating both phases providing an electrically conductive path down the current collector; (c) carbon acting as a “skeleton” which provides both an electrically conductive pathway down to the current collector and prevents material agglomeration during cycling. The possibility of (a) vs. (b) vs. (c) would also depend on the type of carbon added. It is intuitive that crystalline/particulate phases like carbon nanotubes or graphene nanoflakes would be more effective in providing a skeleton, while materials like amorphous carbon would be more effective as coatings and/or binder. At this point in the case of MoS2 there is not enough microstructural evidence to conclusively identify such enhancement mechanisms. There exists a wide range of techniques by which to synthesize electrode-grade molybdenum disulfide of various morphologies. Among these, hydrothermal, assisted hydrothermal, solvothermal, and template assisted techniques are the most successfully employed and will be presently discussed.
7.1 MoS2 nanostructures by hydrothermal techniques
A significant portion of the recent published work uses carbon-templated hydrothermal synthesis to create nanocomposite structures of MoS2 dispersed in a conductive matrix such as graphene, carbon nanotubes (CNTs) or amorphous carbon. The key advantage of hydrothermal methods is that they may be employed to create commercial or near commercial mass loadings (∼10 mg cm−2) on the electrodes.230 Electrically conducting multiwalled carbon nanotubes, possessing large open specific surface areas and excellent chemical and thermal stabilities, are perhaps the most widely employed nanometer-sized templates.190,219–224 Graphene is also becoming a popular choice of support, progressively gaining greater scientific attention as compared to CNTs.225–229 The rationale for this stems from a real cost and scalability advantage of wet methods used to synthesize graphene/graphene oxide versus chemical vapor deposition generally employed to fabricate CNTs.231–233It is well known that, similar to graphene, exfoliated MoS2 often exists in the form of nanosheets due to its layered structure. The presence of 2D graphene sheets in the hydrothermal process could further guide the formation of MoS2 sheets and generate a sheet-on-sheet structure. Despite common literature claims of such structures possessing a long-term benefit (i.e. over numerous charge/discharge cycles), there is little microstructural or electrochemical evidence that suggests they survive past the initial lithiation step. Nevertheless the increased interfacial contact between carbon and Mo/S would promote cycling stability by reducing the rates of material aggregation. Graphene, CNTs and related materials are known to template the growth of various sulfides and oxides from solution, resulting in orders of magnitude reduction in particle sizes as compared to the non-templated baselines.228,234 Since such nanocarbons are very effective in refining the as-synthesized microstructure and hence shortening the lithium diffusion distances, they substantially improve the rate dependent capacity retention. It has also been suggested that during cycling, the electrochemically active surface area of these electrodes can increase due to a gradual breakdown of the graphene and resulting introduction of defect sites.228 These sites serve to trap more lithium ions during intercalation and could explain the gradual increase in specific capacity that is often observed. A highly interspersed carbon phase would also substantially improve the electrical conductivity of the electrode down to the current collector, regardless of the lithium-active phases present.
By introducing graphene nanosheets into the hydrothermal synthesis process for MoS2, authors were able to create a true nanocomposite.46Fig. 4a and b highlight the resulting as-synthesized microstructure, which exhibits significantly improved electrochemical performance over the graphene-free baseline.46Fig. 4c and d show the cycling results, demonstrating a stable reversible capacity of approximately 1290 mA h g−1 and an excellent rate capability. The capacity retention of this material was 99.2% after 50 cycles (current density 100 mA g−1). This is among the best performance, in terms of capacity and cycling stability, reported in literature for an MoS2-based anode. The graphene additive has significantly decreased the size of the MoS2 nanosheets, which likely led to better material utilization during the conversion to Li2S and molybdenum. Authors elaborated upon the synergistic behavior of MoS2 and graphene, and provide a detailed investigation of the electronic and atomic structure of the nanocomposite.235 The work provides evidence that the creation of a graphene–MoS2 nanocomposite improves the overall electrical conductivity of the electrode. Furthermore, the authors discuss the weak van der Waals and electrostatic interactions of the two materials, which would allow for facile expansion at the graphene–MoS2 interface during initial lithiation.235 This may actually influence the subsequent cycling behavior in terms of allowing all the MoS2 to be converted. In contrast, studies repeatedly show that for micro-scale MoS2 much of the material does not react with lithium during the first cycle or afterwards (capacities well below 669 mA h g−1).
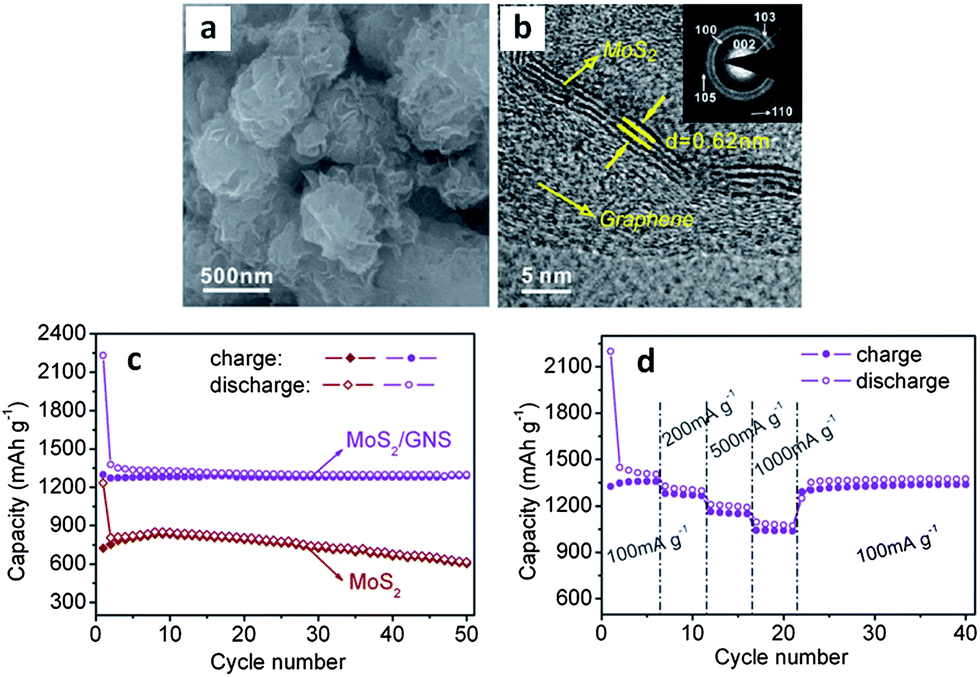 | ||
| Fig. 4 As-synthesized MoS2–graphene nanocomposite. (a and b) SEM and TEM micrographs respectively. (c) Cycling behavior of the nanocomposite (with graphene-free MoS2 as the baseline). (d) Cycling behaviour of MoS2–graphene nanocomposite at various current densities. Adapted from ref. 46 (DOI: 10.1039/c1cc10631g) with permission. | ||
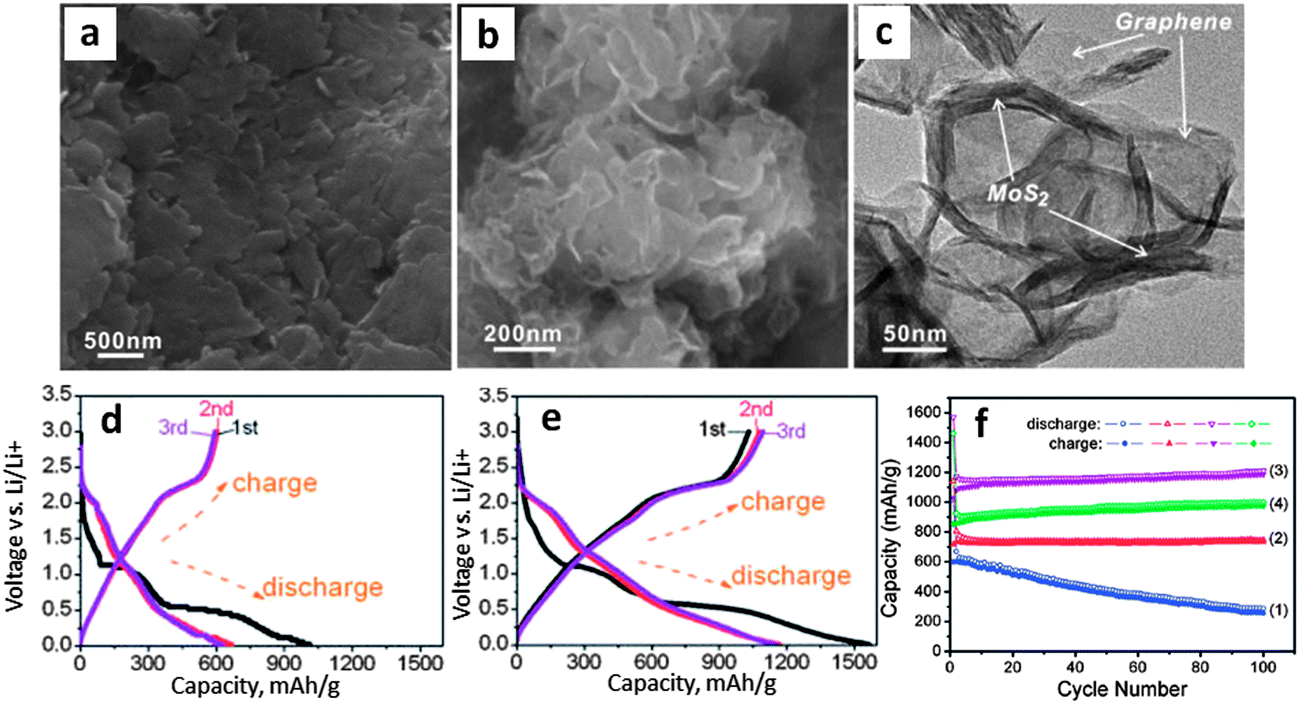
Amorphous carbons formed during the hydrothermal synthesis of MoS2 can also increase the electrode performance. Authors prepared amorphous carbon–MoS2 nanostructures via a hydrothermal/carbonization technique.172Fig. 5a and b display SEM and TEM micrographs of the as-prepared MS-22 nanostructures (MoS2 + 22 wt% carbon). Fig. 5c shows that the capacity retention of this composite had a very strong carbon loading dependence. At a carbon loading of 22 wt%, the stable capacity was approximately 875 mA h g−1 for over 100 cycles. The authors attributed this stability to the MoS2 being fully coated with carbon, which allowed for full material utilization during electrochemical cycling. Moreover, the carbon coating may mitigate the SEI layer formation, though more evidence is needed for this hypothesis. Fig. 5d shows capacity–voltage profiles for charging and discharging, which are typical of MoS2–carbon composites. Here, the first discharge exhibits plateaus at approximately 1.1 and 0.6 V, indicative of the 2H to 1T MoS2 (∼1.1 V) phase transformation, and subsequent conversion to Li2S and molybdenum metal (∼0.6 V). These disappear in subsequent cycles, indicating that this reaction is irreversible. XRD scans from this material (shown in Fig. 3a) suggested the presence of Li2S and molybdenum metal after first discharge. Moreover, the discharge (∼2.0 V) and charging (∼2.3 V) voltage plateaus for the 100th cycle are indicative of the lithium–sulfur redox couple.186,188,210
 | ||
| Fig. 5 Hydrothermally synthesized MoS2–amorphous carbon nanocomposite. (a and b) SEM and TEM micrographs of MS-22 (MoS2 + 22 wt% C). (c) Cycling stability of pure MoS2 and various MoS2–carbon composites, MS-X stands for MoS2 with X wt% C. (d) Voltage capacity profiles for MS-22 (current density 100 mA g−1). Adapted from ref. 172 (DOI: 10.1039/c2jm32468g) with permission. | ||
Researchers have employed polystyrene microspheres to tailor the dispersion and the microscopic assembly of MoS2 nanosheets (MoS2–NS).174 Through a post hydrothermal synthesis annealing treatment in an inert atmosphere, the template polystyrene microspheres were decomposed. The resultant ultrathin MoS2 nanosheet assemblies assumed microsphere morphologies with a wide spacing between the layers. This material had a BET surface area of 36 m2 g−1, with a primarily mesoporous structure due to the stacking of the individual nanosheets. The hierarchical structure of the MoS2–NS microspheres was quite advantageous for battery applications: the high surface area of the nanosheets increased the overall charge storage capacity, while the void space buffered the volumetric changes allowing for facile lithiation/delithiation. Fig. 6a and b show TEM micrographs of these resultant hierarchical structures.174Fig. 6c and d show the cycling performance of the baseline MoS2 flakes (I) and the MoS2–NS microspheres (II) at a current density of 100 mA g−1 (Fig. 6c) and at various current densities (Fig. 6d). The MoS2–NS microspheres consistently outperformed the baseline in terms of overall capacity, cycling capacity retention and rate capability, supporting the authors' argument regarding the essential role of polystyrene microsphere assisted synthesis.
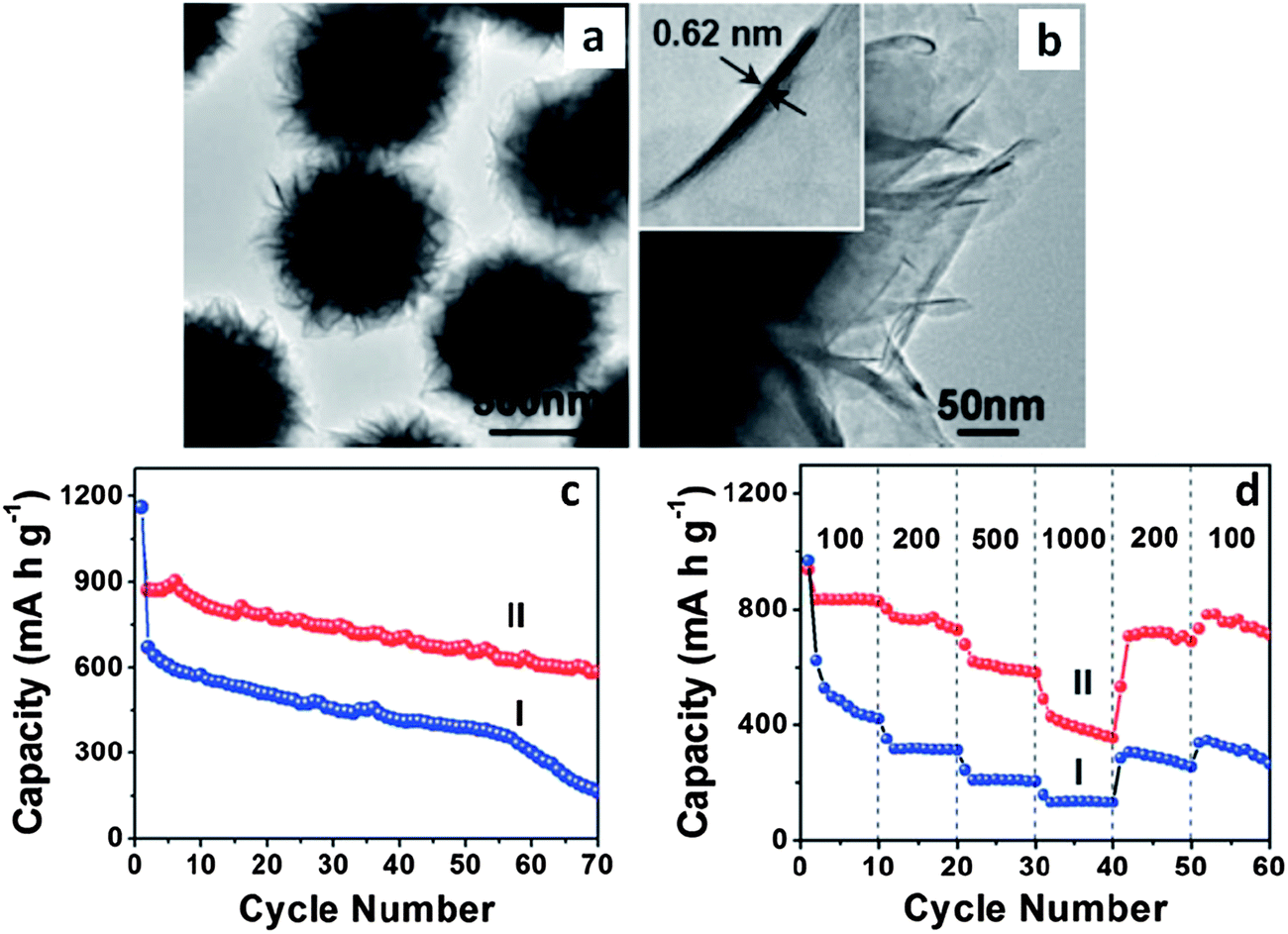 | ||
| Fig. 6 MoS2–nanosheet microspheres. (a) TEM micrograph of as-synthesized MoS2 nanosheet microspheres. (b) HRTEM image of several MoS2 nanosheets; the inset shows an HRTEM image of a single MoS2 nanosheet. (c) Cycling performance of MoS2 flakes (I) and MoS2–NS microspheres (II) at a current density of 100 mA g−1. (d) Cycling performance of MoS2 flakes (I) and MoS2–NS microspheres (II) at different current densities (mA g−1). Adapted from ref. 174 (DOI: 10.1039/c1nr11552a) with permission. | ||
This group also reported similar results using glucose as an additive in the presence of CNTs during hydrothermal synthesis.175 This was shown to significantly decrease the thickness of the MoS2 nanosheets. The glucose adsorbed on the surface of the CNT@MoS2 was further converted into a thin amorphous carbon layer during the calcination process, and hence acted as an additional conductive and perhaps protective coating. The BET surface area was reported at 30 m2 g−1. The glucose-assisted material consistently outperformed the glucose-free baseline (in terms of capacity retention and rate capability). The capacity of the glucose-assisted CNT@MoS2 was nearly 1000 mA h g−1, decreasing to approximately 800 mA h g−1 after 60 cycles.175
Further to their previous work,46 authors present a study on L-cysteine assisted hydrothermal synthesis of MoS2 graphene nanocomposites.170 While biomolecular-assisted synthesis methods have been employed to create other types of sulfide nanostructures, this was the first study of its kind for MoS2. The electrochemical results were very promising. The synthesized materials were true graphene–MoS2 nanocomposites and possessed a synergistic charge capacity of nearly 1200 mA h g−1 which was shown to be dependent on the graphene–MoS2 ratio (optimal was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The strong dependence of the electrode capacity on the amount of graphene added substantiates the argument that it is a major contributor to the net charge storage. The fact that there is an optimum ratio of graphene to MoS2 does not need to be rationalized in terms of any profound electronic effects or a fundamental modification of the MoS2 structure (it does not exist past cycle 1). Rather it can be explained by correlating this ratio to the best microstructural dispersion of the two phases, i.e. a mixture that is the most “nano”. Unfortunately there are no existing literature reports where authors have demonstrated the variations in the key microstructural parameters (i.e. MoS2 crystallite and particle size, total porosity and pore size distribution, electrical conductivity of the composite, degree of encapsulation by the carbon of the MoS2 particles, etc.) with the loading of a given carbon phase. However it is quite reasonable to expect a “volcano” type of electrochemical performance curve versus carbon mass loading, with the peak corresponding to the optimum overall capacity retention and rate capability (e.g. in Fig. 7f). At lower carbon loadings the dispersion would not be optimized due to an insufficient amount of the carbon phase, while at higher mass loading agglomeration would reduce the amount of electrochemically accessible material and drive up the electrode resistivity.
:1). The strong dependence of the electrode capacity on the amount of graphene added substantiates the argument that it is a major contributor to the net charge storage. The fact that there is an optimum ratio of graphene to MoS2 does not need to be rationalized in terms of any profound electronic effects or a fundamental modification of the MoS2 structure (it does not exist past cycle 1). Rather it can be explained by correlating this ratio to the best microstructural dispersion of the two phases, i.e. a mixture that is the most “nano”. Unfortunately there are no existing literature reports where authors have demonstrated the variations in the key microstructural parameters (i.e. MoS2 crystallite and particle size, total porosity and pore size distribution, electrical conductivity of the composite, degree of encapsulation by the carbon of the MoS2 particles, etc.) with the loading of a given carbon phase. However it is quite reasonable to expect a “volcano” type of electrochemical performance curve versus carbon mass loading, with the peak corresponding to the optimum overall capacity retention and rate capability (e.g. in Fig. 7f). At lower carbon loadings the dispersion would not be optimized due to an insufficient amount of the carbon phase, while at higher mass loading agglomeration would reduce the amount of electrochemically accessible material and drive up the electrode resistivity.
 | ||
| Fig. 7
L-cysteine assisted hydrothermal synthesis of graphene–MoS2 nanocomposites. (a) SEM micrograph of the MoS2 baseline. (b and c) SEM and TEM micrographs of the 2:1 by weight graphene–MoS2 nanocomposite. (d and e) Charge–discharge curves for the baseline MoS2 and for the 2:1 nanocomposite, respectively. (f) Cycling stability of the nanocomposites: (1) MoS2 (2) G/MoS2 (1:1) (3) G/MoS2 (2:1) (4) G/MoS2 (4:1). Adapted from ref. 170 (DOI: 10.1021/nn200659w) with permission from the American Chemical Society, Copyright 2011. | ||
Fig. 7 shows SEM and TEM micrographs of their baseline material (a) which was nearly monolithic, and their 2:1 graphene–MoS2 nanocomposite (b and c).170 Though the two materials appear to be well interspersed, further evidence in terms of analytical mapping, Z-contrast imaging, HRTEM, etc. would have been useful. Electrochemical tests (Fig. 7d–f) highlight the significant differences between the baseline MoS2 and the various nanocomposites that were analyzed. Fig. 7f shows that the total capacity and the cycling stability are much better for the 2:1 nanocomposite (marked 3) as compared to the graphene-free MoS2 baseline (marked 1). The 1:1 graphene/MoS2 (marked 2) and 4:1 graphene/MoS2 (marked 4) nanocomposites are both inferior to the 2:1, likely for the reasons previously discussed. There are also significant differences in the voltage–capacity profiles for the baseline (Fig. 7d) and the 2:1 nanocomposite (Fig. 7e). The previously described plateaus at ∼1.1 and 0.6 V vs. Li/Li+ during the first discharge of MoS2 were much less conspicuous for the nanocomposite (Fig. 7e). It is difficult to quantitatively compare the voltage–capacity profiles for the two materials during subsequent cycling, since neither possesses well-defined plateaus. However one can qualitatively state that the voltage profiles did vary with the graphene content, supporting the argument that it had a substantial contribution to the net capacity. Furthermore, the lack of discernible voltage plateaus in subsequent cycles indicates that both materials went through similar phase changes as a result of their initial lithiation. The stable cycling capacity of nearly 1200 mA h g−1 after 100 cycles is among the highest reported in literature for any MoS2-based electrodes. The 2:1 graphene–MoS2 nanocomposite not only displayed over twice the capacity of the MoS2 baseline but was also much more stable. This is shown in Fig. 7f.
It is also important to note that additives such as ionic liquids, glucose and biomolecular compounds show a strong impact on the morphologies of hydrothermally synthesized MoS2. Similar effects have been demonstrated in the hydrothermal synthesis of metal oxides,236–239 where the solution-phase interactions are generally better understood. Hence any comparisons with the carbon-free baselines are further obscured since not only is the resultant carbon content and dispersion different, but also the morphology of the MoS2 phase. Varying the relative amount of precursor would also have an effect on the microstructure of MoS2. For instance, in the previous study170 the microstructure of MoS2 would not only differ between the graphene-free MoS2 baseline and the graphene-MoS2 samples, but also between the 1:1, 2:1 and 4:1 specimens. These types of questions would be better resolved through the application of more robust microscopy analysis on the as-synthesized and post cycled specimens.
7.2 MoS2 prepared by solvothermal synthesis
Despite the success of the MoS2 electrode in a half-cell configuration, published work on a full cell is limited. Cho et al.48 are one of the only groups to successfully test and publish results on such a cell, using a lithium cobalt oxide cathode and an anode consisting of graphene-like MoS2 nanoplates. The nanoplates were synthesized via a liquid phase solvothermal reaction of molybdenum hexacarbonyl (Mo(CO)6) and sulfur in xylene. Fig. 8a and b display SEM and TEM images of the synthesized nanostructures, respectively. The SEM images indicate that the particulate size of the MoS2 is sub-100 nm scale, while the TEM micrographs indicate that the structure is highly disordered. Examining the published micrograph one observes a structure that is quite heterogeneous even on the sub-5 nm scale (dimension of the scale marker), with both crystalline and amorphous regions being present throughout. For the crystalline sections, a variety of contrast fringe spacings are observed. These may not actually be conclusively ascribed to any given set of interplanar spacings due to an unknown orientation of the polycrystalline specimen relative to the electron beam. Moreover some regions of the micrograph show contrast synonymous with Moiré patterns, which are caused by overlapping crystallites. It is not clear as to what kind of feature the marker showing 0.69 nm spacing is referring too, though we believe it to be a Moiré fringe.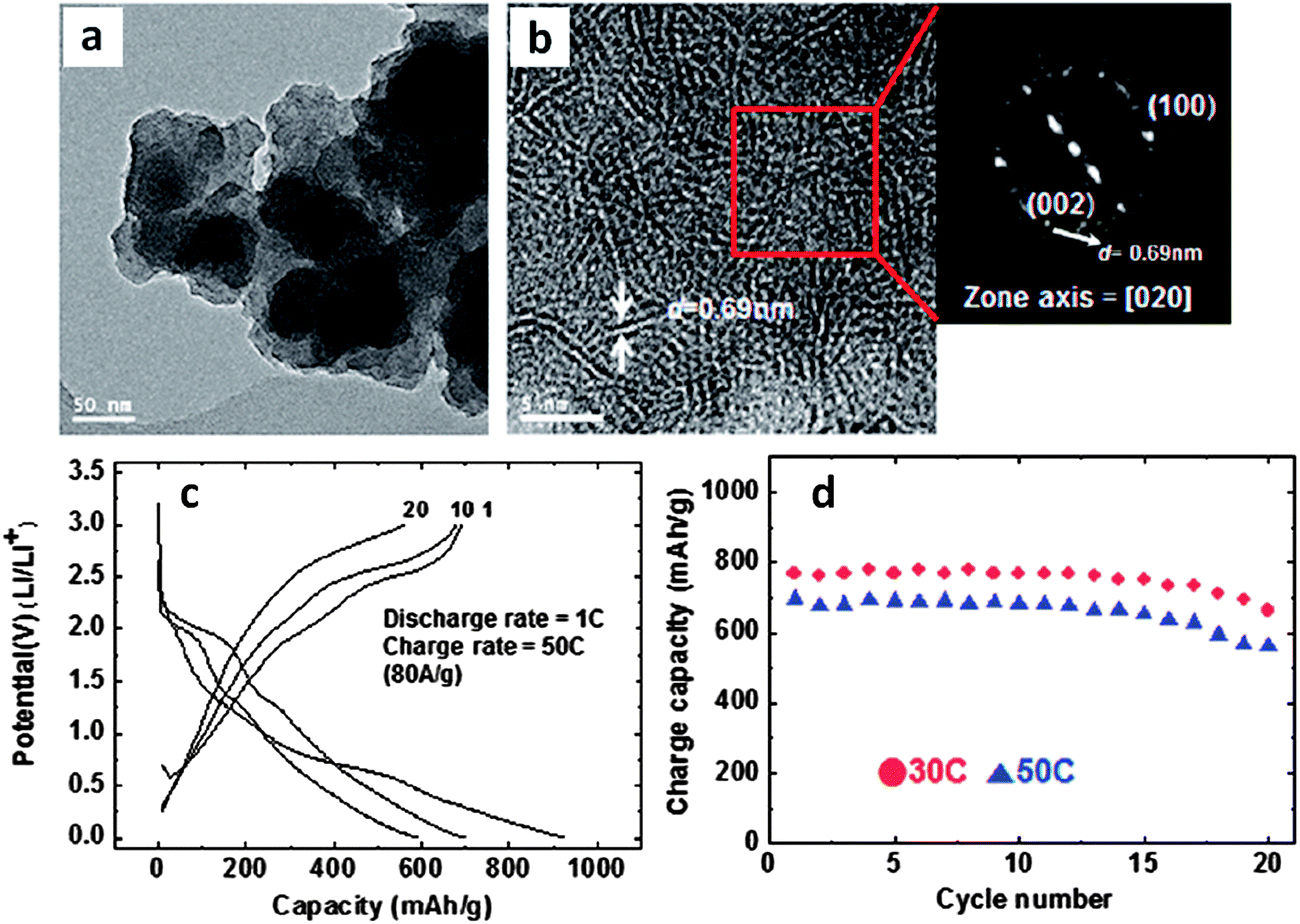 | ||
| Fig. 8 Disordered graphene-like MoS2 achieved via liquid phase solvothermal technique. (a and b) SEM and TEM (with FFT insert) images of the synthesized nanostructures. (c and d) Charging–discharging curves and cycling stability results for the half-cell. Adapted from ref. 48 (DOI: 10.1021/nl202675f) with permission from the American Chemical Society, Copyright 2011. | ||
Fig. 8c and d show the potential–capacity curves and cycling stability results for the half-cell, which are very impressive. The dominant discharge (∼2.0 V) and charge (∼2.3 V) plateaus are consistent with other work172 and are indicative of the lithium–sulfur redox couple. The authors correctly point out that larger interlayer spacing in their nanostructured MoS2 would alter the intercalation thermodynamics and kinetics. However, this effect will only be realized during the first lithiation and cannot contribute to the electrochemical performance in the subsequent cycles. The charge storage capacities are in excess of the theoretical value. Though the authors argued that the structure is only porous MoS2, the residual presence of a substantial amount of carbon from the molybdenum hexacarbonyl precursor cannot be ruled out. Neither TEM nor XRD analysis presented by the authors was sufficiently detailed to negate that possibility. Given the reported surface area of 80 m2 g−1, there may also be a contribution to the net capacity due to the surface adsorption of lithium on metallic molybdenum after the conversion reaction (in addition to the almost certain adsorption of lithium on any residual carbon). The authors' rate dependence results in Fig. 8d (nearly 800 mA h g−1 at 30 C and 700 mA h g−1 at 50 C) may only be realized with a high charge transfer surface area and extremely short diffusion distances, implying that the very fine, high surface area microstructure remains stable throughout the cycling.
7.3 Ordered mesoporous MoS2 through templating
The use of hard templates has been recently and successfully employed to synthesize highly porous MoS2 nanostructures with high surface areas. The surface areas of these materials are large enough to justify lithium ion surface adsorption and lithium metal pore filling (nanoplating) as an important secondary contributor to the net charge storage.177,182,240 In such cases a charge storage capacity beyond 669 mA h g−1 is expected analogous to the high surface area/high porosity carbons that exceed 372 mA h g−1. Fig. 9 highlights one of the more interesting and better performing examples of such an approach.240 The specific surface area and pore volume of mesoporous MoS2 were calculated to be 130 m2 g−1 and 0.24 cm3 g−1, and are expected to further increase after the first lithiation. Fig. 9a shows a low magnification SEM image revealing macroscopic clusters of rod-like interconnected MoS2 nanowires. The mesoporous channels are partially revealed by the TEM micrograph in Fig. 9b, with SAED inset indexed to 2H–MoS2. Fig. 9c shows the excellent cycling stability of the material, retaining a reversible capacity of 876 mA h g−1 after 100 cycles at a current density of 0.1 A g−1. Fig. 9d shows the exceptional rate capability, and capacity recovery of the material, even after cycling at a current density of 10 A g−1. This capability was attributed to the large electrode/electrolyte interface allowed by the mesoporous channels (which had a narrow size distribution). This led to ultrafast lithium intercalation over a large surface area. Similar results have also been obtained for other mesoporous materials.241 The authors comment that the high rate capability is also due to the enhanced layer spacing of their MoS2, however this is doubtful after the first cycle. The high rate capability could only be realized in a highly conductive matrix which suggests the presence of a finely dispersed metallic phase. The authors employed an excessive amount of carbon black in their electrode recipe (70:20:10, active material:carbon black:PVDF binder) which may partially account for the capacity enhancement, though in this case we believe that it is a secondary issue relative to the porosity effect.
 | ||
| Fig. 9 Templated mesoporous MoS2. (a) Low magnification SEM image revealing MoS2 microstructures. (b) TEM micrograph (with SAED inset) revealing the mesoporosity of MoS2 crystallites and wire-like arrays. (c) Cycling performance at a current density of 0.1 A g−1. (d) Cycling performance at different current densities. Adapted from ref. 240 (DOI: 10.1002/aenm.201200087) with permission from Wiley-VHC Verlag GmbH &Co, KGaA, Weinheim, Copyright 2012. | ||
One would expect that the high surface area would lead to excessive SEI layer formation, however the templated electrode is shown to remain quite stable with good coulombic efficiency (97–98%) throughout cycling. This agrees with what is commonly reported in literature for templated carbons with highly ordered porosity, which are also stable upon cycling and demonstrate good coulombic efficiency.206 Apparently after the first cycle, SEI formation is at least partially inhibited. While no post-cycled TEM was completed in this work, others have shown SEI formation on mesoporous MoS2 with partial retention of the mesoporous structure after initial charge and discharge.178 For this material, the pore volume is also expected to accommodate the volume expansions and distortions associated with lithiation and the conversion reaction. The stability of the material suggests that the mesoporous structure may have been partially retained after many cycles, however this was not proven.
Table 2 provides a summary of the electrochemical data from the various literature sources. As can be seen there is a substantial variation not only in the charge storage capacities but also in the coulombic efficiencies between the studies. Interestingly, nominally similar techniques, e.g. hydrothermal synthesis, can result is radically different electrochemical performance outcomes. The implication of this is that both subtle changes in the experimental synthesis parameters, the electrode/cell fabrication techniques, and the morphology of the as-synthesized material can all play a major role in determining how well the battery performs.
| Material | Synthesis method | First discharge capacity (mA h g−1) | First charge capacity (mA h g−1) | Reversible capacity after (X) cycles (mA h g−1) | Coulombic efficiency after (Y) cycles (%) | Current density | Highest current density tested | Reference |
|---|---|---|---|---|---|---|---|---|
| a * – indicates a value estimated from a published graph. | ||||||||
| MoS2–PEO (plate-like particles) | Exfoliation/hydrolysis | 1131 | 822* | 890 (50) | 95* (50) | 50 mA g−1 | 50 mA g−1 | 180 |
| MoS2–GNS–PEO (nanoparticles) | Exfoliation/hydrolysis | 1130 | 830* | 1000 (180) | 93* (180) | 50 mA g−1 | 10 A g−1 | 181 |
| MoS2–GNS (nanoparticles) | Hydrothermal | 2200* | 1300 | 1290 (50) | 99.2 (50) | 100 mA g−1 | 1000 mA g−1 | 46 |
| MoS2–GNS (nanoparticles) | Hydrothermal | 1571 | 1031 | 1187 (100) | 99* (100) | 100 mA g−1 | 1000 mA g−1 | 170 |
| MoS2–CNTs (nanosheets) | Hydrothermal | 1434 | 862 | 698 (60) | 94 (3) | 100 mA g−1 | 1000 mA g−1 | 174 |
| MoS2–CNTs (nanosheets) | Hydrothermal | 710* | 390* | 390 (50) | 98 (50) | 0.6 mA cm−2 | 0.6 mA cm−2 | 171 |
| MoS2–amC (nanosheets) | Hydrothermal | 1175 | 870* | 852 (40) | 94* (40) | 60 mA g−1 | 60 mA g−1 | 159 |
| MoS2–amC (nanoparticles) | Hydrothermal | 1340* | 869 | 633 (50) | 65 (1) | 100 mA g−1 | 400 mA g−1 | 173 |
| MoS2–amC (nanoparticles) | Hydrothermal | 1160 | 791 | 585 (70) | 95 (3) | 100 mA g−1 | 1000 mA g−1 | 174 |
| MoS2–amC (nanoparticles) | Hydrothermal | 2100* | 930* | 912 (100) | 99* (100) | 100 mA g−1 | 100 mA g−1 | 169 |
| MoS2-amC (nanowires) | Template-assisted | 880 | 625 | 630 (20) | 98.5 (20) | 33 mA g−1 | 669 mA g−1 | 177 |
| MoS2@CMK-3 (nanorods) | Template-assisted | 1056 | 824 | 602 (100) | 97* (100) | 250 mA g−1 | 2000 mA g−1 | 182 |
| MoS2–AB (nanorods) | Template-assisted | 1060* | 1052 | 876 (100) | 98* (100) | 100 mA g−1 | 10 A g−1 | 240 |
| MoS2–amC (nanoparticles) | Solvothermal | 1062 | 917 | 907 (50) | 87 (1) | 1062 mA g−1 | 53.1 A g−1 | 48 |
8 Promising MoS2 nanomaterials not investigated for lithium storage
As a nanostructured material, MoS2 can exist in a diverse range of morphologies and microstructures. These include fullerene-like MoS2 (layered onion-like nanospheres),200,201,242–247 MoS2 nanotubes,194–196,248–252 MoS2 nanowires with various stoichiometries,253–255 nanoribbons and nanosheets.256–262 As summarized in previous review papers,197,263,264 these nanostructures can be synthesized via a wide range of methods. At this stage the vast majority of these structures have not been investigated as electrode materials for lithium storage. We believe that these MoS2 nanostructures could hold great promise for electrode applications as many of the synthesis techniques offer opportunities for nanoscale carbon incorporation. In this section, we will give a brief overview of the nanostructures achieved using two of the most scalable methods: template-assisted and gas-phase synthesis techniques. These techniques are already widely utilized in sectors such as microelectronics and thin films coatings for various industrial applications, making the technology mature and transferrable to the energy storage sector. Moreover the described approaches yield arrays of interconnected nanostructures that offer direct electron conductions paths down to the current collectors; a key advantage over techniques that result in isolated crystallites or particles.8.1 Various MoS2 nanostructures synthesized through templating
Many MoS2 nanostructures can be synthesized through the templating strategy. Authors performed seminal work on template-assisted synthesis of monodispersed microscale MoS2 nanofibers and nanotubes.265 They utilized thermal decomposition of two different ammonium thiomolybdate precursors within the confines of a porous aluminum oxide template. Their technique resulted in a dense fibrous network of MoS2 nanotubes that extended parallel to the substrate. This is appealing for lithium storage due to the high degree of interconnectivity and potential for flexibility.266 Additional work describe routes to MoS2 nanotube and fullerene synthesis.267,268 Authors describe a procedure to synthesize MoS2 nanotubes of different chiralities, similar to carbon nanotubes.267 While drawing comparisons the authors mentioned that the unique sandwich-like structure of MoS2 may offer greater resistance to nanotube buckling and kinking than for CNTs. This fact may prove useful for the creation of flexible lithium ion batteries that can withstand large amounts of deformation.Researchers have also developed an alternate template-assisted technique to create a coaxial–binary system of graphene and MoS2 nanotubes.269 Since it is known that a capacity enhancement is accompanied by the coordination of graphene with MoS2 nanosheets, the creation of a coaxial–binary system of MoS2 and carbon nanotubes may exhibit excellent electrochemical performance. Additionally, a template-assisted method using silica for producing mesoporous MoS2 has also been successfully completed.270 Here, the authors have developed a method of synthesizing tubular mesoporous domains of MoS2 which are highly layered and nanoscale. They go on to describe a dimensional tunability, which is difficult to achieve with other synthesis techniques and could therefore be useful for creating MoS2 nanostructures with controllable size for lithium storage. This technique may be useful for quantifying the dimensional effect of MoS2 on charge capacity.
8.2 MoS2 nanostructures synthesized though gas-phase techniques
Gas-phase synthesis techniques are amongst the most intriguing since they yield a wide range of unique microstructures and offer a high degree of versatility. Authors describe methods which involve aerosol assisted CVD processes that form MoS2via the decomposition of a single source precursor gas.271,272 These methods have led to some very distinctive MoS2 microstructures, which exhibit a plate-like morphology that was found to vary with annealing temperature. Although complex, the result was the deposition of nanoscale MoS2 structures over centimeter square areas which may be useful for conformal coatings on complex geometries.271 Additionally, researchers also report on a novel CVD method of synthesizing MoS2 monolayers over large areas.273 These procedures are advantageous since many of the methods previously discussed yield interconnected particulates of MoS2 that are difficult to deposit as large-area coatings. While these coatings are usually evaluated for anti-friction applications, their adaptation for lithium storage on complex geometries could be beneficial, since electrode templates often have intricate, high surface area morphologies.Authors have shown that it is also possible to synthesize high surface area molybdenum disulfide nanotubes directly from a reaction of molybdenum metal and sulfur powder together with iodine flakes reacted in a glass ampoule at 850 °C.274 In their method, C60 was added at 5 wt% and used as a growth catalyst in their reactions but was removed in subsequent processing steps. Others have reported an electrochemical enhancement by adding C60.84 These tubes were observed to have a high defect density along their length and be of relatively uniform diameter. Furthermore, they demonstrate that it is possible to grow vertically aligned MoS2 nanotube forests across a substrate surface, similar to CNTs.263
Researchers have achieved physical vapor deposition of MoS2 thin films using reactive magnetron sputtering of a solid molybdenum target and magnetron sputtering a solid MoS2 target.198,275 In this work the authors noticed that a significant portion of crystallites would form with their c-axis parallel to the substrate, which would present dangling bonds in the form of edge vacancies to the outer surface as well as provide potential inter-planar diffusion pathways to incident lithium ions. Authors describe a technique for the synthesis of MoS2 nanoparticles using pulsed laser ablation.276 Here pure, fullerene-like nanoparticles with a very uniform size distribution were synthesized by the ablation of a target in water. This technique could be adapted as a simple way to fabricated nanocomposites with carbon, via carbon incorporation into the pressed molybdenum disulfide target pellet. The propensity for unique facile nanostructures involving carbon encapsulation and incorporation seems plausible with this technique.
9 Concluding thoughts
While LiMoS2 cathodes may offer little in the way of capacity enhancement compared to LiCoO2, the material is still worth considering due to its exceptional rate performance and cycling stability. As an anode, the capacity enhancement of MoS2 over graphite has been well demonstrated. There is still significant debate regarding the actual lithiation reaction sequence during charging/discharging. However, there is progressively more evidence to support what is currently the minority view, that the primary lithium active phase is elemental sulfur beyond the first cycle. Metallic molybdenum appears to have a secondary albeit very important role of both enhancing the electrical conductivity of the electrode and perhaps stabilizing the shuttling of polysulfides that are known to be the source of premature failure in Li–S batteries. However for the case of Li–MoS2, the role of polysulfides and their interaction both with the molybdenum and the various nanocarbon phases interspersed within the electrodes remains quite poorly understood. This brings up the second outstanding scientific issue: the experimentally reported reversible charge storage capacities for MoS2-based architectures are consistently above the theoretical capacity of the literature-proposed lithiation conversion reactions. These involve either MoS2 or sulfur (keeping in mind the weight contribution of the “inactive” molybdenum). While scenarios such as nanoscale molybdenum actually being electro-active towards lithium are possible and should be further explored, we believe that the charge capacity of the nanodispersed carbons present in the composites is often underestimated.References
- Q. H. Wang, K. Kalantar-zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- L. Rapoport, N. Fleischer and R. Tenne, J. Mater. Chem., 2005, 15, 1782 RSC.
- T. Le Mogne, C. Donnet, J. M. Martin, A. Tonck and N. Millard-Pinard, J. Vac. Sci. Technol., A, 1994, 12(4), 1998 CAS.
- H. W. Wang, P. Skeldon and G. E. Thompson, Surf. Coat. Technol., 1997, 91, 200 CrossRef CAS.
- A. P. M. Barboza, H. Chacham, C. K. Oliveira, T. F. D. Fernandes, E. H. M. Ferreira, B. S. Archanjo, R. J. C. Batista, A. B. de Oliveira and B. R. A. Neves, Nano Lett., 2012, 12, 2313 CrossRef CAS PubMed.
- H. Zhang, K. P. Loh, C. H. Sow, H. Gu, X. Su, C. Huang and Z. K. Chen, Langmuir, 2004, 20, 6914 CrossRef CAS PubMed.
- C. Zhu, Z. Zeng, H. Li, F. Li, C. Fan and H. Zhang, J. Am. Chem. Soc., 2013, 135, 5998 CrossRef CAS PubMed.
- H. Li, Z. Yin, Q. He, H. Li, X. Huang, G. Lu, D. W. H. Fam, A. L. Y. Tok, Q. Zhang and H. Zhang, Small, 2012, 8(1), 63 CrossRef CAS PubMed.
- Q. He, Z. Zeng, Z. Yin, H. Li, S. Wu, X. Huang and H. Zhang, Small, 2994, 8(19) Search PubMed.
- N. Barreau and J. C. Bernede, J. Phys. D: Appl. Phys., 2002, 35, 1197 CrossRef CAS.
- J. Feng, X. Qian, C.-W. Huang and J. Li, Nat. Photonics, 2012, 6, 866 CrossRef CAS.
- C.-J. Liu, S.-Y. Tai, S.-W. Chou, Y.-C. Yu, K.-D. Chang, S. Wang, F. S.-S. Chien, J.-Y. Lin and T.-W. Lin, J. Mater. Chem., 2012, 22, 21057 RSC.
- S.-Y. Tai, C.-J. Liu, S.-W. Chou, F. S.-S. Chien, J.-Y. Lin and T.-W. Lin, J. Mater. Chem., 2012, 22, 24753 RSC.
- M. Shanmugam, T. Bansal, C. W. Durcan and B. Yu, Appl. Phys. Lett., 2012, 100, 153901 CrossRef.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- L. P. Hansen, Q. M. Ramasse, C. Kisielowski, M. Brorson, E. Johnson, H. Topsoe and S. Helveg, Angew. Chem., Int. Ed., 2011, 50, 10153 CrossRef CAS PubMed.
- J. P. Wilcoxon, J. Phys. Chem. B, 2000, 104, 7334 CrossRef CAS.
- W. Ho, J. C. Yu, J. Lin, J. Yu and P. Li, Langmuir, 2004, 20, 5865 CrossRef CAS.
- W. Han, P. Yuan, Y. Fan, G. Shi, H. Liu, D. Bai and X. Bao, J. Mater. Chem., 2012, 22, 25340 RSC.
- X. Chu, G. Yao, A. T. S. Wee and X.-S. Wang, Nanotechnology, 2012, 23, 375603 CrossRef PubMed.
- X. Zhong, H. Yang, S. Guo, S. Li, G. Guo, Z. Niu, Z. Dong, Y. Lei, J. Jin, R. Li and J. Ma, J. Mater. Chem., 2012, 22, 13925 RSC.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262 RSC.
- A. B. Laursen, S. Kegnaes, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577 CAS.
- T. Wang, L. Liu, Z. Zhu, P. Papakonstantinou, J. Hu, H. Liu and M. Li, Energy Environ. Sci., 2013, 6, 625 CAS.
- Y. Yan, B. Xia, X. Qi, H. Wang, R. Xu, J.-Y. Wang, H. Zhang and X. Wang, Chem. Commun., 2013, 49, 4884 RSC.
- A. B. Laursen, P. C. K. Vesborg and I. Chorkendorff, Chem. Commun., 2013, 49, 4965 RSC.
- X. Huang, Z. Zeng, S. Bao, M. Wang, X. Qi, Z. Fan and H. Zhang, Nat. Commun., 2013, 4, 1444 CrossRef PubMed.
- J. Liu, Z. Zeng, X. Cao, G. Lu, L. H. Wang, Q. L. Fan, W. Huang and H. Zhang, Small, 2012, 8(22), 3517 CrossRef CAS PubMed.
- Advances in Lithium-Ion Batteries, ed. W. A. van Schalkwijk and B. Scrosati, Klumer Academic Publishers, New York, USA, 2002, pp. 1–5 Search PubMed.
- Lithium Batteries, ed. J. P. Gabano, Academic Press, New York, USA, 1983, pp. 1–12 Search PubMed.
- Linden's Handbook of Batteries, ed. D. Linden and T. B. Reddy, McGraw Hill, NewYork, USA, 2011, p. 27.1 Search PubMed.
- W. S. Harris, Ph.D. Thesis, UCRL-8381, University of California, Berkeley, 1958, pp. 1–37.
- W. Rudorff, Angew. Chem., 1959, 71, 487 CrossRef CAS.
- W. Rudorff and H. H. Sick, Angew. Chem., 1959, 71, 127 CrossRef CAS.
- M. S. Whittingham and F. R. Gamble Jr, Mater. Res. Bull., 1975, 10, 363 CrossRef CAS.
- M. S. Whittingham, Science, 1976, 192, 1126 CAS.
- M. S. Whittingham, J. Electrochem. Soc., 1976, 123(3), 315 CrossRef CAS PubMed.
- M. S. Whittingham, Prog. Solid State Chem., 1978, 12, 41 CrossRef CAS.
- B. G. Silbernagel and M. S. Whittingham, J. Chem. Phys., 1976, 64(9), 3670 CrossRef CAS.
- G. C. Farrington and J. L. Briant, Science, 1979, 204, 1371 CAS.
- M. B. Dines, Mater. Res. Bull., 1975, 10, 287 CrossRef CAS.
- J. B. Goodenough, Proc. R. Soc. London, Ser. A, 1984, 393, 215 CrossRef CAS.
- Y. Xie and C. Wu, Dalton Trans., 2007, 45, 5235 RSC.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294 RSC.
- K. Chang and W. Chen, Chem. Commun., 2011, 47, 4252 RSC.
- A. Kohandehghan, P. Kalisvaart, M. Kupsta, B. Zahiri, B. S. Amirkhiz, Z. Li, E. L. Memarzadeh, L. A. Bendersky and D. Mitlin, J. Mater. Chem. A., 2013, 1, 1600–1612 CAS.
- H. Hwang, H. Kim and J. Cho, Nano Lett., 2011, 11, 4826 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Chio, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577 CrossRef CAS PubMed.
- L.-F. Cui, L. Hu, J. W. Choi and Y. Cui, ACS Nano, 2010, 4(7), 3671 CrossRef CAS PubMed.
- J. Xie, X. Yang, S. Zhou and D. Wang, ACS Nano, 2011, 5(11), 9225 CrossRef CAS PubMed.
- R. R. Haering; J. A. R. Stiles and K. Brandt. US Pat., 4,224,390, 23 September 1980.
- C. Julien, S. I. Saikh and G. A. Nazri, Mater. Sci. Eng., B, 1992, 15, 73 CrossRef.
- Y. Miki, D. Nakazato, H. Ikuta, T. Uchida and M. Wakihara, J. Power Sources, 1995, 54, 508 CrossRef CAS.
- V. Yufit, M. Nathan, D. Golodnitsky and E. Peled, J. Power Sources, 2003, 122, 169 CrossRef CAS.
- A. V. Murugan, M. Quintin, M.-H. Delville, G. Campet, C. S. Gopinath and K. Vijayamohanan, J. Power Sources, 2006, 156, 615 CrossRef CAS PubMed.
- E. Shembel, R. Apostolova, I. Kirsanova and V. Tysyachny, J. Solid State Electrochem., 2008, 12, 1151 CrossRef CAS PubMed.
- M. G. Kim, M. Jo, Y.-S. Hong and J. Cho, Chem. Commun., 2008, 219 Search PubMed.
- H. Chen, X. Qiu, W. Zhu and P. Hagenmuller, Electrochem. Commun., 2002, 4, 488 CrossRef CAS.
- L. Xia, S.-L. Li, X.-P. Ai, H.-X. Yang and Y.-L. Cao, Energy Environ. Sci., 2011, 4, 2845 CAS.
- Z. Hong, X. Zheng, X. Ding, L. Jiang, M. Wei and K. Wei, Energy Environ. Sci., 2011, 4, 1886 CAS.
- R. Koksbang, J. Barker, H. Shi and M. Y. Saidi, Solid State Ionics, 1996, 84, 1 CrossRef CAS.
- Z. Chen and J. R. Dahn, Electrochim. Acta, 2004, 49, 1079 CrossRef CAS PubMed.
- Y. Lu, S. K. Das, S. S. Moganty and L. A. Archer, Adv. Mater., 2012, 24, 4430 CrossRef CAS PubMed.
- C.-H. Lai, M.-Y. Lu and L.-J. Chen, J. Mater. Chem., 2012, 22, 19 RSC.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986 RSC.
- X. Huang, Z. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934 RSC.
- S. Balendhran, S. Walia, H. Nili, J. Z. Ou, S. Zhuiykov, R. B. Kaner, S. Sriram, M. Bhaskaran and K. Kalantar-zadeh, Adv. Funct. Mater., 2013, 23, 3952 CrossRef CAS.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294 RSC.
- A. Manthiram, A. V. Murugan, A. Sarkar and T. Muraliganth, Energy Environ. Sci., 2008, 1, 621 CAS.
- P. Balaya, Energy Environ. Sci., 2008, 1, 645 CAS.
- X. Fan, H. Zhang, N. Du, P. Wu, X. Xu, Y. Li and D. Yang, Nanoscale, 2012, 4, 5343 RSC.
- A.-H. Lim, H.-W. Shim, S.-D. Seo, G.-H. Lee, K.-S. Park and D.-W. Kim, Nanoscale, 2012, 4, 4694 RSC.
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater, 2007, 19, 2336 CrossRef CAS.
- S. Ding and X. W. Lou, Nanoscale, 2011, 3, 3586 RSC.
- S. J. R. Pabakar, Y.-H. Hwang, E.-G. Bae, S. Shim, D. Kim, M. S. Lah, K.-S. Sohn and M. Pyo, Adv. Mater, 2013, 25, 3307 CrossRef PubMed.
- D. S. Sun and R. Schlogl, ChemSusChem, 2010, 3, 136 CrossRef PubMed.
- R. Song, H. Song, J. Zhou, X. Chen, B. Wu and H. Y. Yang, J. Mater. Chem., 2012, 22, 12369 RSC.
- E. Frackowiak, S. Gautier, H. Gaucher, S. Bonnamy and F. Beguin, Carbon, 1999, 37, 61 CrossRef CAS.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638 CAS.
- J. Chen, J. Z. Wang, A. I. Minett, Y. Liu, C. Lynam, H. Liu and G. Wallace, Energy Environ. Sci., 2009, 2, 393 CAS.
- D. Pan, S. Wang, B. Zhao, M. Wu, H. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136 CrossRef CAS.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049 CrossRef CAS PubMed.
- E. Yoo, J. Kim, E. Hosono, H. S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8(8), 2277 CrossRef CAS PubMed.
- T. Bhardwaj, A. Antic, B. Pavan, V. Barone and B. D. Fahlman, J. Am. Chem. Soc., 2010, 132, 12556 CrossRef CAS PubMed.
- M. Pumera, Energy Environ. Sci., 2011, 4, 668 CAS.
- C.-X. Zu and H. Li, Energy Environ. Sci., 2011, 4, 2614 CAS.
- Z. Li, Z. Xu, X. Tan, H. Wang, C. M. B. Holt, T. Stephenson, B. C. Olsen and D. Mitlin, Energy Environ. Sci., 2013, 6, 871 CAS.
- P. Wu, N. Du, H. Zhang, J. Yu, Y. Qi and D. Yang, Nanoscale, 2011, 3, 746 RSC.
- B. G. Choi, S.-J. Chang, Y. B. Lee, J. S. Bae, H. J. Kim and Y. S. Huh, Nanoscale, 2012, 4, 5924 RSC.
- S. K. Das, S. Darmakolla and A. J. Bhattacharyya, J. Mater. Chem., 2010, 20, 1600 RSC.
- H. Han, T. Song, J.-Y. Bae, L. F. Nazar, H. Kim and U. Paik, Energy Environ. Sci., 2012, 4, 4532 Search PubMed.
- Z. Yang, G. Du, Q. Meng, Z. Guo, X. Yu, Z. Chen, T. Guo and R. Zeng, J. Mater. Chem., 2012, 22, 5848 RSC.
- G.-N. Zhu, Y.-G. Wang and Y.-Y. Xia, Energy Environ. Sci., 2012, 5, 6652 CAS.
- F. Xia, X. Hu, Y. Sun, W. Luo and Y. Huang, Nanoscale, 2012, 4, 4707 RSC.
- A. Bhaskar, M. Deepa and T. N. Rao, Appl. Mater. Inferfaces, 2013, 5, 2555 CrossRef CAS PubMed.
- Y. Sun, X. Hu, J. C. Yu, Q. Li, W. Luo, L. Yuan, W. Zhang and Y. Huang, Energy Environ. Sci., 2011, 4, 2870 CAS.
- X. H. Huang, J. P. Tu, C. Q. Zhang, X. T. Chen, Y. F. Yuan and H. M. Wu, Electrochim. Acta, 2007, 52, 4177 CrossRef CAS PubMed.
- Y. Zou and Y. Wang, Nanoscale, 2011, 3, 2615 RSC.
- H. Liu and W. Yang, Energy Environ. Sci., 2011, 4, 4000 CAS.
- X. Zhou, Y.-X. Yin, L.-J. Wan and Y.-G. Guo, Chem. Commun., 2012, 48, 2198 RSC.
- E. L. Memarzadeh, W. P. Kalisvaart, A. Kohandehghan, B. Zahiri, C. M. B. Holt and D. Mitlin, J. Mater. Chem., 2012, 22, 6655 RSC.
- L. Ji and X. Zhang, Energy Environ. Sci., 2010, 3, 124 CAS.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56 CAS.
- C. Du, C. Gao, G. Yin, M. Chen and L. Wang, Energy Environ. Sci., 2011, 4, 1037 CAS.
- H. T. Nguyen, M. R. Zamfir, L. D. Duong, Y. H. Lee, P. Bondavalli and D. Pribat, J. Mater. Chem., 2012, 22, 24618 RSC.
- J.-W. Seo, J.-T. Jang, S.-W. Park, C. Kim, B. Park and J. Cheon, Adv. Mater., 2008, 20, 4269 CrossRef CAS.
- B. Luo, Y. Fang, B. Wang, J. Zhou, H. Song and L. Zhi, Energy Environ. Sci., 2012, 5, 5226 CAS.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821 RSC.
- K. Cai, M.-K. Song, E. J. Cairns and Y. Zhang, Nano Lett., 2012, 12, 6474 CrossRef CAS PubMed.
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. V. Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS.
- R. Marom, S. F. Amalraj, N. Leifer, D. Jacob and D. Aurbach, J. Mater. Chem., 2011, 21, 9938 RSC.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Energy Mater., 2010, 22, E170 CrossRef CAS PubMed.
- X. H. Liu and J. Y. Huang, Energy Environ. Sci., 2011, 4, 3844 CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 3243 CAS.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287 CAS.
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176 CrossRef CAS.
- J. Liwen, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682 Search PubMed.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526 RSC.
- R. G. Dickinson and L. Pauling, J. Am. Chem. Soc., 1923, 45, 1466 CrossRef CAS.
- R. E. Bell and R. E. Herfert, J. Am. Chem. Soc., 1957, 79(13), 3351 CrossRef CAS.
- R. J. Traill, Can. Mineral., 1963, 7, 524 CAS.
- J. C. Wildervanck and F. Jellinek, Z. Anorg. Allg. Chem., 1964, 328, 309 CrossRef CAS.
- Y. Takeuchi and W. Nowacki, Schweiz. Mineral. Petrogr. Mitt., 1964, 44, 105 CAS.
- F. E. Wickman and D. K. Smith, Am. Mineral., 1970, 55, 1843 CAS.
- J. A. Wilson and A. D. Yoffe, Adv. Phys., 1969, 18, 193 CrossRef CAS.
- G. A. N. Connell, J. A. Wilson and A. D. Yoffe, J. Phys. Chem. Solids, 1969, 30, 287 CrossRef CAS.
- R. J. J. Newberry, Am. Mineral., 1979, 64, 758 CAS.
- B. Schonfeld, J. J. Huang and S. C. Moss, Acta Crystallogr., 1983, B39, 404 Search PubMed.
- K. D. Bronsema, J. L. De Boer and F. Jellinek, Z. Anorg. Allg. Chem., 1986, 540–541, 15 CrossRef CAS.
- P. D. Fleischauer, J. R. Lince, P. A. Bertrand and R. Bauer, Langmuir, 1989, 5, 1009 CrossRef CAS.
- S. V. Didziulis, J. R. Lince, D. K. Shuh, T. D. Durbin and J. A. Yarmoff, J. Electron Spectrosc. Relat. Phenom., 1992, 60, 175 CrossRef CAS.
- F. Wypych and R. Schollhorn, J. Chem. Soc., Chem. Commun., 1992, 1386 RSC.
- C. Rovira and M.-H. Whangbo, Inorg. Chem., 1993, 32, 4094 CrossRef CAS.
- J. B. Goodenough, G. Dutta and A. Manthiram, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 10170 CrossRef CAS.
- D. Guay, Chem. Mater., 1994, 6, 614 CrossRef CAS.
- O. El Beqqali, I. Zorkani, F. Rogemond, H. Chermette, R. Ben Chaabane, M. Gamoudi and G. Guillaud, Synth. Met., 1997, 90, 165 CrossRef CAS.
- F. Wypych, T. Weber and R. Prins, Chem. Mater., 1998, 10, 723 CrossRef CAS.
- F. Wypych, C. Solenthaler, R. Prins and T. Weber, J. Solid State Chem., 1999, 144, 430 CrossRef CAS.
- E. Benavente, M. A. Santa Ana, F. Mendizabal and G. Gonzalez, Coord. Chem. Rev., 2002, 224, 87 CrossRef CAS.
- L. F. Mattheiss, Phys. Rev. B: Condens. Matter Mater. Phys., 1973, 8, 3719 CrossRef CAS.
- P. Lu, X. Wu, W. Guo and X. C. Zeng, Phys. Chem. Chem. Phys., 2012, 14, 13035 RSC.
- Y. Zhou, P. Yang, H. Zu, F. Gao and X. Zu, Phys. Chem. Chem. Phys., 2013, 15, 10385 RSC.
- M. A. Py and R. R. Haering, Can. J. Phys., 1983, 61, 76 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1931, 53, 1367 CrossRef CAS.
- R. Hultgren, Phys. Rev., 1932, 40, 891 CrossRef CAS.
- L. S. Selwyn, W. R. McKinnon, U. von Sacken and C. A. Jones, Solid State Ionics, 1987, 22, 337 CrossRef CAS.
- C. A. Papageorgopoulos and W. Jaegermann, Surf. Sci., 1995, 338, 83 CrossRef CAS.
- N. Imanishi, M. Toyoda, Y. Takeda and O. Yamamoto, Solid State Ionics, 1992, 58, 333 CrossRef CAS.
- G. Gonzalez, M. A. Santa Ana, E. Benavente, J. P. Donoso, T. J. Bonagamba, N. C. Mello and H. Panepucci, Solid State Ionics, 1996, 85, 225 CrossRef CAS.
- K. E. Dungey, M. D. Curtis and J. E. Penner-Hahn, Chem. Mater., 1998, 10, 2152 CrossRef CAS.
- A. Zak, Y. Feldman, V. Lyakhovitskaya, G. Leitus, R. Popovitz-Biro, E. Wachtel, H. Cohen, S. Reich and R. Tenne, J. Am. Chem. Soc., 2002, 124, 4747 CrossRef CAS PubMed.
- D. Tonti, C. Pettenkofer and W. Jaegermann, J. Phys. Chem. B, 2004, 108, 16093 CrossRef CAS.
- J. Chen, Z.-L. Tao and S.-L. Li, Angew. Chem., Int. Ed., 2003, 42, 2147 CrossRef CAS PubMed.
- C. M. Julien, Mater. Sci. Eng., R, 2003, 40, 47 CrossRef.
- Y. Kim and J. B. Goodenough, J. Phys. Chem. C, 2008, 112, 15060 CAS.
- C. Feng, J. Ma, H. Li, R. Zeng, Z. Guo and H. Liu, Mater. Res. Bull., 2009, 44, 1811 CrossRef CAS PubMed.
- K. Chrissafis, M. Zamani, K. Kambas, J. Stoemenos, N. A. Economou, I. Samaras and C. Julien, Mater. Sci. Eng., B, 1989, 3, 145 CrossRef.
- Y. Li, D. Wu, Z. Zhou, C. R. Cabrera and Z. Chen, J. Phys. Chem. Lett., 2012, 3, 2221 CrossRef CAS.
- D. Yang, S. J. Sandoval, W. M. R. Divigalpitiya, J. C. Irwin and R. F. Frindt, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43(14), 12053 CrossRef CAS.
- Y. V. Zubavichus, Y. L. Slovokhotov, P. J. Schilling, R. C. Tittsworth, A. S. Golub, G. A. Protzenko and Y. N. Novikov, Inorg. Chim. Acta, 1998, 280, 211 CrossRef CAS.
- J. Heising and G. Kanatzidis, J. Am. Chem. Soc., 1999, 121, 11720 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Science, 1995, 270, 590 CAS.
- P. J. Mulhern, Can. J. Phys., 1989, 67, 1049 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, Internet Version, ed. W. M. Hayes and D.R. Lide, CRC Press: Boca Raton, Florida, edn. 92nd, 2012, pp. 9–50, 12-11, http://www.hbcpnetbase.com Search PubMed.
- G. Du, Z. Guo, S. Wang, R. Zeng, Z. Chen and H. Liu, Chem. Commun., 2010, 46, 1106 RSC.
- K. Chang, W. Chen, L. Ma, H. Li, H. Li, F. Huang, Z. Xu, Q. Zhang and J.-Y. Lee, J. Mater. Chem., 2011, 21, 6251 RSC.
- K. Chang and W. Chen, ACS Nano, 2011, 5(6), 4720 CrossRef CAS PubMed.
- Q. Wang and J. Li, J. Phys. Chem. C, 2007, 111, 1675 CAS.
- S. K. Das, R. Mallavajula, N. Jayaprakash and L. A. Archer, J. Mater. Chem., 2012, 22, 12988 RSC.
- H. Li, W. Li, L. Ma, W. Chen and J. Wang, J. Alloys Compd., 2009, 471, 442 CrossRef CAS PubMed.
- S. Ding, D. Zhang, J. S. Chen and X. W. Lou, Nanoscale, 2012, 4, 95 RSC.
- S. Ding, J. S. Chen and X. W. Lou, Chem.–Eur. J., 2011, 17, 13142 CrossRef CAS PubMed.
- J.-H. Kwon, H.-J. Ahn, M.-S. Jeon, K.-W. Kim, I.-S. Ahn, J.-H. Ahn, G. Wang and H.-S. Ryu, Res. Chem. Intermed., 2010, 36, 749 CrossRef CAS PubMed.
- X. Fang, X. Guo, Y. Mao, C. Hua, L. Shen, Y. Hu, Z. Wang, F. Wu and L. Chen, Chem.–Asian J., 2012, 7, 1013 CrossRef CAS PubMed.
- X. Fang, X. Yu, S. Liao, Y. Shi, Y. S. Hu, Z. Wang, G. D. Stucky and L. Chen, Microporous Mesoporous Mater., 2012, 151, 418 CrossRef CAS PubMed.
- X. Fang, C. Hua, X. Guo, Y. Hu, Z. Wang, X. Gao, F. Wu, J. Wang and L. Chen, Electrochim. Acta, 2012, 81, 155 CrossRef CAS PubMed.
- J. Xiao, D. Choi, L. Cosimbescu, P. Koech, J. Liu and J. P. Lemmon, Chem. Mater., 2010, 22, 4522 CrossRef CAS.
- J. Xiao, X. Wang, X. Q. Yang, S. Xun, G. Liu, P. K. Koech, J. Liu and J. P. Lemmon, Adv. Funct. Mater., 2011, 21, 2840 CrossRef CAS.
- X. Zhou, L.-J. Wan and Y.-G. Guo, Nanoscale, 2012, 4, 5868 RSC.
- R. D. Rauh, K. M. Abraham, G. F. Pearson, J. K. Surprenant and S. B. Brummer, J. Electrochem. Soc., 1979, 126, 523 CrossRef CAS PubMed.
- H. Yamin and E. Peled, J. Power Sources, 1983, 9, 281 CrossRef CAS.
- C. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8(6), 500 CrossRef CAS PubMed.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531 CAS.
- X.-P. Gao and H.-X. Yang, Energy Environ. Sci., 2010, 3, 174 CAS.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691 CrossRef CAS.
- J.-J. Chen, Q. Zhang, Y.-N. Shi, L.-L. Qin, Y. Cao, M.-S. Zheng and Q.-F. Dong, Phys. Chem. Chem. Phys., 2012, 14, 5376 RSC.
- M. S. Park, J.-S. Yu, K. J. Kim, G. Jeong, J.-H. Kim, Y.-N. Jo, U. Hwang, S. Kang, T. Woo and Y.-J. Kim, Phys. Chem. Chem. Phys., 2012, 14, 6796 RSC.
- S. Dorfler, M. Hagen, H. Althues, J. Tubke, S. Kaskel and M. J. Hoffmann, Chem. Commun., 2012, 48, 4097 RSC.
- W. Ahn, K.-B. Kim, K.-N. Jung, K.-H. Shin and C.-S. Jin, J. Power Sources, 2012, 202, 394 CrossRef CAS PubMed.
- A. N. Enyashin and A. L. Ivanovskii, Semiconductors, 2007, 41(1), 81 CrossRef CAS.
- F. L. Deepak and M. Jose-Yacaman, Isr. J. Chem., 2010, 50, 426 CrossRef CAS.
- H. Pan and Y.-W. Zhang, J. Mater. Chem., 2012, 22, 7280 RSC.
- A. Enyashin, S. Gemming and G. Seifert, Eur. Phys. J.: Spec. Top., 2007, 149, 103 CrossRef.
- W. Weiss, W. Bohne, J. Rohrich, E. Strub, U. Bloeck, I. Sieber, K. Ellmer, R. Mientus and F. Prosch, J. Appl. Phys., 2004, 95(12), 7665 CrossRef.
- Y. Chan and J. M. Hill, Nanoscale Res. Lett., 2011, 6, 203 CrossRef PubMed.
- J. Kibsgaard, J. V. Lauritsen, E. Laegsgaard, B. S. Clausen, H. Topsoe and F. Besenbacher, J. Am. Chem. Soc., 2006, 128, 13950 CrossRef CAS PubMed.
- A. N. Enyashin, M. Bar-Sadan, J. Sloan, L. Houben and G. Seifert, Chem. Mater., 2009, 21, 5627 CrossRef CAS.
- A. Tuxen, J. Kibsgaard, H. Gobel, E. Laegsgaard, H. Topsoe, J. V. Lauritsen and F. Besenbacher, ACS Nano, 2010, 4(8), 4677 CrossRef CAS PubMed.
- C. F. Castro-Guerrero, F. L. Deepak, A. Ponce, J. Cruz-Reyes, M. D. Valle-Granados, S. Fuentes-Moyado, D. H. Galvan and M. Jose-Yacaman, Catal. Sci. Technol., 2011, 1, 1024 CAS.
- D. I. Kochubei, V. A. Rogov, V. P. Babenko, S. V. Bogdanov and V. I. Zaikovskii, Kinet. Catal., 2003, 44, 135 CrossRef CAS.
- N. Balke, S. Jesse, A. N. Morozovska, E. Eliseev, D. W. Chung, Y. Kim, L. Adamczyk, R. E. Garcia, N. Dudney and S. V. Kalinin, Nat. Nanotechnol., 2010, 5, 749 CrossRef CAS PubMed.
- Y. Mao, H. Duan, B. Xu, L. Zhang, Y. Hu, C. Zhao, Z. Wang, L. Chen and Y. Yang, Energy Environ. Sci., 2012, 5, 7950 CAS.
- Y.-X. Wang, L. Huang, L.-C. Sun, S.-Y. Xie, G.-L. Xu, S.-R. Chen, Y.-F. Xu, J. T. Li, S.-L. Chou, S.-X. Dou and S.-G. Sun, J. Mater. Chem., 2012, 22, 4744 RSC.
- R. Elazari, G. Salitra, A. Garsuch, A. Panchenko and D. Aurbach, Adv. Mater., 2011, 23, 5641 CrossRef CAS PubMed.
- Y.-S. Su and A. Manthiram, Chem. Commun., 2012, 48, 8817 RSC.
- X. Li, Y. Cao, W. Qi, L. V. Saraf, J. Xiao, Z. Nie, J. Mietek, J.-G. Zhang, B. Schwenzer and J. Liu, J. Mater. Chem., 2011, 21, 16603 RSC.
- J. Jamnik and J. Maier, Phys. Chem. Chem. Phys., 2003, 5, 5215 RSC.
- Y. Yang, M. T. McDowell, A. Jackson, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2010, 10, 1486 CrossRef CAS PubMed.
- Y. Li, B. Tan and Y. Wu, Nano Lett., 2008, 8, 265 CrossRef CAS PubMed.
- L. Qie, W.-M. Chen, Z.-H. Wang, Q.-G. Shao, X. Li, L.-X. Yuan, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, Adv. Mater., 2012, 24, 2047 CrossRef PubMed.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J.-M. Tarascon, J. Electrochem. Soc., 2002, 149(5), A627 CrossRef CAS PubMed.
- R. Dedryvere, S. Laruelle, S. Grugeon, P. Poizot, D. Gonbeau and J.-M. Tarascon, Chem. Mater., 2004, 16, 1056 CrossRef CAS.
- Y. Shi, B. Guo, S. A. Corr, Q. Shi, Y.-S. Hu, K. R. Heier, L. Chen, R. Seshardri and G. D. Stucky, Nano Lett., 2009, 9(12), 4215 CrossRef CAS PubMed.
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13(8), 621 CrossRef CAS.
- C. Zhai, N. Du, H. Zhang, J. Yu, P. Wu, C. Xiao and D. Yang, Nanoscale, 2011, 3, 1798 RSC.
- K. Bindumadhavan, S. K. Srivastava and S. Mahanty, Chem. Commun., 2013, 49, 1823 RSC.
- A. Marschilok, C.-Y. Lee, A. Subramanian, K. J. Takeuchi and E. S. Takeuchi, Energy Environ. Sci., 2011, 4, 2943 CAS.
- G. Zhou, D.-W. Wang, P.-X. Hou, W. Li, N. Li, C. Liu, F. Li and H.-M. Cheng, J. Mater. Chem., 2012, 22, 17942 RSC.
- S.-K. Park, S.-H. Yu, S. Woo, B. Quan, D.-C. Lee, M. K. Kim, Y.-E. Sung and Y. Piao, Dalton Trans., 2013, 42, 2399 RSC.
- S. W. Lee, B. M. Gallant, H. R. Byon, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 1972 CAS.
- X. Cao, Y. Shi, W. Shi, X. Rui, Q. Yan, J. Kong and H. Zhang, Small, 2013, 9, 3433 CrossRef CAS PubMed.
- J. Xi, M. L. Long, L. Tang, D. Wang and Z. Shuai, Nanoscale, 2012, 4, 4348 RSC.
- X. Jia, J. Campos-Delgado, M. Terrones, V. Meunier and M. S. Desselhaus, Nanoscale, 2011, 3, 86 RSC.
- X. Zhou, L.-J. Wan and Y.-G. Guo, Chem. Commun., 2013, 49, 1838 RSC.
- K. Chang, D. Geng, X. Li, J. Yang, Y. Tang, M. Cai, R. Li and X. Sun, Adv. Funct. Mater., 2013, 3, 839 CAS.
- H. Wang, C. M. B. Holt, Z. Li, X. Tan, B. S. Amirkhiz, Z. Xu, B. C. Olsen, T. Stephenson and D. Mitlin, Nano Res., 2012, 5, 605–617 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80(6), 1339 CrossRef CAS.
- W. Chen, Z. Zhu, S. Li, C. Chen and L. Yan, Nanoscale, 2012, 4, 2124 RSC.
- S. Malik, A. Vijayaraghavan, R. Erni, K. Arida, I. Khalakhan and J. P. Hill, Nanoscale, 2010, 2, 2139 RSC.
- Z. Xu, Z. Li, X. Tan, C. M. B. Holt, L. Zhang, B. Shalchi-Amirkhiz and D. Mitlin, RSC Adv., 2012, 2, 2753 RSC.
- Y. Ma, Y. Dai, M. Guo, C. Niu and B. Huang, Nanoscale, 2011, 3, 3883 RSC.
- H. Jiang, T. Zhao, C. Yan, J. Ma and C. Li, Nanoscale, 2010, 2, 2195 RSC.
- F.-F. Cao, Y.-G. Guo and L.-J. Wan, Energy Environ. Sci., 2011, 4, 1634 CAS.
- H.-G. Jung, S.-T. Myung, C. S. Yoon, S.-B. Son, K. H. Oh, K. Amine, B. Scrosati and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 1345 CAS.
- D. Lei, M. Zhang, B. Qu, L. Chen, Y. Wang, E. Zhang, Z. Xu, Q. Li and T. Wang, Nanoscale, 2012, 4, 3422 RSC.
- H. Liu, D. Su, R. Zhou, B. Sun, G. Wang and S. Z. Qiao, Adv. Energy Mater., 2012, 2, 970 CrossRef CAS.
- K. Saravanan, K. Ananthanarayanan and P. Balaya, Energy Environ. Sci., 2010, 3, 939 CAS.
- D. J. Srolovitz, S. A. Safran, M. Homyonfer and R. Tenne, Phys. Rev. Lett., 1995, 74(10), 1779 CrossRef CAS.
- Y. Xiong, Y. Xie, Z. Li, X. Li and R. Zhang, Chem. Phys. Lett., 2003, 382, 180 CrossRef CAS PubMed.
- I. Alexandrou, N. Sano, A. Burrows, R. R. Meyer, H. Wang, A. I. Kirkland, C. J. Kiely and G. A. J. Amaratunga, Nanotechnology, 2003, 14, 913 CrossRef CAS.
- M. Bar-Sadan, A. N. Enyashin, S. Gemming, R. Popovitz-Biro, S. Y. Hong, Y. Prior, R. Tenne and G. Seifert, J. Phys. Chem. B, 2006, 110, 25399 CrossRef CAS PubMed.
- L. Chang, H. Yang, W. Fu, J. Zhang, Q. Yu, H. Zhu, J. Chen, R. Wei, Y. Sui, X. Pang and G. Zou, Mater. Res. Bull., 2008, 43, 2427 CrossRef CAS PubMed.
- T. Li and G. Galli, J. Phys. Chem. C, 2007, 111, 16192 CAS.
- G. L. Frey and R. Tenne, J. Mater. Res., 1998, 13(9), 2412 CrossRef CAS.
- G. Seifert, H. Terrones, M. Terrones, G. Jungnickel and T. Frauenheim, Phys. Rev. Lett., 2000, 85(1), 146 CrossRef CAS.
- R. Tenne and C. N. R. Rao, Philos. Trans. R. Soc., A, 2004, 362, 2099 CrossRef CAS PubMed.
- R. Tenne and G. Seifert, Annu. Rev. Mater. Res., 2009, 39, 387 CrossRef CAS.
- N. Zibouche, A. Kuc and T. Heine, Eur. Phys. J. B, 2012, 85(49), 1 Search PubMed.
- A. Hassanien, M. Tokumoto, A. Mrzel, D. Mihailovic and H. Kataura, Physica E., 2005, 29, 684 CrossRef CAS PubMed.
- A. Hassanien, G. Leintschnig, A. Mrzel, M. Tokumoto and H. Kataura, Surf. Interface Anal., 2006, 38, 1530 CrossRef CAS.
- J. Kibsgaard, A. Tuxen, M. Levisen, E. Laegsgaard, S. Gemming, G. Seifert, J. V. Lauritsen and F. Besenbacher, Nano Lett., 2008, 8(11), 3928 CrossRef CAS PubMed.
- J. Heising and M. G. Kanatzidis, J. Am. Chem. Soc., 1999, 121, 638 CrossRef CAS.
- Q. Li, J. T. Newberg, E. C. Walter, J. C. Hemminger and R. M. Penner, Nano Lett., 2004, 4(2), 277 CrossRef CAS.
- Y. Li, Z. Zhou, S. Zhang and Z. Chen, J. Am. Chem. Soc., 2008, 130, 16739 CrossRef CAS PubMed.
- J. Brivio, D. T. L. Alexander and A. Kis, Nano Lett., 2011, 11, 5148 CrossRef CAS PubMed.
- Z. Wang, H. Li, Z. Liu, Z. Shi, J. Lu, K. Suenaga, S.-K. Joung, T. Okazaki, Z. Gu, J. Zhou, Z. Gao, G. Li, S. Sanvito, E. Wang and S. Iijima, J. Am. Chem. Soc., 2010, 132, 13840 CrossRef CAS PubMed.
- H. S. S. R. Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., Int. Ed., 2010, 49, 4059 CrossRef CAS PubMed.
- S. Balendhran, J. Z. Ou, M. Bhaskaran, S. Sriram, S. Ippolito, Z. Vasic, E. Kats, S. Bhargava, S. Zhuiykov and K. Kalatar-zadeh, Nanoscale, 2012, 4, 461 RSC.
- M. Remskar, A. Mrzel, Z. Skraba, A. Jesih, M. Ceh, J. Demsar, P. Stadelmann, F. Levy and D. Mihailovic, Science., 2001, 292, 479 CrossRef CAS PubMed.
- H. Terrones and M. Terrones, New J. Phys., 2003, 5, 126 CrossRef.
- C. M. Zelenski and P. K. Dorhout, J. Am. Chem. Soc., 1998, 120, 734 CrossRef CAS.
- X. Jia, Z. Chen, A. Suwarnasarn, L. Rice, X. Wang, H. Sohn, Q. Zhang, B. M. Wu, F. Wei and Y. Lu, Energy Environ. Sci., 2012, 5, 6845 CAS.
- W. K. Hsu, B. H. Chang, Y. Q. Zhu, W. Q. Han, H. Terrones, M. Terrones, N. Grobert, A. K. Cheetham, H. W. Kroto and D. R. M. Walton, J. Am. Chem. Soc., 2000, 122, 10155 CrossRef CAS.
- Y. Feldman, G. L. Frey, M. Homyonfer, V. Lyakhovitskaya, L. Margulis, H. Cohen, G. Hodes, J. L. Hutchison and R. Tenne, J. Am. Chem. Soc., 1996, 118, 5362 CrossRef CAS.
- C. R. S. German, P. Santiago, J. A. Ascencio, U. Pal, M. Perez-Alvarez, L. Rendon and D. Mendoza, J. Phys. Chem. B, 2005, 109, 17488 CrossRef PubMed.
- Y. Shi, Y. Wan, R. Liu, B. Tu and D. Zhao, J. Am. Chem. Soc., 2007, 129, 9522 CrossRef CAS PubMed.
- A. Adeogun, M. Afzaal and P. O'Brien, Chem. Vap. Deposition, 2006, 12, 597 CrossRef CAS.
- J. Zhang, J. M. Soon, K. P. Loh, J. Yin, J. Ding, M. B. Sullivian and P. Wu, Nano Lett., 2007, 7(8), 2370 CrossRef CAS PubMed.
- Y. H. Lee, X.-Q. Zhang, W. Zhang, M.-T. Chang, C.-T. Lin, K.-D. Chang, Y.-C. Yu, J. T.-W. Wang, C.-S. Chang, L.-J. Li and T.-W. Lin, Adv. Mater., 2012, 24, 2320 CrossRef CAS PubMed.
- M. Remskar, A. Mrzel, M. Virsek, M. Godec, M. Krause, A. Kolitsch, A. Singh and A. Seabaugh, Nanoscale Res. Lett., 2011, 6(26), 1 Search PubMed.
- P. D. Fleischauer, Thin Solid Films, 1987, 154, 309 CrossRef CAS.
- H. Wu, R. Yang, B. Song, Q. Han, J. Li, Y. Zhang, Y. Fang, R. Tenne and C. Wang, ACS Nano, 2011, 5(2), 1276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ee42591f |
| This journal is © The Royal Society of Chemistry 2014 |