Energy applications of ionic liquids
Douglas R.
MacFarlane
*a,
Naoki
Tachikawa
a,
Maria
Forsyth
b,
Jennifer M.
Pringle
b,
Patrick C.
Howlett
b,
Gloria D.
Elliott
c,
James H.
Davis
Jr.
d,
Masayoshi
Watanabe
e,
Patrice
Simon
f and
C. Austen
Angell
g
aAustralian Centre for Electromaterials Science, School of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: douglas.macfarlane@monash.edu
bInstitute for Frontier Materials, Deakin University, Burwood, Victoria, Australia
cDepartment of Mechanical Engineering and Engineering Sciences, University of North Carolina, Charlotte 9201 University City Blvd., Charlotte, NC 28223-0001, USA
dDepartment of Chemistry, University of South Alabama, Mobile, Al 36688, USA
eDepartment of Chemistry & Biotechnology, Yokohama National University, Yokohama 240-8501, Japan
fUniversité Paul Sabatier, Toulouse III, CIRIMAT UMR-CNRS 5085, 118 Route de Narbonne, 31062 Toulouse Cedex, France
gDepartment of Chemistry and Biochemistry, Arizona State University, AZ 85287-1604, USA
First published on 15th August 2013
Abstract
Ionic liquids offer a unique suite of properties that make them important candidates for a number of energy related applications. Cation–anion combinations that exhibit low volatility coupled with high electrochemical and thermal stability, as well as ionic conductivity, create the possibility of designing ideal electrolytes for batteries, super-capacitors, actuators, dye sensitised solar cells and thermo-electrochemical cells. In the field of water splitting to produce hydrogen they have been used to synthesize some of the best performing water oxidation catalysts and some members of the protic ionic liquid family co-catalyse an unusual, very high energy efficiency water oxidation process. As fuel cell electrolytes, the high proton conductivity of some of the protic ionic liquid family offers the potential of fuel cells operating in the optimum temperature region above 100 °C. Beyond electrochemical applications, the low vapour pressure of these liquids, along with their ability to offer tuneable functionality, also makes them ideal as CO2 absorbents for post-combustion CO2 capture. Similarly, the tuneable phase properties of the many members of this large family of salts are also allowing the creation of phase-change thermal energy storage materials having melting points tuned to the application. This perspective article provides an overview of these developing energy related applications of ionic liquids and offers some thoughts on the emerging challenges and opportunities.
1. Introduction
Sustainability of energy supply has become one the great challenges of our time. The combination of concerns about the contribution of fossil fuels to greenhouse gas induced climate change, as well as the long term sustainability of their supply, has necessitated an increasingly urgent development of alternative approaches to energy generation and storage. Similarly, air quality related environmental concerns, as well as energy security, is driving the development of alternate energy sources for transportation including passenger vehicles. As increasingly larger solar and wind energy capacity is installed around the world, the intermittency of these sources is creating the need for large-scale energy storage solutions and variable-load applications, including fuel generation. At the same time, there is huge effort being applied to the minimisation of the impact of fossil fuel use via carbon capture technologies.In all of this development activity, there is enormous potential for the discovery and application of new materials that offer significant improvements in the way that energy is generated, stored and delivered. Ionic liquids (ILs) are one such family of materials that are beginning to have an impact on a broad swath of energy technologies, offering a range of properties that can be tuned to optimise their performance in a variety of contexts. As liquid salts, dominated by strong electrostatic forces between their molecular ions, the key properties that certain members of this huge family of compounds offer is low volatility/flammability and high chemical and electrochemical stability. This makes them potentially ideal as solvents and electrolytes, and in electrochemical applications their intrinsic ionic conductivity is also an important feature.
However, the properties of ILs vary enormously as a function of their molecular structure and considerable effort has been devoted to identifying and understanding those that have superior properties in any given application. Our goal in this article therefore is to provide a perspective on how, and why, ionic liquids are impacting on a range of electrochemical technologies, including advanced batteries, dye sensitised solar cells, double layer capacitors, actuators, fuel cells, thermo-cells and water splitting, as well as in non-electrochemical areas including carbon capture and in emerging thermal energy storage applications. Since the literature is voluminous we have only made reference to some of the key, recent and exemplary papers in each field, sufficient to provide some background and in the hope that these will provide a starting point for the interested reader to follow.
2. Ionic liquids in advanced battery technologies
Lithium and lithium–ion cells
The attractive properties of ILs, in particular the high electrochemical stability of some cation and anion types, lend themselves to application in high-energy electrochemical devices such as lithium batteries. One of the key advantages that they offer as electrolytes is low volatility and flammability, hence offering the possibility of enhanced safety and stability. For an excellent summary of the properties and development of lithium ion rechargeable batteries generally, the reader is referred to a recent perspective article by Goodenough and Park.1 Early reports of ILs offering sufficient stability for lithium electrochemistry in quaternary ammonium and phosphonium [NTf2]− ILs2,3 sparked the interest of battery researchers (structures and nomenclature of some commonly used ILs are summarised in Fig. 1). Cyclic pyrrolidinium and piperidinium [NTf2]− ILs, which are typically less viscous and slightly more cathodically stable than the aliphatic cations, were then shown to support highly efficient lithium cycling.4 A large number of publications have subsequently explored variations of IL cation (dominated by small aliphatic and cyclic ammonium cations) and anion (dominated by [NTf2]− and [BF4]−) combinations with a range of lithium battery anodes and cathodes.5 Matsumoto6 has reported high rate cycling of a Li|LiCoO2 cell incorporating a pyrrolidinium FSI IL, demonstrating the ability of this smaller amide anion to promote higher transport rates. In addition this anion avoided the need for additives7 to prevent irreversible interaction of the IL cation at graphitic carbon electrodes, indicating a probable chemical role in forming a surface layer on the electrode.8 Application of small phosphonium cations (with [NTf2]−) has also been reported,9 including their use with high voltage cathodes.10 Within this relatively narrow group of ILs, reliable cell cycling with other high-capacity and/or high-voltage electrodes including sulfur,11 silicon,12 polyaniline CNTs,13 LiMn1.5Ni0.5O4 (ref. 14) and LiVPO4F (ref. 15) has also been reported recently. | ||
| Fig. 1 Common ionic liquid ion families appearing in energy applications and their commonly used acronym systems. In acronymns such as [Cnmpyr]+ the subscript “n” indicates the number of carbons in the alkyl substituent. | ||
Throughout the evolution of ionic liquid electrolytes for lithium batteries the importance of the fate of the dissolved lithium ion and its speciation in the bulk liquid has been recognised, both in terms of its influence on ion transport and on interactions at the electrode surface. Design of both the cation and anion functionality (e.g., alkoxy and cyano functionality) and the use of additives and diluents (e.g., vinylene carbonate) have been explored as methods to influence the electrolyte properties in this regard.16–20 The formation of protective surface films (also known as the solid electrolyte interphase – SEI)4,21 and the importance of the role of IL interfacial and bulk structuring on electrochemical stability, ion transport and charge transfer has been discussed.22,23 Lane recently reported a detailed assessment of the cation reduction reactions occurring at negative electrodes for the most common ILs.24 Taken together, these studies (and others beyond the scope of this article) now provide an understanding of the role of the specific interactions in each IL system (e.g. cation and anion association, speciation, electrochemical stability, decomposition products and interfacial structuring) that influence lithium ion transport and the electrode interactions that occur in an operating lithium cell. However, uncertainty still remains as to the mechanism by which Li salt addition results in an extension of the cathodic reduction limit.25 The relative importance of the chemical breakdown of the IL constituents to form a SEI versus cation–anion interfacial structuring and speciation to exclude reactive constituents, is not clearly understood and further research in this area is needed to better inform the design of new IL electrolytes.
The growing need for large-scale energy storage to ameliorate the intermittency aspect of renewable energy installations will provide further stimulus for IL electrolyte development, with requirements for high energy density becoming less important compared to safety and robustness, very long cycle life and an ability to operate at elevated temperatures. This provides an excellent opportunity for ILs to come to the fore, with their intrinsic stability offering possible solutions to the shortcomings of current technology in this regard. Recent efforts to develop prototype batteries for this type of application have highlighted the feasibility of IL electrolytes, for example in combination with photovoltaic panels where thermal stability is important and rate limitations are less so.26
However, despite such extensive study and demonstrated wide applicability of a variety of different ILs in various Li cell configurations of different energy and power capabilities, no commercial application of batteries incorporating IL electrolytes has yet been developed. It would seem this is due in part to the prohibitive expense of ILs in comparison to the conventional carbonate solvents, in particular of some of the fluorinated amide anions. If we accept that the inherent cost of current IL electrolytes is prohibitive for the development of commercial devices, then strategies to either reduce cost or increase value are required. The most obvious strategy is to continue to explore and develop lower cost cations and anions; the recent demonstration of Li cells incorporating a pyrrolidinium dicyanamide IL, Fig. 2, demonstrates some progress in this direction by avoiding the use of fluorinated anions.27 Another avenue will emerge from the inherent stability of ILs, aiming to develop batteries capable of very large numbers of charge–discharge cycles, particularly under adverse thermal conditions. This has been initially demonstrated for devices based on stable intercalation electrodes (i.e., Li4Ti5O12 and LiFePO4) where substantial efforts have been made to demonstrate prototype devices with high cycle stability.28
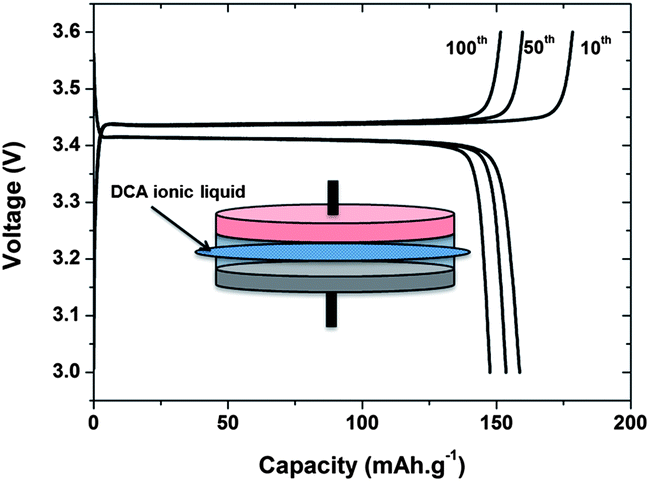 | ||
| Fig. 2 Charge–discharge profiles at the 10th, 50th, and 100th cycle of a Li|LiFePO4 cell using a [dca]− IL at 80 °C (redrawn from ref. 27). | ||
The other significant factor limiting application relates to the relatively low rate capability displayed by IL based batteries, owing mainly to the greater viscosity of the electrolyte, particularly after Li salt addition which contributes to stronger ion association in the electrolyte. Progress on this issue has emerged recently from the FSI family of ILs, where very high lithium ion contents have been found to support substantial charge–discharge rates,6 although this has come at the cost of reduced stability.29 Clearly these concerns about thermal stability and safety of ILs of this anion need to be further investigated.29,30
More recently, to overcome both the issues of cost and rate capability (particularly at low temperature), studies which investigate blended systems of ILs with conventional carbonate based electrolytes have become increasingly common.31–33 In this case, the ILs are incorporated to increase the ionic strength of the electrolyte and hence manipulate ion dynamics, interfacial stability and, most importantly, flammability.
Recently, research focus has turned to high-energy electrode combinations such as Li–O2 and Li–S rechargeable cells to address the limitations of current Li–ion devices. Particularly in the case of Li–O2, problems associated with electrolyte decomposition at the cathode, and the need for high efficiency Li cycling at the anode requires significant focus on electrolyte development.34 Mizuno et al.35 assessed the stability of a range of electrolytes based on the calculated Mulliken charge for each atom. It was determined that a piperidinium [NTf2]− IL should possess stability towards the O2− radical anion and this was confirmed by demonstrating a cell with reduced cathodic polarization compared to a carbonate electrolyte. Higashi et al.36 subsequently showed improved performance by the incorporation of an ether functionalised cation (N,N-diethyl-N-methyl-N-(2-methoxyethyl) ammonium, [DEME], Fig. 1). Allen et al.37 reported a strong correlation between the oxygen reduction reaction products and the ionic charge density of the IL cation, which was rationalised in terms of the Lewis acidity of the cation present in the electrolyte, indicating that ‘soft’ IL cations could stabilise the superoxide anion in the presence of hard acids such as Li+. The demonstration of stabilised superoxide species in a phosphonium IL in the presence of water further supports this notion.38 Also highlighting the relevance of the hydrophobic nature of some ILs, Zhang and Zhou39 recently reported the development of single-walled carbon nanotube cross-linked network IL gels for use as air cathodes.
Although the vast majority of research investigating ILs for lithium batteries has been devoted to their use as electrolytes, it may eventuate that their most prominent and valuable contribution will come from their use as a solvent for the synthesis of inorganic electrode materials. The development of ionothermal synthetic methods40–42 has offered unprecedented opportunities to expand the way in which inorganic materials can be synthesised. These lower temperature procedures create the possibility of reduced costs and the ability of ILs to stabilise metastable phases offers a range of attractive new low-cost/high energy materials that were previously inaccessible.
Sodium and sodium–ion cells
In recent years concerns about the sustainability of lithium supplies, and therefore its cost, have prompted researchers to consider other anodic metals such as sodium, magnesium and zinc that are significantly more abundant, environmentally friendly and safer (in the case of Mg and Zn in particular) than lithium. Increasing numbers of electric vehicles and the application of lithium based batteries for large scale stationary energy storage, involving load levelling and storage for intermittent renewable electricity generation, will undoubtedly put pressure on the supply and cost of lithium. Thus other technologies, including Na–ion, Mg–air and Zn–air batteries, whilst not providing the same energy density as lithium, may be potential alternatives for certain applications. IL electrolytes are an obvious choice for these devices for all of the reasons that they are useful in lithium devices – i.e., large electrochemical window, low volatility and high thermal stability. To date there are only a few publications detailing IL electrolytes studied in these emerging applications. In the case of Na batteries, Buchner et al.43 have reported conductivity, dielectric properties and viscosity of NaBF4 in 1-butyl-3-methyl imidazolium ILs and shown that, unlike lithium, the sodium salt has only a minor influence on the transport properties of the IL. On the other hand Egashira et al. have suggested that NaBF4 is not very soluble in tetralkylammonium based ILs and have explored the ternary mixture with a methyl ether capped polyether as a coordinating agent.44 Recently an investigation of [C4mpyr][NTf2], doped with NaNTf2 has been reported;45 in addition to describing the conductivity, viscosity and ion association in these electrolytes, reversible sodium deposition and stripping was demonstrated as shown in Fig. 3. On the other hand, inorganic IL systems based on NaFSI–KFSI and NaNTf2–CsNTf2 binary mixtures have been reported by Nohira and co-workers46,47 as intermediate-temperature ionic liquid systems. These ILs possess wide electrochemical windows, up to 5.2 V at 363 K, and Na battery devices were found to perform well at operating temperatures between 333 and 393 K.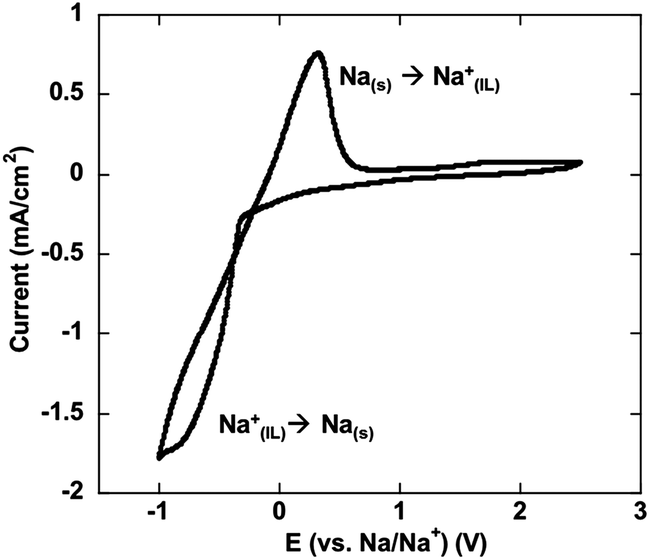 | ||
| Fig. 3 Reversible cycling of sodium in a [C4mpyr][NTf2] ionic liquid electrolyte (from ref. 45). | ||
It is clear from this emerging work that ILs may offer the same advantages as electrolytes for sodium based battery systems as they do for lithium, however it would be a mistake to assume that the SEI related transport phenomena will be identical and therefore further study of the formation and nature of SEI layer formation on sodium in ILs is needed.
Magnesium batteries
In the case of Mg anodes, the use of ionic liquid mixtures have been reported recently as electrolytes for both primary magnesium–air devices48,49 as well as in reversible Mg devices.50,51 The literature remains unclear in regard to the reversibility of Mg electrochemistry and this area is in need of considerable further investigation. Whilst reduction of Mg from an electrolyte onto a metal substrate can be achieved quite readily, its subsequent dissolution is less apparent. Early work from Wang and co-workers52 suggested that mixtures of [C4mim][BF4] and N-methyl-N-propylpiperidinium[NTf2], in a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and in the presence of 0.3 M MgNTf2 allowed the reversible deposition and dissolution of magnesium metal. The evidence supporting this was, however, not clear as cyclic voltammograms were not reported and instead the cycling of a cell consisting of a magnesium counter-electrode and a polished silver working electrode was studied. The data showed that initially there was instability in the deposition–dissolution cycles which the authors attributed to complex interfacial chemistry, but eventually a stable reversible signal was obtained and they suggested that this was evidence for 100% reversible cycling of Mg. In our experience, in ILs based on fluorinated anions, Mg and its alloys readily passivate53,54 and therefore it is more likely that the data presented by Wang et al. is due to a short circuit that has developed in the cell due to cycling, which is also consistent with the very low overpotentials measured. Furthermore, in these experiments, the use of a Ag working electrode in their work complicates study of reversible Mg cycling due to alloying with the Ag. Thus the high efficiency reversible cycling of Mg in an IL remains an important goal. The electrochemical behaviour of magnesium perchlorate in ILs has been investigated by Sutto and Duncan using Co3O4 (ref. 55) or RuO2 (ref. 56) as the working electrode and ILs based on substituted imidazolium cations and [NTf2]− anions. The capacity of devices assembled from these components showed poor stability upon cycling and the authors attributed this to interactions of Mg2+ with the cathodes; however, given the reports of passivation of Mg metal with [NTf2]− based ILs53,54 we should also consider that decreased performance of the device may be related to this factor.
:1, and in the presence of 0.3 M MgNTf2 allowed the reversible deposition and dissolution of magnesium metal. The evidence supporting this was, however, not clear as cyclic voltammograms were not reported and instead the cycling of a cell consisting of a magnesium counter-electrode and a polished silver working electrode was studied. The data showed that initially there was instability in the deposition–dissolution cycles which the authors attributed to complex interfacial chemistry, but eventually a stable reversible signal was obtained and they suggested that this was evidence for 100% reversible cycling of Mg. In our experience, in ILs based on fluorinated anions, Mg and its alloys readily passivate53,54 and therefore it is more likely that the data presented by Wang et al. is due to a short circuit that has developed in the cell due to cycling, which is also consistent with the very low overpotentials measured. Furthermore, in these experiments, the use of a Ag working electrode in their work complicates study of reversible Mg cycling due to alloying with the Ag. Thus the high efficiency reversible cycling of Mg in an IL remains an important goal. The electrochemical behaviour of magnesium perchlorate in ILs has been investigated by Sutto and Duncan using Co3O4 (ref. 55) or RuO2 (ref. 56) as the working electrode and ILs based on substituted imidazolium cations and [NTf2]− anions. The capacity of devices assembled from these components showed poor stability upon cycling and the authors attributed this to interactions of Mg2+ with the cathodes; however, given the reports of passivation of Mg metal with [NTf2]− based ILs53,54 we should also consider that decreased performance of the device may be related to this factor.
On the other hand, Yoshimoto et al. have shown that electrochemical deposition and dissolution of Mg can be achieved in organic solutions based on alkylmagnesium bromides in THF (i.e., Grignard type reagents) with various ILs, in particular imidazolium cations substituted in the 1, 2 and 3 positions to avoid excessive reactivity with the Grignard reagent.51 It thus appears that the Grignard chemistry approach to design of the IL electrolyte may be one of the ways forward in the Mg battery area.
Metal–air batteries – primary and secondary
Interest in metal–air primary batteries has accelerated recently driven by concepts that envisage a rapid exchange of an electric vehicle battery pack at a changeover station, as a way of solving the intrinsically slow recharge rate problem associated with most rechargeable battery technologies (to put this recharge problem into perspective it is worth noting that most petrol bowsers deliver energy to the vehicle at the rate of about 40 MJ s−1, or 40 MW, – a rate extraordinarily difficult to match with electrical technologies). IL based electrolytes consisting of quaternary phosphonium chlorides have been shown recently to support magnesium discharge with current densities up to 1 mA cm−2 and to sustain a Mg–air primary device for up to 3 weeks with a stable cell voltage of about 1.5–1.6 V.48,49 Water is an intrinsic feature of the oxygen reduction reaction in a practical air electrode and it was shown that an electrolyte with 8 mol% water in the IL gave the best performance. It was hypothesized that this led to an amorphous gel-like interphase that supported magnesium ion transport, in addition to improving the fluidity of the electrolyte and hence improving ion transport. In this context, the oxygen reduction reaction generally requires a proton source which, in some instances, has been shown to be abstracted from the IL cation57 leading to a 1e− reduction process for O2. However, the presence of water or other protic additives can support either the 1e−,38 2e−, or even the 4e− reduction process which is more efficient, leading to a lower overpotential and higher current densities.58 Continuing work is investigating IL mixtures whereby the ionic components can be designed to control the interfacial properties and solubility of Mg2+ complexes after discharge. Given the importance of the interfacial layer in the successful cycling of Li anodes and the apparent role of the interphase in these Mg–air devices, it may be possible to design the interphase on the Mg electrode to enable reversibility in a Mg–air device. This would require the formation and retention of complexes in the interphase that could readily be reduced. Such a rechargeable Mg–air device would be a significant advance in the energy storage field.IL electrolytes have also recently been shown to support reversible deposition and dissolution of Zn for application in secondary zinc batteries.59,60 Zn–air primary cells are used extensively at present with an electrolyte based on a 14 M KOH aqueous solution. These devices, while having a relatively low cell voltage (1.2 V) have high energy densities (500 W h kg−1) and are based on abundant, safe and inexpensive materials. A reversible Zn–air cell would offer a viable alternative to Li–ion technology. Early work by Deng et al.61 demonstrated that quasi-reversible Zn2+/Zn0 electrochemistry could be supported by [C4mpyr][dca]. However, only modest current densities were obtained when zinc was deposited on a magnesium alloy electrode at potentials as negative as −2.3 V vs. Fc0/+. Recently, it was shown that the nature of the anion in the Zn2+ salt added to [C2mim][dca] had a dramatic effect on the Zn2+ electrochemistry;60 when Zn[dca]2 was used in place of ZnCl2, higher currents and more positive potentials for deposition were observed. Spectroscopic data suggests that the favourable electrochemistry and transport properties are probably due to the formation of a Zn/dca− complex anions in the mixture.59 Reversible deposition/dissolution of Zn was also recently demonstrated in ILs based on quaternary alkoxy alkylammonium cations with varying oligo-ether side chain and the [NTf2]− anion.62 The cation was designed to act as a chelating species for Zn2+ which would have the two-fold effects of (i) avoiding the formation of neutral species or even anionic species due to association of Zn2+ with the anion and (ii) forming cationic complexes between the IL cation and the Zn2+. Both of these factors would contribute to improved electrochemical performance in a device. The work thus far shows a promising future for reversible Zn–air batteries based on ILs, although the reversible oxygen reduction reaction still remains a major challenge in this family of electrolytes. However much further work investigating and understanding speciation in multi-valent metal ion–IL mixtures, of the type pioneered by Rocher et al.,63 is desperately needed.
3. Application of ionic liquids to advanced fuel cell concepts
Until the last decade or so, it seems that, despite the 150 year history of fuel cell studies, no-one had considered, or at least succeeded in using, proton carriers other than hydronium ions or hydroxide ions in a fuel cell. This of course served to confine fuel cell science to the use of aqueous electrolytes of acid or basic character. In 1998, Kreuer et al.64 made an attempt to use the imidazole molecule as a proton transfer agent in polymer membranes, but the polymers of choice were too limiting in the transport process for much success to be achieved. A more promising result was obtained by Sun et al.65 by substituting protic imidazolium ILs, and mixtures of these with imidazole, for the water in a Nafion membrane. The Watanabe group, using protic ILs of the type being synthesized by the Ohno group66 demonstrated proton transport in a concentrated non-aqueous solution of imidazolium [NTf2]− in excess imidazole67 and reported the successful implementation of this principle in a simple fuel cell (Fig. 4).68 Parallel efforts of the Angell group concentrated on the use of ethylammonium nitrate for a similar purpose, and succeeded in being granted the first patent issued on IL electrolyte-based fuel cells.69 Apart from the novelty, the excitement in the fuel cell field created by the Susan et al. publication67 arose partly because of the possibility of creating electrolytes that were not limited by the solvent water, but could be highly variable, including neutral, in character, while remaining highly conducting. Furthermore, the anhydrous and high temperature conditions of operation permitted by such electrolytes raised the possibility of working under conditions that would eliminate both carbon monoxide poisoning, and noble metal catalyst corrosion.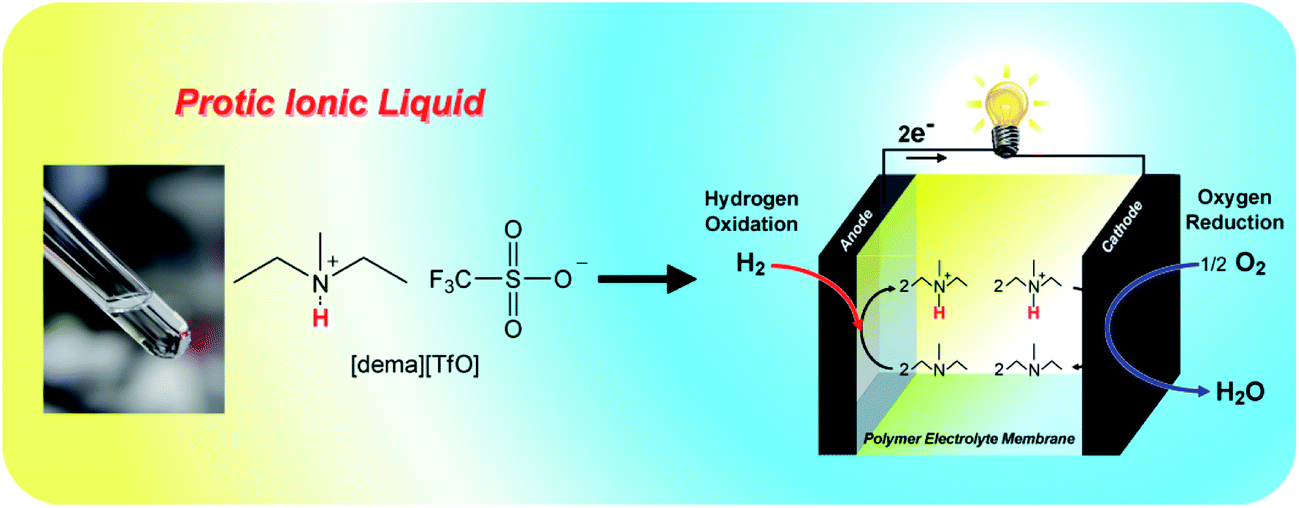 | ||
| Fig. 4 Application of ILs as proton carrier in a non-humidifying intermediate temperature fuel cell. Reproduced from ref. 71 with permission from The Royal Society of Chemistry. | ||
A further step away from the confines of aqueous solution fuel cell electrolytes was taken more recently by Belieres et al.70 who recognized that the small cation NH4+ might serve the same purpose as H3O+. They described liquid electrolytes based on a number of ammonium salt mixtures that had eutectic melting temperatures below 100 °C (hence qualified as inorganic ILs) and that could yield polarization curves competitive with those of hydrated phosphoric acid in the mid-temperature range (100–200 °C). Systems containing the nitrate anion prove to have narrow ranges of current density in which there appears to be almost no energy barrier to oxygen reduction. The reasons for this are still not clarified, though they are presumably related to some instability of the nitrate anion. This work revealed a systematic trend with increasing anion pKa to maximize the open circuit voltage of the fuel cell at an intermediate pKa value. A more detailed study of this effect by Miran et al.71 confirms the general trend, and refines its quantitative aspects.
A wide range of ammonium cation based protic ionic liquids (PILs) were recently studied and characterised for their potential fuel cell applications by Lee et al.72 The important but difficult step of incorporating the preferred PIL electrolyte, diethylmethylammonium triflate ([dema][TfO]), into a suitable membrane electrode assembly MEA was then undertaken by the same group.73
This field of application of ILs in medium temperature range fuel cells clearly has an important potential and further electrochemical and property studies are needed in the >100 °C range. One of the interesting perspectives that this field highlights is that applications of liquid salts is not limited to <100 °C and that salts of both organic and inorganic nature having melting points around and above 100 °C are equally of interest in both this application and potentially many others.
4. Dye sensitised solar cells
The dye-sensitised solar cell (DSSC)74 is a photo-electrochemical device that has the potential to be significantly cheaper to manufacture than traditional semiconductor photovoltaics. These devices can also be flexible, lightweight, different colours, and may offer better performance under low light (e.g. indoor) conditions. However, this technology is yet to be widely commercialised, with improvements in efficiency, stability and cost still required. The highest confirmed DSSC solar conversion efficiency reported to date is 11.9 ± 0.4% using an acetonitrile-based electrolyte,75 but this drops significantly upon use of an IL electrolyte.76 As for most of the other energy applications discussed in this article, the interest in using ILs as non-volatile electrolytes in DSSCs stems from the desire to develop longer lasting devices. The repeated exposure of DSSCs to solar heating (>70 °C for up to 10% of their operating life77), makes the development of stable devices particularly challenging and the volatility of traditional aprotic electrolytes is a significant issue.The components of a traditional “sandwich structure” DSSC, including a number of the materials variations that have been explored in IL-based devices, is shown in Fig. 5. Generally, IL electrolytes for DSSCs utilise a combination of a redox active salt with a low viscosity IL, to optimise the diffusion rate of the redox couple.78 A range of additives then optimise the kinetics of the electron transfer processes occurring at the photoanode.79 The two most efficient IL-based systems reported to-date utilise the I−/I3− redox couple with either: (i) a eutectic melt of two imidazolium iodide salts and [C2mim][B(CN)4],76 which produces good stability and 8.2% efficiency, or (ii) a [C2mim][N(CN)2] system,80 which yields 8.4% efficiency. This latter electrolyte uses a more available IL anion, but also contains 4-tert-butylpyridine that could result in lower long-term stability. One of the important features of this report, however, is the facile adjustment of the position of the titania conduction band through gradual addition of a lithium salt, thus allowing the kinetics of the electron injection from the dye to the TiO2 to be optimised.80 The nature of the IL, in particular basic anions such as dicyanamide, can also significantly affect the position of the conduction band edge and thus the open circuit voltage (Voc) of the device.78,81
 | ||
| Fig. 5 The “sandwich structure” DSSC, with some of the different components that have been used in combination with ionic liquid electrolytes. (TCO = transparent conductive oxide, ITO–PEN = indium tin oxide–poly(ethylene naphthalate), McMT = 2-mercapto-5-methyl-1,3,4-thiadiazole, BMT = 5,5′-dimethyl-2,2′-dithiodi-1,3,4-thiadiazole). | ||
The reasons for the high efficiency of these two IL electrolyte systems are manyfold and highlight the general requirements for DSSC IL-redox electrolytes: (i) a high redox couple diffusion rate (for the I−/I3− couple this can be augmented by an additional Grotthuss mechanism82), (ii) fast electron donation by the I− to regenerate the dye, (iii) slow charge recombination between the I3− and the injected electron at the photoanode, (iv) good electrochemical reversibility (which will also depend on the nature of the electrocatalyst), and (v) larger electron diffusion lengths, as a result of screening of the photoinjected electrons in the TiO2 film by the IL.76
There are a number of limitations of these redox electrolyte systems that suggest important areas for further development. The energy level of the redox electrolyte should be close enough to the dye to ensure good electron transfer (ca. 0.2–0.3 eV (ref. 83)) but not so close that the Voc of the device is unduly limited. Of the plethora of alternative redox couples explored to replace I−/I3−,84,85 few have been successfully utilised in ILs. The recent report of a high efficiency cobalt-based redox couple that can produce an efficiency of 12.3% when used with a molecular solvent,86 highlights the advances that can be made in the redox couple area.87 However, the use of a Co redox couple has not yet been reported in IL electrolytes. There is also considerable scope for the counter ion in these salts to be tuned to suit the IL ions (similar developments are discussed further in the thermoelectrochemical cells section below), which would help to address mass transport limitations resulting from the high viscosity of the ILs.
The influence of the DSSC components on the different photovoltaic parameters is, to a large extent, inter-related and high efficiency IL-based devices require specific optimisation of each component.78,79,83,88,89 For example, the larger extinction coefficients of organic or phorphyrin dyes compared to Ru-based dyes can allow the use of thinner photoanode films, helping address any problems of shorter electron diffusion lengths and lifetimes and slow ionic diffusion through the TiO2 film that can be present with IL electrolytes. The limited studies of porphyrin dyes thus far have produced 4.9% efficiencies with [C2mim][B(CN)4]-based IL electrolytes.90
The benefits of using ILs may be more compelling in the development of flexible DSSCs on plastic substrates.91 These devices are more suitable for large scale manufacture via roll-to-roll processes, are lighter and have a wider range of possible applications. The use of ILs would reduce problems of solvent evaporation through the plastic substrates, thus potentially increasing long-term performance. However, these substrates are also permeable to H2O and O2, which may enter the cell during sustained use, and the ITO layer can be corroded by electrolyte additives such as LiI.92 The impact of these effects on the performance of IL-based flexible DSSCs is still to be clarified and thorough studies on the long-term performance of these devices is urgently needed.
Few of the alternative counter electrode materials reported93,94 have yet been studied with ILs and this is clearly an important area of future work. Electrochemically deposited CoS nanoparticles can exhibit electrocatalytic activity equivalent to Pt in [C2mim][B(CN)4] electrolyte; 6.5% efficiency was achieved and 85% of this is retained after 1000 hours light soaking at 60 °C.95 This type of accelerated lifetime testing is extremely important for device development and is an area of research that is still significantly lacking in the field. Other alternative electrode materials that have shown promise with ILs include graphene nanoplatelets, which can have lower charge transfer resistance for I−/I3− in an IL electrolyte than in a molecular solvent.96 However, the charge transfer resistance still needs to be reduced by an order of magnitude in order to achieve good performance at full light intensity. PEDOT electrocatalysts can also give lower charge transfer resistances than Pt when used with ILs, possibly as a result of a larger surface area that is more suitable for the high I−/I3− concentrations used in IL electrolytes.97,98
For the ultimate IL-based DSSC, it would be preferable to utilise the ionic electrolyte in a quasi-solid state form, to reduce leakage, as long as the diffusion rate of the redox couple could be maintained. Options include ILs physically gelled through the addition of polymers or nanoparticles.78,79,89 Organic ionic plastic crystals (OIPCs) can also allow sufficient rates of I−/I3− diffusion to support device efficiencies of >5%,99 and good performance at high temperatures.100 These solid-state analogues of ILs contain some degree of structural disorder, with either rotational or translational motion of the ions possible, leading to high solid state ionic conductivities. There is significant scope for further developing these materials for DSSC applications as there is a plethora of ion types available, the nature of which can be chosen to directly improve the photovoltaic performance.78
In the future the development of an array of different DSSC device structures, utilising ILs to ensure maximum device lifetimes, may be key to improving their widespread utility. For example monolithic cells, which use a single conducting glass substrate, with a porous carbon counter electrode, thus reducing the cost and enabling large scale fabrication;101 when used with a [C2mim][B(CN)4]-based electrolyte these devices show good stability during storage in the dark at 85 °C for 1000 hours,102 but more accelerated lifetime testing under constant illumination is needed. Similarly, recently described wire-shaped DSSCs,103 for applications such as electronic textiles,104 will ultimately require a non-volatile IL type of electrolyte and represent an exciting new direction in the field.
5. Thermo-electrochemical cells
Thermo-electrochemical cells are the electrochemical equivalents of traditional semiconductor-based thermoelectric devices. The cells can directly convert thermal energy into electrical energy and offer the possibility of harvesting useful amounts of energy from available heat sources, for example from power station waste heat, or geothermal sources. The electrolyte requirements of these cells are very similar to the DSSCs discussed above and much of that science is now being transferred to this emerging field. As shown in Fig. 6, the cell is based on an electrochemical redox couple, such as the [Fe(CN)6]3−/[Fe(CN)6]4−, in an ionically conducting phase, with the two electrodes held at different temperatures.105,106 The temperature difference between the two electrodes generates a voltage difference according to the Seebeck coefficient, Se = ∂E(T)/∂T, where E(T) is the electrode potential of the redox couple. Thermo-electrochemical cells can be regarded as renewable energy systems because they can produce a constant voltage with no overall change in electrolyte composition as long as there is a constant supply of thermal energy. However, until recently, these devices have predominantly utilised water-based electrolytes,105,106 which restricts their operating temperatures to less than 100 °C, severely limiting their range of application. | ||
| Fig. 6 Schematic of a thermo-electrochemical cell using the [Fe(CN)6]3−/[Fe(CN)6]4− redox couple in aqueous electrolyte. | ||
Thermodynamically, the temperature dependence of the electrode potential is associated with the reaction entropy of the redox couple, ΔSrc, such that ∂E(T)/∂T = nFΔSrc, where n is the number of electrons involved in the redox reaction and F is Faraday's constant.107 Thus, it is anticipated that the Seebeck coefficient is correlated with the structural changes of the redox species as well as the surrounding solvent, during the redox process.107
As discussed above, many ionic liquids such as [C4mpyr][NTf2] display a wide liquid temperature range. This is a key property prompting their use in thermo-electrochemical cells, allowing access to a wider temperature range of heat sources and producing higher output cell voltages due to the ability to utilise larger temperature differences between the two electrodes. In addition, the devices would have longer service lifetimes as a result of the negligible vapour pressure of the IL. Importantly, in some cases the IL may produce higher Seebeck coefficients via the different solvent environment they represent. Thus an understanding of how the IL influences the reaction entropy for different redox couples is fundamental to this application. Migita and co-workers have reported the Seebeck coefficients for Fe and Cr metal complexes in [C4mpyr][NTf2];108Fig. 7 shows the Seebeck coefficient plotted as a function of the charge density of the redox couple in their work. Among them, [Fe(CN)6]3−/[Fe(CN)6]4− showed the highest Seebeck coefficient, 1.49 mV K−1,108 which is similar to the value in aqueous systems.106 The Seebeck coefficients of [Fe(bpy)3]3+/[Fe(bpy)3]2+ and [Cr(bpy)3]3+/[Cr(bpy)3]2+ (bpy = 2,2′-bipyridine), whose charge densities are close to each other, are similar and both positive. On the other hand, the value and even the sign of the Seebeck coefficients for other redox couples vary widely. From the linear dependence of the Seebeck coefficient on (ZOx2 − ZRed2)/r, where Zox and ZRed are the charge numbers of the oxidized and reduced forms and r is the effective radius of the redox couple, the authors suggest that the electrostatic interaction of the metal complexes with the ions of the IL is a dominant factor determining the Seebeck coefficient.108 A recent report by Yamato et al.,109 who studied a range of Fe, Ru and Ni redox couples in different ILs, has also highlighted the importance of the strength of these interactions and also the polarization and/or steric shape of the ions. Abraham et al.110,111 have recently investigated the Seebeck coefficient of the I3−/I− redox couple in a series of ILs, the results, ranging from 0.03 to 0.26 mV K−1, depending on the nature of both the cation and anion. While the Seebeck coefficients of this redox couple are not high compared to the metal complexes with high charge density, a high output current is produced, potentially due to the very high diffusion rates produced by the Grotthuss-like ion transport of I3−/I−.82
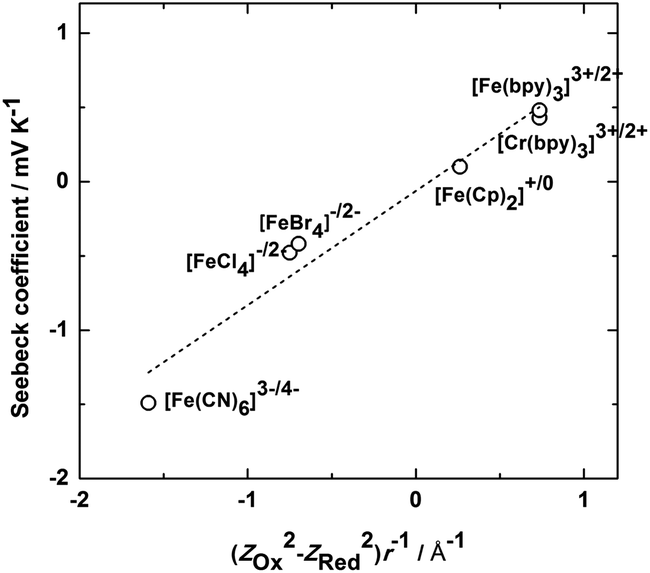 | ||
| Fig. 7 Dependence of the Seebeck coefficients on (ZOx2 − ZRed2)/r for various iron and chromium complexes in [C4mpyr][NTf2].108 | ||
The thermal conductivity of the IL is also a key parameter determining the efficient conversion of thermal to electrical energy in these devices. The reported values of the thermal conductivity of ILs are typically significantly lower than that of water (for example, 0.2 W m−1 K−1 for [C2mim][BF4] compared to 0.6 W m−1 K−1 for water112,113). This is advantageous for efficient thermo-electrochemical cell applications, because materials with a low thermal conductivity suppress thermal energy loss across the cell and allow a larger temperature difference to be maintained between the two electrodes. Further work in understanding the role of ion structure in determining the thermal conductivity would be of significant assistance in this field.
Abraham and co-workers have demonstrated an IL-based thermoelectrochemical cell operating at 70 °C/10 °C for the hot/cold electrodes, respectively.114 The [Fe(CN)6]3−/[Fe(CN)6]4− redox couple dissolved in choline[H2PO4], with various amounts of water, was used as the electrolyte. The electrodes for both the hot and cold side used single-walled carbon nanotubes (SWNTs) to achieve a high surface area. This is the first report of a functioning thermo-electrochemical device using an ionic liquid.114 More recently the same group has described an operating cell based on a novel, high Seebeck coefficient Co(II)/Co(III) redox couple operating in various ILs.115
To fully realise the potential of ionic liquids in thermo-electrochemical applications, future research is desperately needed in a number of areas: (i) exploration of a wider variety of redox couples, to identify those with the largest ΔSrc, (ii) further understanding via theoretical and MD simulations of how the ionic liquid influences this entropy change, (iii) investigation of alternative electrode/electrocatalytic materials to optimise surface kinetics and reduce cost and (iv) further development of prototype devices and thorough investigation of the factors limiting their performance.
6. Supercapacitors
Electrochemical capacitors (ECs) or supercapacitors are electrochemical energy storage devices that deliver high energy for short times – up to a few seconds. Different to the battery systems discussed above, they store the charge by fast surface processes, namely ion adsorption from an electrolyte into either (i) the double layer on the electrode in Electrical Double Layer Capacitors (EDLCs), or (ii) fast surface redox reactions in pseudo-capacitive materials. These systems are already commercialized in numerous applications, where high power delivery is needed for a few seconds, such as in power electronics and in tramways, aircraft, cars and energy harvesting.116 Thanks to their attractive properties of significance to these devices, including high electrochemical stability, thermal stability, melting points below 20 °C and low vapour pressure,117 ILs have attracted much interest in the past decade for use in supercapacitors.22 They were first used in EDLCs as salts in solvent-based electrolytes to produce highly conducting electrolytes,118,119 thanks to the high solubility of ionic liquids in most of the organic solvents used in supercapacitors.120,121 However, despite noticeable improvements, for example as reported by Balducci and Krause,122 the cell voltage is limited by the electrochemical voltage window of the solvent.123 Thus, these incremental improvements have to be put in perspective with the breakthrough that could be generated by ILs in terms of energy density. Energy in a capacitor is related to electrode voltage via E = 1/2CV2 where E is the energy (J), C the capacitance (F) and V the cell voltage (V). Being proportional to V2, significant improvement in the energy density can be achieved by increasing the cell voltage.116 Typically, the cell voltage is mainly limited by the electrochemical stability of the electrolyte used. In aqueous electrolytes, cell voltage cannot go beyond 1.0 V in most cases. In non-aqueous electrolytes, cell voltage reaches 2.7 V thanks to the use of organic solvents like acetonitrile or propylene carbonate; cell voltages up to 2.85 V or 3 V have been reported using acetonitrile. Beyond that, irreversible redox reactions occur resulting in a constant degradation of the cell performance. According to the above equation, moving from 2.7 V to 3.7 V would lead in a 50% energy increase; this is where ILs have a key role to play.Safe operation at elevated temperatures may also be achieved by using IL electrolytes in EC systems, and one of the first demonstrations of this was that of Mastragostino's group.124–126 Using activated carbon as the negative electrode and polymethylthiophene as the positive electrode in a pyrrolidinium-based IL electrolyte, they demonstrated a voltage window up to 3.5 V, resulting in enhanced stability and energy density.125 However, the limited ionic conductivity below room temperature, as well as the high viscosity of these electrolytes constrained the operating temperature of the systems to >50 °C. Similar results were obtained using carbon/carbon systems in neat IL electrolytes.127–129 Despite the large voltage window (3.5 V) and high capacitance retention during cycling, the power capability was strongly affected by a decrease of the operating temperature below 10 °C.127 Another detrimental effect was the limited accessibility of the IL to the small pores of the high surface area activated carbons, resulting in only modest capacitance.
Other strategies are thus needed for exploiting the exceptional properties of IL electrolytes. One approach involves adding a pseudocapacitive contribution like that observed in sulphuric acid electrolytes. Thus, Protic Ionic Liquids (PILs) were investigated. As a result, an important capacitance increase beyond 120 F g−1 has been reported for porous carbons at room temperature, with good low temperature behaviour.130 However, the easy proton transfer narrows the electrochemical voltage windows because of redox reactions, resulting in maximum voltage of about 2 V (see Fig. 8)131 and current work is directed towards the design of stable PILs at high voltage. The control of the water content is also a major challenge.
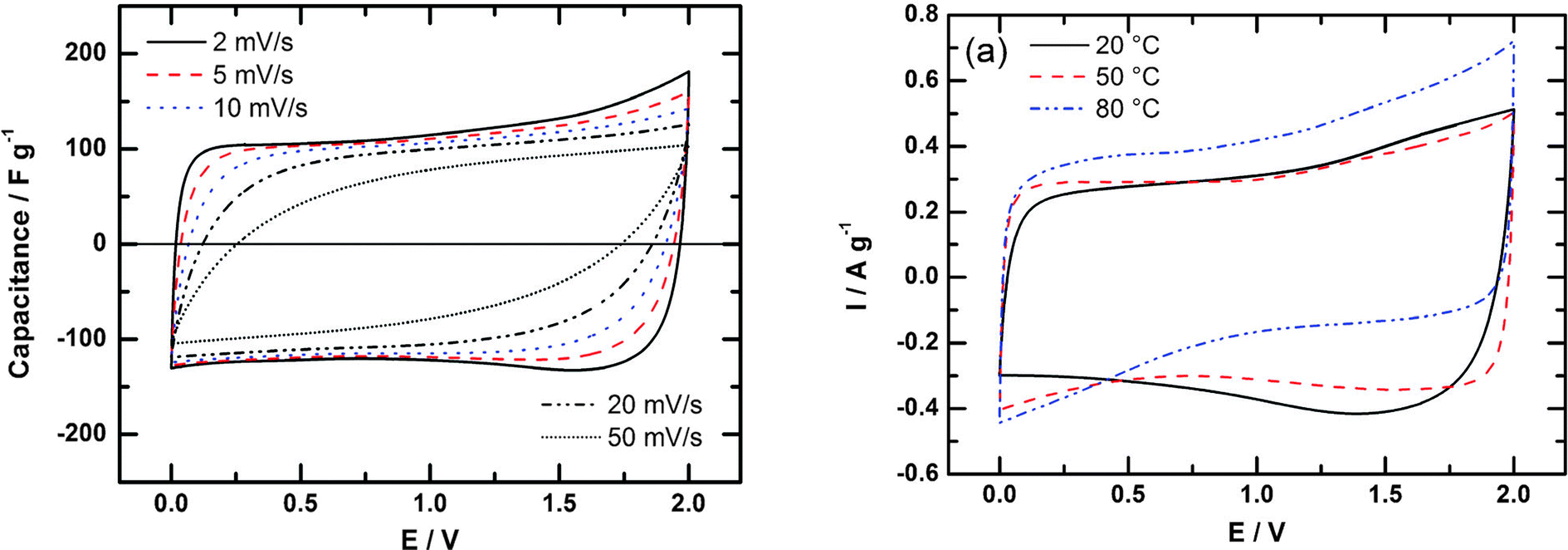 | ||
| Fig. 8 Cyclic voltammograms for a two-electrode cell with activated carbon in [Et3NH][NTf2] at different scan rates, at 20 °C (left) and at different temperatures (right). Reproduced from ref. 131 with permission from the PCCP Owner Societies. | ||
Recently, another approach was proposed to tackle the low conductivity and high viscosity issues of ILs. It was shown that the right combination of nanostructured carbon (nanotubes, onions or graphene) electrodes and eutectic mixture ILs can dramatically extend the temperature range of electrical energy storage, thus defying the conventional wisdom that ILs can only be used as electrolytes above room temperature, Fig. 9.132 Following pioneering work of Passerini's group,133,134 it was possible to prevent an ordered arrangement and crystallization of the IL by selecting a combination of cations with the same anion, thereby inhibiting the formation of the crystalline lattice.132 A eutectic mixture (1:1 by weight or mole ratio) of N-methyl-N-propylpiperidinium[FSI] and [C4mpyr][FSI] was designed accordingly. Using activated graphene as the active material, the operating temperature of a supercapacitor cell was extended to the −50–100 °C range, while the cell voltage was increased up to 3.7 V. Carbon capacitance reached 170 F g−1 at room temperature, still maintaining 100 F g−1 at −50 °C.135 The challenge is now improvement of the electrode areal capacitance (F cm−2).
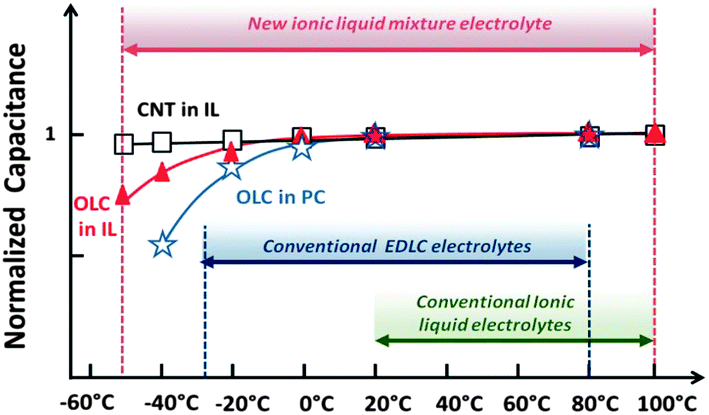 | ||
| Fig. 9 Normalized capacitance ((C/C20 °C) for carbon nanotubes and Onion Like Carbon (OLC) electrodes) in aprotic and in IL mixture electrolytes. The IL mixture extends the temperature range for supercapacitors to −50–100 °C while conventional electrolytes are limited to the −30–80 °C window. Reprinted with permission from ref. 132. Copyright 2011 American Chemical Society. | ||
While ILs are today extensively studied for energy storage devices, they have also been used as model materials/electrolytes by theoreticians for addressing fundamental concerns raised by the capacitance increase and the ion transfer in carbon nanopores.116 Revisiting the double layer charging mechanism, several new models describing the double layer structure in such confined environments have been successfully proposed. Starting from the concept of over-screening proposed by Kornyshev's group at the planar graphite electrode,136,137 Shim and Kim proposed the exclusion of counter ions in small pores.138 Also, several groups reported a capacitance oscillation from constructive superposition of ion densities139–141 or the decrease of the approach distance of the ions to the carbon surface.142 As a result, this field is now expanding and needless to say forthcoming results will be of great help in improving our basic understanding of these fundamental issues, with applications both in, and beyond, the energy storage area.
Research on the design of IL electrolytes for supercapacitor applications has seen a tremendous increase during the past few years, stimulated by growing large scale applications of these devices. Different from battery applications where the electrolyte composition and stability must fit with the requirements of SEI formation, electrochemical kinetics and electrode processes, there is potentially more room for breakthroughs in supercapacitor applications. Designing the electrolyte in conjunction with the nano-structure of the carbon is certainly an important way forward for developing the next generation of high energy supercapacitors that can operate over a large temperature range. The search for new electrolytes based on ILs could also offer an opportunity to standardize the electrochemical tests and methods used in laboratories world-wide for evaluating electrolyte stabilities for supercapacitor applications. Moving from basic constant-current charge–discharge tests at room temperature to more revealing constant potential experiments at elevated temperature (e.g. 60 °C) for hundreds of hours is needed for a proper assessment of the electrolyte stability.143
7. Actuators
The electro-mechanical actuator is an electrochemical device that converts electrical energy into mechanical energy; its electrochemical operation benefits from the same advantages of IL electrolytes over volatile solvent based electrolytes, as all of the other devices discussed above. Electroactive polymer (EAP) actuators are expected to be the next generation of driving parts because they are easily controllable and have unique features such as soft motion, light weight, and ease and flexibility of processing.144–153 The EAP actuators are classified into two groups: one is the electronic EAP actuator and the other is the ionic EAP actuator (Fig. 10).144,145 The electronic EAP is driven by coulombic attraction between two flexible electrodes deposited on both sides of an insulating elastomer membrane.146 The application of a high voltage (>1 kV) generates attractive force between the two electrodes, which collapses the elastomer. On the other hand, the ionic EAP actuators are driven by migration or diffusion of ions in the electrolyte and generally exhibit large deformation at low voltages (<5 V), as seen in conducting polymer,148–150 ionic–polymer metal composite,151,152 and carbon nanotube based variations.153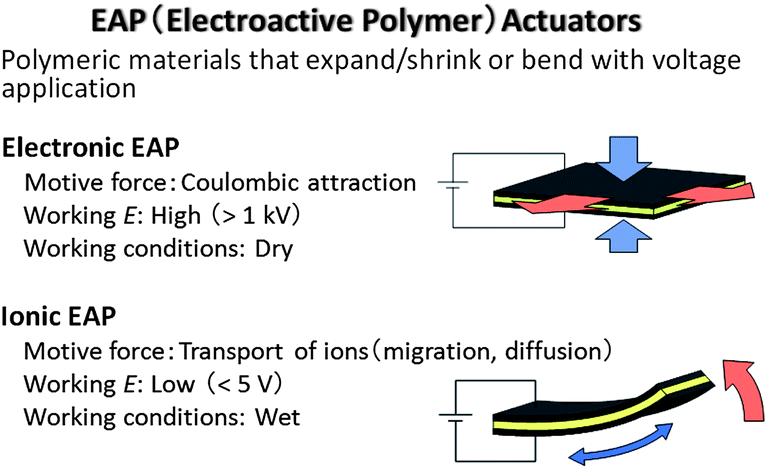 | ||
| Fig. 10 Classification of electroactive polymer (EAP) actuators and their motive force and working voltage and conditions. | ||
However, conventional ionic EAP actuators suffer a drawback in durability under open atmosphere conditions owing to the evaporation of solvents contained in the actuators. Moreover, the narrow potential windows of the solvents limit the applied voltage and frequently induce side reactions, resulting in the deterioration of actuators. Thus ionic liquid electrolytes have become of importance to the success of these devices.154–160 The first study of the use of ILs for ionic EAP actuators was reported on conducting polymer actuators.156 The actuator of this type is driven by doping and undoping of π-conjugated polymers caused by the redox reactions, which induces expansion or contraction of the conducting polymers due to the insertion or expulsion of dopant ions.148–150 Mattes et al. reported156 that conducting polymers are electrochemically cycled (doped and undoped) in ILs with enhanced lifetimes without failure and fast cycle switching speeds.
The conducting polymer actuators involve electrochemical reactions for the actuation, which frequently shorten the life time and lengthen the response time. Ionic EAP actuators, operating on a basis similar to the electric double layer capacitors described in the previous section, appear to offer significant advantages in terms of the simplicity and durability of actuator structure. Ionic–polymer metal composites151,152 and carbon nanotube actuators153 belong to this class. Such actuators generally have a trilaminar structure consisting of a polymer electrolyte membrane sandwiched between electrode layers and exhibit a bending motion (Fig. 10). Nafion™ membranes have been widely used to construct this type of actuator which can be driven in aqueous solutions or under wet conditions. Polymer electrolytes containing ILs exhibit high ionic conductivity under dry conditions and even under reduced pressures,161 and consequently they are quite promising materials in this context. Such polymer electrolytes are easily available by free-radical polymerization (gelation) in the presence of ILs,155,162,163 ionogels formed in ILs from silica networks164 or the sol/gel transition of crystalline fluorinated copolymers in ILs,165,166 and self-organization of amphiphilic block copolymers167–170 and ionomers171,172 in ILs.
Amongst these, the utilization of block copolymers is of great interest since this methodology may have the potential to afford easily processable and mechanically strong polymer electrolytes by utilizing self-assembly of the block copolymers. ABA-triblock copolymers, polystyrene-block-poly(methyl methacrylate)-block-polystyrene (SMS), were synthesized by successive atom-transfer radical polymerizations (Fig. 11).173 Polymer electrolytes consisting of SMS and [C2mim][NTf2] were then self-assembled into a micro phase-separated structure, where polystyrene (PS) is phase-separated to form sphere domains that serve as physical crosslinking points because PS is not compatible with [C2mim][NTf2], while a continuous poly(methyl methacrylate) (PMMA) phase dissolved in [C2mim][NTf2] is formed to serve as the ion conduction path.173 The polymer electrolytes were then used as an electrolyte of an ionic EAP actuator, along with composite carbon electrodes; by applying low voltages (<3.0 V) to the electrodes, the actuator exhibited a soft bending motion.173
 | ||
| Fig. 11 Preparation of polymer electrolytes by self-assembly of ABA-type block copolymer and ionic liquid and application to ionic polymer actuators. Reprinted with permission from ref. 173. Copyright 2012 American Chemical Society. | ||
A simple model of the actuation mechanisms was proposed by taking the difference in ionic mobility (transference number) and ionic size between the cations and anions of the IL into consideration.174 This model indicates that the magnitude of deformation is in proportion to the accumulated charge in the electric double layer and discriminates the behavior of the actuators in terms of the products of transference numbers and ionic volumes.174
Future challenges in this field revolve around the need for faster response times, which rely on high conductivity in the IL. Hydrophobicity of the IL is also a valuable feature that ensures that higher voltage operation is not impaired by water absorption from the atmosphere.
8. Hydrogen generation by water splitting
Ionic liquids have recently made a significant impact on a quite different aspect of electrochemical energy generation and storage – the generation of hydrogen as a fuel from water (also frequently referred to as “water splitting”) by direct solar energy input, or by electrolysis from renewable sources of electricity. Research has focussed on two distinct aspects of this application: (i) the use of (hydrated) ILs as the solvent/electrolyte in the water splitting process and (ii) the use of ILs as the solvent/electrolyte in the electrosynthesis of the electro-catalytic materials that drive the reaction, in particular for the water oxidation reaction. Interestingly it is catalytic materials produced from ILs that have subsequently proven to produce fascinating and surprising outcomes when used in water splitting from an IL electrolyte, so we will begin by discussing these new catalysts here.Water splitting involves the simultaneous oxidation and reduction of water. The four electron water oxidation reaction to oxygen, 2H2O = O2 + 4H+ + 4e−, (also known as the ‘oxygen evolution reaction’, OER) is by far the more sluggish, compared to the water reduction reaction. Typically more than 450 mV overpotential is required to drive the OER at useful rates. This represents a very significant energy loss (as heat) compared to the available energy content of the hydrogen produced (more than one quarter of the total energy input to the reaction). Thus tremendous effort has focussed recently on developing and understanding water oxidation electrocatalysts, to an extent taking some inspiration from Nature since photosynthesis in plants involves a similar OER process and has evolved an oxo-manganese cluster as the catalytic centre for this process. Thus manganese oxide catalysts, MnOx (where x is typically between 1.5 and 2), have been intensively investigated using a variety of synthetic strategies, including classical aqueous electrodeposition. Following on from some original semiconductor electrodeposition studies of Izgorodin,175 Zhou et al.176 investigated the use of ethylammonium nitrate ionic liquid as the electrolyte to carry out the electrosynthesis of MnOx layers at elevated temperatures (100–150 °C). One of the main features of the IL was to enable the use of a reactant amount of water in the mixture at these elevated temperatures without the need for a pressure vessel. Characterisation of the electrodeposit reveals a composition similar to that of the mineral Birnessite in which the ethylammonium cation is entrained in the structure in place of the alkali cation. The electrochemical performance of these catalysts in the OER is extremely good, producing significantly higher currents at lower overpotentials than other known MnOx catalysts (Fig. 12). Extending this work to the other major family of high activity OER electrocatalysts based on earth abundant elements, i.e. the cobalt oxides, showed similar results,177 exhibiting some of the highest OER activity levels ever observed at room temperature and equalling that of the champion precious metal oxides. Exploring the role of the cation in this electrodeposition reaction reveals that among the primary alkylammonium cations, the small ethylammonium cation appears to be close to optimal, possibly because in other cases the increasing hydrophobicity of the alkyl chain at longer chain lengths alters the compatibility of the cation with the oxide structure. Nonetheless there is much yet to learn about the role of the IL cation in these materials and potential for further improvement in their properties.
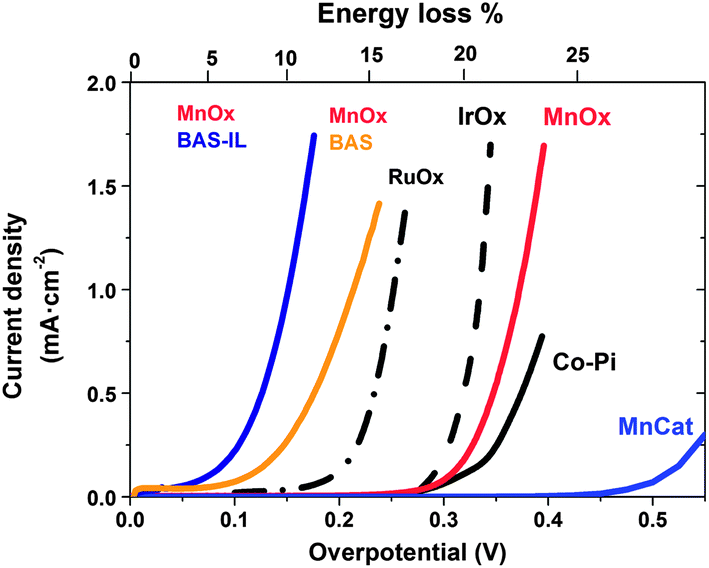 | ||
| Fig. 12 Performance of various oxide electro-catalysts in the water oxidation reaction in 1 M NaOH, except BAS = 0.4 M di(butylammonium) sulfate pH10, BAS-IL = 2 M di(butylammonium) sulfate pH10. The top axis shows the impact of overpotential as an energy loss. | ||
ILs also have the potential to play a role as electrolyte and solvent in the water splitting reaction itself, the reactant water being simply a solute in the reaction mixture. One hypothesis proposes that if dissolving the water molecules into an appropriate IL has the effect of breaking hydrogen bonds and thereby increasing their free energy, then a smaller energy input would be required in the water splitting reaction. The free energy might approach that of gaseous water which requires around 10 kJ mol−1 lower free energy input. Such a process must thermodynamically involve a water dissolution process that is endothermic (the energy of H-bond disruption). In fact, such ionic liquids exist – [C2mim][B(CN)4] is a good example; we are not aware that its ΔH of mixing has ever been quantified and such data are desperately needed, but the endothermic water addition to the IL is very easily observed. However, water electrolysis in this ionic liquid only produced a narrowing of the potential window, from its usual 1.23 V, at elevated temperatures around 150 °C.178
On the other hand protic ionic liquids have emerged as fascinating players in this endeavour. Ground-breaking work by Izgorodin179 showed that when the MnOx electrocatalysts carry out the water oxidation reaction in a protic IL buffer–water mixture, the product is diverted into the two electron oxidation product, hydrogen peroxide, at impressively low over-potentials (150–250 mV); data are shown for the case of basic butyl ammonium sulphate buffer (BAS)–water mixtures in Fig. 12.
Ab initio calculations of possible H-bonded solvation structures of H2O2 in the PIL–buffer showed that the H-bonding environment, Fig. 13, is more stable than would normally be the case in an aqueous electrolyte. This suggest that the H2O2 is solvated into the IL and thereby removed from the electrode where further oxidation might occur. The energy difference calculated was sufficient to explain the shift in redox potential for the H2O/H2O2 couple that would be required to place it at lower potential than the OER on these electrodes.
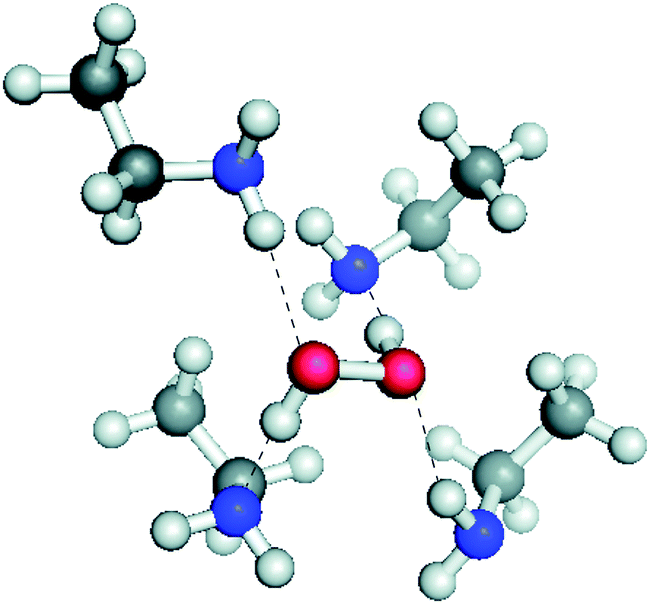 | ||
| Fig. 13 Ab initio calculated structure of the solvation environment around H2O2 in an protic ionic liquid buffer medium. Energy calculations show that this solvation is of lower energy than the analogous solvation in water. Reproduced from ref. 179 with permission from The Royal Society of Chemistry. | ||
That the water oxidation reaction can be carried out at low overpotential is an extremely important step in achieving the goal of an energy efficient route to hydrogen via water splitting since the H2O2 can be decomposed into oxygen in a separate step as shown conceptually in Fig. 14. Investigation of the role of IL structure and the IL buffering action180 of the excess base that is present in these electrolytes can surely produce improvements on these early studies and we commend the attention of the IL electrochemical field to this significant area. It is also important to note that advances in the water oxidation area are also key to the reversible metal–air battery technology discussed above, since the charge reaction in such batteries must inevitably involve a water oxidation step and this is currently one of the major sources of inefficiency in these batteries.
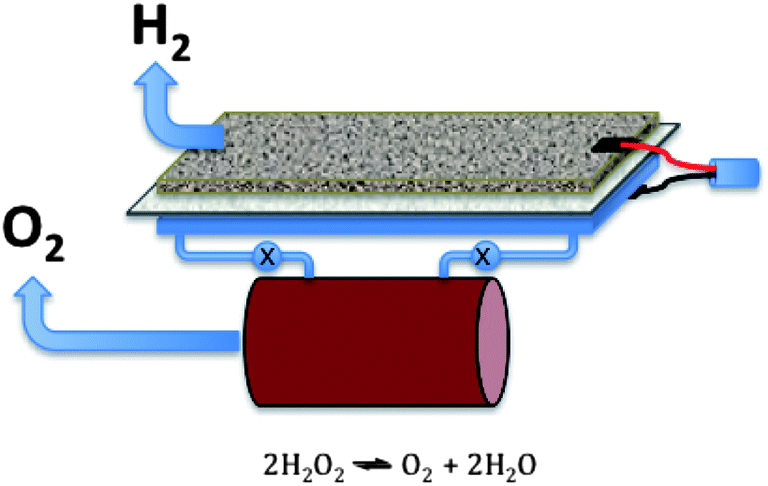 | ||
| Fig. 14 High energy-efficiency water splitting electrolyser concept using a two electron oxidation to H2O2 prior to decomposition to oxygen in a different flow reactor. | ||
9. Thermal storage applications
In the final sections of this article we turn our attention to two recently emerging areas of application of ILs in energy technologies that are not of an electrochemical nature. In both cases the usual advantageous properties of ILs are important, however the function is one stemming from tuneable chemical or phase properties of the liquid. In the first of these we discuss emerging thermal energy transfer and storage applications.The development of solar troughs and solar towers has enabled thermal energy to be concentrated to the extent that solar energy can be used to drive traditional steam cycles, thus providing alternatives to the use of fossil fuels.181 Material advances that are fundamental to the success of solar thermal power generation include the heat transfer fluids that allow for adequate thermal energy absorption and transfer to the power loop, along with thermal storage media to enable a supply of thermal energy when solar radiation levels drop. While concentrated solar thermal loops can be interfaced to traditional high temperature steam power cycles, Organic Rankine Cycles (ORCs) are also an emerging option for lower temperature operation in small scale and remote area applications. These ORCs thus generate interest in thermal energy storage at temperatures in the 75–200 °C range that is not easily achieved with traditional materials. Phase change materials store thermal energy in the latent heat of a phase transition, and materials with a high enthalpy of fusion can absorb (or release) significant amounts of energy in the vicinity of the phase change temperature. ILs and organic salts having melting points above 75 °C clearly have significant potential in this context; their enormously broad range of cation and anion choices offers the ability to design-in both a desirable solid–liquid temperature range and also the necessary thermal properties. However, fundamental data to guide their use in emerging thermal energy technologies remains somewhat limited. The key properties of liquids that are used as a heat transfer and storage media are density, heat capacity, thermal conductivity, phase change temperatures, enthalpy of fusion, ΔHf, thermal decomposition temperature, flash point, and viscosity. Although the range of ILs that has been fully characterized is somewhat narrow, certain design rules for thermal properties are starting to emerge as we discuss below.
One of the desirable features of ILs and organic salts in thermal energy storage is their expanded liquidus range (we expand our vision here to include a broader range of salts that have melting points above 100 °C). The melting point can easily be adjusted by choice of the cation or anion, for example as demonstrated by Terasawa et al.182 Zhu et al.183 have recently reported enthalpy of fusion values for a family of alkylimidazolium bromide ionic liquids, and demonstrated that this value could be systematically increased by lengthening the alkyl chain. The largest ΔHf that they reported, 153 J g−1, was for [C16mim]Br at Tm = 372 K. More recently Vijayaraghavan et al.184 have described a series of protic salts that have ΔHf as high as 190 J g−1 (=260 MJ m−3) which places them amongst the best known phase change materials in this medium temperature range. This ability to tailor the melting transition in this type of organic salt can potentially provide great design flexibility for niche applications. In addition to latent energy storage, which depends on the heat of fusion, sensible energy storage, dependent on the heat capacity (as discussed below), can be beneficial in supporting these thermal energy storage systems.
In heat transfer applications the heat capacity of the fluid is one of the key properties; Paulechka et al. have reviewed heat capacity measurements in ILs recently.185 As expected for such a diverse range of compounds, the molar heat capacity has wide ranging values. For this application, however, the more relevant quantity is specific heat per unit volume, and in these terms a typical IL value is ∼2 J K−1 cm−3, similar to common polyols and hydrocarbons, indicating a possible role for ILs in this context where their thermal stability is a key property.
In both thermal energy storage and transfer, the thermal conductivity of the fluid is of significance. The ILs characterized to date appear to have slightly higher thermal conductivities than those of other organic molecular liquids such as methanol or toluene;186,187 for example values of approximately 0.2 W m−1 K−1 have been measured for [C4mim][BF4] and [C2mim][BF4],115 which is higher than many conventionally used heat transfer fluids. The temperature dependence of thermal conductivity was observed to be minimal over the range studied (300–400 K). Studies conducted on imidazolium compounds show that small structural changes in the IL can significantly impact the thermal conductivity and this effect is worthy of considerable further study. There is also relatively little known about the structure dependence of thermal conductivity in the solid phases of these materials.
10. Carbon dioxide capture and separation
Using ionic liquids to assist in separating CO2 from other gases has become a topic of considerable activity in both academic and industrial spheres. As an approach to capturing CO2, post combustion, from power station flue gases it has become an important goal world-wide to improve the sustainability of the use of coal in power generation. In recent years many publications have appeared dealing with IL–CO2 interactions of one sort or another and readers interested in technical surveys of IL–CO2 research are urged to peruse some of the excellent recent reviews as a starting point for this body of work.188–190 The Davis group was one of the first to report the synthesis of ILs specifically targeted towards the chemical capture of CO2, and to demonstrate their capacity to do so.191 Because this type of CO2 capture involves formation of a carbamate from the CO2 and an amine functionality in the IL (Fig. 15) it can be accomplished at low partial pressures; this is especially attractive for use in the post-combustion situation, which is probably the largest currently conceived end-use of the technology. Indeed, the intervening decade has seen an intense interest in these materials form the research community, as well as a large number of prospective end-users, most of them commercial enterprises.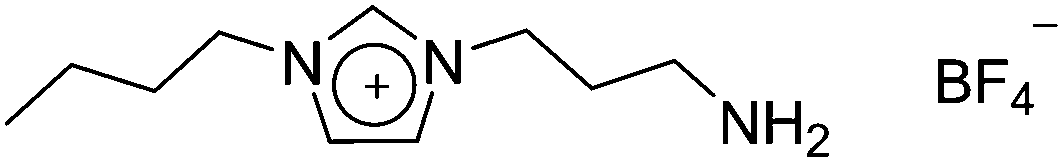 | ||
| Fig. 15 Structure of the first ionic liquid designed and proven capable of reversible, covalent capture of CO2. One mole of this IL captures 0.5 moles of CO2, by way of carbamate salt formation. This mechanism is the same as that by which CO2 capture occurs using monoethanolamine, the industry-standard reagent. | ||
One of the key issues in this application of ILs is the economics of the process. Carbon capture is not a profit center for large generators of CO2 such as electric power utilities, which are being increasingly pressured or even legally mandated to engage in the activity. Indeed, carbon capture will be a net cost (even if the captured CO2 is sold, for example for enhanced oil recovery), and these costs will be passed along to consumers. No matter how well-conceived and well-intended, carbon credits and cap-and-trade schemes of various sorts will not eliminate the costs of carbon capture, they will only shift the burden between the generators, government and the consumer. As a consequence, it is important that any new IL proposed must be as inexpensive to make and use as possible.192 In particular the volumes required to accomplish CO2 capture using ILs of the type in Fig. 15 would, on the scales envisioned, be prohibitively expensive. Indeed, it is quite possible to conceive of a situation where the required quantity of the IL would cost more to buy and use than the cost of fines imposed for failing to capture the CO2. Thus recent research efforts have set out to design and synthesize effective CO2-capturing ILs that were made from the most basic and inexpensive building blocks that be could make to work.193 Variations that tether the functional groups to the anion194 have also been explored. In general, these compounds capture CO2 in higher per-mole quantities than the IL in Fig. 15. Several of the ILs reported have segued now into more advanced stages of evaluation and development, and are even commercially manufactured.195 A representative example of this type is shown in Fig. 16.
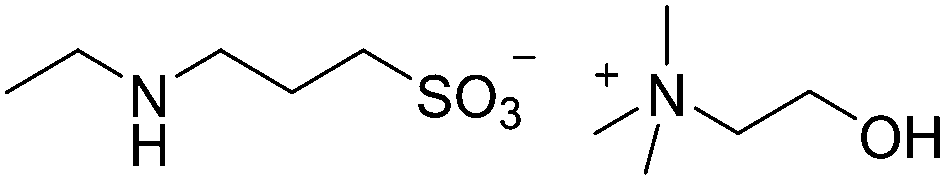 | ||
| Fig. 16 A second-generation CO2-reactive IL. This type of compound is rapidly prepared in a process that uses inexpensive commodity chemicals, has low energy demand, and gives high yields.193 Further, the anions of these compounds belong to an ion class (taurine–homotaurine) that is generally non-toxic, and cations can be chosen (shown is choline) which are similarly likely to have innocuous or at least comparatively tolerable toxicological characteristics. | ||
Clearly, any IL proposed for CO2 capture should be designed with its potential toxicological effects in mind. If the ILs or other components of IL-containing systems contrived to accomplish CO2 capture are themselves easily introduced into the environment (especially in sizeable amounts), then we may end up replacing one environmental issue with another. While the whole CO2-reactive IL effort is rationally rooted in the subversion of the evaporative discharge of the capture material into the environment, other avenues of its introduction – liquid spills, especially – must be conceived of as possible (perhaps even likely given the number of potential point sources at which they may be deployed). We must strive to design ILs that are effective, but as likely as possible to be biologically innocuous.
The energetics of the CO2 capture and release must also be considered carefully. The flue gas stream to be treated may be at temperatures ranging from 70 °C to 120 °C. The IL must bind tightly enough, thermodynamically, to the CO2 to capture it at this temperature. At the same time, the IL must be recycled by removing (and concentrating) the captured CO2. This is most commonly achieved via heating. As a consequence, a perverse dynamic exists in which IL design factors favouring one part of the process (absorption) can disfavour the other (desorption). Walking a thermodynamic tightrope in this regard is unavoidable, and it needs to be done with care as a large part of the cost to industry of the overall carbon-capture process is the energy used to recycle the capture agent. A further confounding factor that is often overlooked in the literature is the role of the ever-present water vapour in this process. Post-combustion gas streams contain about equal amounts of CO2 and water and the capture mechanism must be capable of working under various conditions of water level.
Even in best-case scenarios, the volumes of ILs needed to accomplish flue gas CO2 capture are enormous. Note that the size of the vessels needed to contain them will increase with their volumes, as will the heat required for recycling, and recycle residence times due to heat conduction factors. Accordingly, when it comes to IL design, one needs to think small. There may be a great design in mind for an IL that will capture one mole of CO2 per mole of IL, but if the IL has a molar mass of 1000 amu, the mass and volume of the compound per ton of CO2 absorbed will be prohibitively large.
An alternative approach to the CO2 capture problem is via selective membrane based separation from the flue gases.196,197 A number of ionic liquid containing polymer blends and poly-ionic liquid (also known as polyelectrolyte) materials have been investigated for this purpose.198–200 In this approach the function of the membrane is to selectively absorb and transport CO2 across the membrane and the ionic liquid component can serve as either or both the absorbent or as a transport facilitator. Low volatility is a vital property in this application as loss of IL components from the membrane is ultimately destructive to its function. The IL component offers a particularly useful approach to tuning the selective absorption of CO2 based on the functionality that can be introduced into either or both of the ions, although in this case strong binding is undesirable as it impedes desorption on the other side of the membrane. As for liquid absorbents, water in the gas stream is a feature of the situation and membrane materials need to be able to work in a distinctly high water vapour pressure environment.
Industrial interest in these technologies is clearly strong, and based upon both patent publications and anecdotal indicators, growing. Nonetheless there is considerable further research needed to develop and optimise IL functionality for both liquid and membrane absorption functions, in particular focussed on the economic issues of this very high volume application and delving more deeply into the factors that control the thermodynamics of the absorption/desorption process.
11. Challenges, prospects and opportunities
Because of the almost limitless tunability of ILs, there is enormous scope to explore structure–property relationships that will support the development of enhanced electrochemical and thermal performance. Depending on the property being investigated, changes in the chemical character of the cation and anion (size, charge distribution, metal complexing, lipophilicity, etc.) and the nature of substituted functional groups can have markedly different effects. This offers various different design points to construct ions with a specific subset of properties. A particular property requirement that emerges at many points in the discussion above is “low-cost” and there is therefore a strong need for the development of ILs that are orders of magnitude less expensive than first generation ILs, while still retaining the basic properties that make them attractive in the energy applications.Considerable work is also needed to more deeply characterize the IL families of known interest in the energy sciences. Understanding this relationship between IL structure and the thermochemical and electrochemical properties of these fluids – in particular by applying quantum chemical, molecular dynamics and quantitative structure property relationship methods – has significant potential to support further breakthroughs in energy applications and we strongly recommend the theoretical community to these important computational tasks. Quantitatively understanding the link between molecular structure and electrochemical and transport properties is one of the grand challenges of this field. The challenge becomes even greater when the speciation of electroactive metal ions in the IL is of vital significance, as is almost certainly the case in all of the battery applications discussed here. Further understanding of these speciation equilibria in ILs is desperately needed. In all of the electrochemical applications the interlayer that is formed at the charged electrode plays an important role and this needs to be more thoroughly explored, whether it be a double layer structure or a more complex, chemically distinct layer. However the distinction between those two is possibly less clear-cut than is often assumed and further advances in investigating the compositional details of these more complex interlayers at the nano level would enable significant advances in engineering their structure.
In conclusion, it is our view that ionic liquids have a strong contribution to make in the energy sciences to address some of the most pressing issues of our time. However, much remains to be done and we encourage researchers to take up these challenges with a clear and enduring focus on what is genuinely of benefit in each area.
Acknowledgements
DRM and MF are grateful to the Australian Research Council for Australian Laureate Fellowships and, along with JP and PH, project support through its Discovery and Centre of Excellence schemes. NT acknowledges the support of the JSPS (Japan Society for the Promotion of Science) postdoctoral fellowship for research abroad. MW is grateful for a Grant-in-Aid for Scientific Research in the Priority Area “Science of Ionic Liquids” from the MEXT of Japan.References
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- H. Matsumoto, M. Yanagida, K. Tanimoto, M. Nomura, Y. Kitagawa and Y. Miyazaki, Chem. Lett., 2000, 29, 922–923 CrossRef.
- A. I. Bhatt, I. May, V. A. Volkovich, M. E. Hetherington, B. Lewin, R. C. Thied and N. Ertok, J. Chem. Soc., Dalton Trans., 2002, 4532–4534 RSC.
- P. C. Howlett, D. R. MacFarlane and A. F. Hollenkamp, Electrochem. Solid-State Lett., 2004, 7, A97–A101 CrossRef CAS.
- B. Garcia, S. Lavalle, G. Perron, C. Michot and M. Armand, Electrochim. Acta, 2004, 49, 4583–4588 CrossRef CAS.
- H. Matsumoto, H. Sakaebe, K. Tatsumi, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 160, 1308–1313 CrossRef CAS.
- M. Ishikawa, T. Sugimoto, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 162, 658–662 CrossRef CAS.
- M. Holzapfel, C. Jost and P. Novak, Chem. Commun., 2004, 2098–2099 RSC.
- K. Tsunashima, F. Yonekawa and M. Sugiya, Chem. Lett., 2008, 37, 314–315 CrossRef CAS.
- K. Tsunashima, F. Yonekawa and M. Sugiya, Electrochem. Solid-State Lett., 2009, 12, A54–A57 CrossRef CAS.
- L. Yuan, J. Feng, X. Ai, Y. Cao, S. Chen and H. Yang, Electrochem. Commun., 2006, 8, 610–614 CrossRef CAS.
- V. Baranchugov, E. Markevich, E. Pollak, G. Salitra and D. Aurbach, Electrochem. Commun., 2007, 9, 796–800 CrossRef CAS.
- S. Sivakkumar, D. R. MacFarlane, M. Forsyth and D.-W. Kim, J. Electrochem. Soc., 2007, 154, A834–A838 CrossRef CAS.
- V. Borgel, E. Markevich, D. Aurbach, G. Semrau and M. Schmidt, J. Power Sources, 2009, 189, 331–336 CrossRef CAS.
- L. S. Plashnitsa, E. Kobayashi, S. Okada and J.-i. Yamaki, Electrochim. Acta, 2011, 56, 1344–1351 CrossRef CAS.
- S. Seki, Y. Kobayashi, H. Miyashiro, Y. Ohno, Y. Mita, A. Usami, N. Terada and M. Watanabe, Electrochem. Solid-State Lett., 2005, 8, A577–A578 CrossRef CAS.
- M. Egashira, M. Tanaka-Nakagawa, I. Watanabe, S. Okada and J.-i. Yamaki, J. Power Sources, 2006, 160, 1387–1390 CrossRef CAS.
- T. Sato, T. Maruo, S. Marukane and K. Takagi, J. Power Sources, 2004, 138, 253–261 CrossRef CAS.
- G. H. Lane, A. S. Best, D. R. MacFarlane, M. Forsyth, P. M. Bayley and A. F. Hollenkamp, Electrochim. Acta, 2010, 55, 8947–8952 CrossRef CAS.
- P. M. Bayley, G. H. Lane, N. M. Rocher, B. R. Clare, A. S. Best, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2009, 11, 7202–7208 RSC.
- M. Yoshizawa-Fujita, D. R. MacFarlane, P. C. Howlett and M. Forsyth, Electrochem. Commun., 2006, 8, 445–449 CrossRef CAS.
- D. R. Macfarlane, M. Forsyth, P. C. Howlett, J. M. Pringle, J. Sun, G. Annat, W. Neil and E. I. Izgorodina, Acc. Chem. Res., 2007, 40, 1165–1173 CrossRef CAS PubMed.
- D. R. MacFarlane, J. M. Pringle, P. C. Howlett and M. Forsyth, Phys. Chem. Chem. Phys., 2010, 12, 1659–1669 RSC.
- G. H. Lane, Electrochim. Acta, 2012, 83, 513–528 CrossRef CAS.
- N. Byrne, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Adv. Mater., 2005, 17, 2497–2501 CrossRef CAS.
- G.-T. Kim, S. Jeong, M.-Z. Xue, A. Balducci, M. Winter, S. Passerini, F. Alessandrini and G. Appetecchi, J. Power Sources, 2012, 199, 239–246 CrossRef CAS.
- H. Yoon, G. Lane, Y. Shekibi, P. Howlett, M. Forsyth, A. Best and D. MacFarlane, Energy Environ. Sci., 2013, 6, 979–986 CAS.
- A. Balducci, S. Jeong, G. Kim, S. Passerini, M. Winter, M. Schmuck, G. Appetecchi, R. Marcilla, D. Mecerreyes and V. Barsukov, J. Power Sources, 2011, 196, 9719–9730 CrossRef CAS.
- R. Vijayaraghavan, M. Surianarayanan, V. Armel, D. MacFarlane and V. Sridhar, Chem. Commun., 2009, 6297–6299 RSC.
- Y. Wang, K. Zaghib, A. Guerfi, F. F. Bazito, R. M. Torresi and J. Dahn, Electrochim. Acta, 2007, 52, 6346–6352 CrossRef CAS.
- A. Guerfi, M. Dontigny, P. Charest, M. Petitclerc, M. Lagac, A. Vijh and K. Zaghib, J. Power Sources, 2010, 195, 845–852 CrossRef CAS.
- J. A. Choi, E.-G. Shim, B. Scrosati and D.-W. Kim, Bull. Korean Chem. Soc., 2010, 31, 3190–3194 CrossRef CAS.
- L. Lombardo, S. Brutti, M. A. Navarra, S. Panero and P. Reale, J. Power Sources, 2013, 227, 8–14 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- F. Mizuno, S. Nakanishi, A. Shirasawa, K. Takechi, T. Shiga, H. Nishikoori and H. Iba, Electrochemistry, 2011, 79, 876–881 CrossRef CAS.
- S. Higashi, Y. Kato, K. Takechi, H. Nakamoto, F. Mizuno, H. Nishikoori, H. Iba and T. Asaoka, J. Power Sources, 2013, 240, 14–17 CrossRef CAS.
- C. J. Allen, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. Abraham, J. Phys. Chem. Lett., 2011, 2, 2420–2424 CrossRef CAS.
- C. Pozo-Gonzalo, A. A. Torriero, M. Forsyth, D. R. MacFarlane and P. C. Howlett, J. Phys. Chem. Lett., 2013, 4, 1834–1837 CrossRef CAS PubMed.
- T. Zhang and H. Zhou, Angew. Chem., 2012, 124, 11224–11229 CrossRef.
- N. Recham, J.-N. Chotard, J.-C. Jumas, L. Laffont, M. Armand and J.-M. Tarascon, Chem. Mater., 2009, 22, 1142–1148 CrossRef.
- N. Recham, J.-N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand and J.-M. Tarascon, Nat. Mater., 2009, 9, 68–74 CrossRef PubMed.
- J.-M. Tarascon, N. Recham, M. Armand, J.-N. Chotard, P. Barpanda, W. Walker and L. Dupont, Chem. Mater., 2009, 22, 724–739 CrossRef.
- V. A. Nikitina, A. Nazet, T. Sonnleitner and R. Buchner, J. Chem. Eng. Data, 2012, 57, 3019–3025 CrossRef CAS.
- M. Egashira, T. Tanaka, N. Yoshimoto and M. Morita, Electrochemistry, 2012, 80, 755–758 CrossRef CAS.
- S. A. M. Noor, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2013 Search PubMed , submitted.
- T. Yamamoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2012, 217, 479–484 CrossRef CAS.
- T. Nohira, T. Ishibashi and R. Hagiwara, J. Power Sources, 2012, 205, 506–509 CrossRef CAS.
- T. Khoo, P. C. Howlett, M. Tsagouria, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2011, 58, 583–588 CrossRef CAS.
- T. Khoo, A. Somers, A. A. J. Torriero, D. R. MacFarlane, P. C. Howlett and M. Forsyth, Electrochim. Acta, 2013, 87, 701–708 CrossRef CAS.
- T. Kakibe, J. Y. Hishii, N. Yoshimoto, M. Egashira and M. Morita, J. Power Sources, 2012, 203, 195–200 CrossRef CAS.
- N. Yoshimoto, M. Matsumoto, M. Egashia and M. Morita, J. Power Sources, 2010, 195, 2096–2098 CrossRef CAS.
- P. Wang, Y. NuLi, J. Yang and Z. Feng, Surf. Coat. Technol., 2006, 201, 3783–3787 CrossRef CAS.
- N. Birbilis, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Surf. Coat. Technol., 2007, 201, 4496–4504 CrossRef CAS.
- M. Forsyth, P. C. Howlett, S. K. Tan, D. R. MacFarlane and N. Birbilis, Electrochem. Solid-State Lett., 2006, 9, B52–B55 CrossRef CAS.
- T. E. Sutto and T. T. Duncan, Electrochim. Acta, 2012, 80, 413–417 CrossRef CAS.
- T. E. Sutto and T. T. Duncan, Electrochim. Acta, 2012, 79, 170–174 CrossRef CAS.
- E. E. Switzer, R. Zeller, Q. Chen, K. Sieradzki, D. A. Buttry and C. Friesen, J. Phys. Chem. C, 2013, 117, 8683–8690 CAS.
- C. e. a. Pozo-Gonzalo, Electrochim. Acta, 2013 Search PubMed , submitted.
- T. J. Simons, P. C. Howlett, A. A. J. Torriero, D. R. MacFarlane and M. Forsyth, J. Phys. Chem. C, 2013, 117, 2662–2669 CAS.
- T. J. Simons, A. A. J. Torriero, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochem. Commun., 2012, 18, 119–122 CrossRef CAS.
- M. J. Deng, P. C. Lin, J. K. Chang, J. M. Chen and K. T. Lu, Electrochim. Acta, 2011, 56, 6071–6077 CrossRef CAS.
- M. Kar, B. Winther-Jensen, M. Forsyth and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2013, 15, 7191–7197 RSC.
- N. M. Rocher, E. I. Izgorodina, T. Ruther, M. Forsyth, D. R. MacFarlane, T. Rodopoulos, M. D. Horne and A. M. Bond, Chem.–Eur. J., 2009, 15, 3435–3447 CrossRef CAS PubMed.
- K. D. Kreuer, A. Fuchs, M. Ise, M. Spaeth and J. Maier, Electrochim. Acta, 1998, 43, 1281–1288 CrossRef CAS.
- J. Z. Sun, L. R. Jordan, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2001, 46, 1703–1708 CrossRef CAS.
- M. Yoshizawa, W. Ogihara and H. Ohno, Electrochem. Solid-State Lett., 2001, 4, E25–E27 CrossRef CAS.
- A. Noda, A. B. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS.
- M. Susan, A. Noda, S. Mitsushima and M. Watanabe, Chem. Commun., 2003, 938–939 RSC.
- C. A. Angell, W. Xu, J.-P. Belieres and M. Yoshizawa, U.S. Patent WO2004114445A1, 2004.
- J. P. Belieres, D. Gervasio and C. A. Angell, Chem. Commun., 2006, 4799–4801 RSC.
- M. S. Miran, T. Yasuda, M. A. B. H. Susan, K. Dokko and M. Watanabe, RSC Adv., 2013, 3, 4141 RSC.
- S.-Y. Lee, A. Ogawa, M. Kanno, H. Nakamoto, T. Yasuda and M. Watanabe, J. Am. Chem. Soc., 2010, 132, 9764–9773 CrossRef CAS PubMed.
- T. Yasuda, S. Nakamura, Y. Honda, K. Kinugawa, S.-Y. Lee and M. Watanabe, ACS Appl. Mater. Interfaces, 2012, 4, 1783–1790 CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2013, 21, 1–11 Search PubMed.
- Y. Bai, Y. Cao, J. Zhang, M. Wang, R. Li, P. Wang, S. M. Zakeeruddin and M. Grätzel, Nat. Mater., 2008, 7, 626–630 CrossRef CAS PubMed.
- R. Harikisun and H. Desilvestro, Sol. Energy, 2011, 85, 1179–1188 CrossRef CAS.
- J. M. Pringle and V. Armel, Int. Rev. Phys. Chem., 2011, 30, 371–407 CrossRef CAS.
- S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2009, 19, 2187–2202 CrossRef CAS.
- Y. Bai, J. Zhang, Y. Wang, M. Zhang and P. Wang, Langmuir, 2011, 27, 4749–4755 CrossRef CAS PubMed.
- M. Zhang, J. Zhang, Y. Bai, Y. Wang, M. Su and P. Wang, Phys. Chem. Chem. Phys., 2011, 13, 3788–3794 RSC.
- R. Kawano and M. Watanabe, Chem. Commun., 2003, 330–331 RSC.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- M. Wang, C. Grätzel, S. M. Zakeeruddin and M. Grätzel, Energy Environ. Sci., 2012, 5, 9394–9405 CAS.
- H. Tian and L. Sun, J. Mater. Chem., 2011, 21, 10592–10601 RSC.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- M. K. Kashif, J. C. Axelson, N. W. Duffy, C. M. Forsyth, C. J. Chang, J. R. Long, L. Spiccia and U. Bach, J. Am. Chem. Soc., 2012, 134, 16646–16653 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- M. Gorlov and L. Kloo, Dalton Trans., 2008, 2655–2666 RSC.
- V. Armel, J. M. Pringle, M. Forsyth, D. R. MacFarlane, D. L. Officer and P. Wagner, Chem. Commun., 2010, 46, 3146–3148 RSC.
- G. Hashmi, K. Miettunen, T. Peltola, J. Halme, I. Asghar, K. Aitola, M. Toivola and P. Lund, Renewable Sustainable Energy Rev., 2011, 15, 3717–3732 CrossRef CAS.
- M. Ikegami, J. Suzuki, K. Teshima, M. Kawaraya and T. Miyasaka, Sol. Energy Mater. Sol. Cells, 2009, 93, 836–839 CrossRef CAS.
- M. Wu, X. Lin, Y. Wang, L. Wang, W. Guo, D. Qi, X. Peng, A. Hagfeldt, M. Grätzel and T. Ma, J. Am. Chem. Soc., 2012, 134, 3419–3428 CrossRef CAS PubMed.
- M. Wu and T. Ma, ChemSusChem, 2012, 5, 1343–1357 CrossRef CAS PubMed.
- M. Wang, A. M. Anghel, B. Marsan, N.-L. Cevey Ha, N. Pootrakulchote, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2009, 131, 15976–15977 CrossRef CAS PubMed.
- L. Kavan, J.-H. Yum and M. Grätzel, ACS Appl. Mater. Interfaces, 2012, 4, 6999–7006 CAS.
- Y. Saito, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Photochem. Photobiol., A, 2004, 164, 153–157 CrossRef CAS.
- J. M. Pringle, V. Armel, M. Forsyth and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 348–352 CrossRef CAS.
- V. Armel, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2011, 4, 2234–2239 CAS.
- Q. Li, J. Zhao, B. Sun, B. Lin, L. Qiu, Y. Zhang, X. Chen, J. Lu and F. Yan, Adv. Mater., 2012, 24, 945–950 CrossRef CAS PubMed.
- K. Fredin, M. Gorlov, H. Pettersson, A. Hagfeldt, L. Kloo and G. Boschloo, J. Phys. Chem. C, 2007, 111, 13261–13266 CAS.
- S. Ito and K. Takahashi, Int. J. Photoenergy, 2012, 915352 Search PubMed.
- X. Sun, T. Chen, Z. Yang and H. Peng, Acc. Chem. Res., 2013, 46, 539–549 CrossRef CAS PubMed.
- Y. Fu, M. Peng, Z. Lv, X. Cai, S. Hou, H. Wu, X. Yu, H. Kafafy and D. Zou, Nano Energy, 2013, 2, 537–544 CrossRef CAS.
- T. I. Quickenden and Y. Mua, J. Electrochem. Soc., 1995, 142, 3985–3994 CrossRef CAS.
- R. C. Hu, B. A. Cola, N. Haram, J. N. Barisci, S. Lee, S. Stoughton, G. Wallace, C. Too, M. Thomas, A. Gestos, M. E. dela Cruz, J. P. Ferraris, A. A. Zakhidov and R. H. Baughman, Nano Lett., 2010, 10, 838–846 CrossRef CAS PubMed.
- J. T. Hupp and M. J. Weaver, Inorg. Chem., 1984, 23, 3639–3644 CrossRef CAS.
- T. Migita, N. Tachikawa, Y. Katayama and T. Miura, Electrochemistry, 2009, 77, 639–641 CrossRef CAS.
- Y. Yamato, Y. Katayama and T. Miura, J. Electrochem. Soc., 2013, 160, H309–H314 CrossRef CAS.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Chem. Commun., 2011, 47, 6260–6262 RSC.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Electrochim. Acta, 2013 Search PubMed , accepted.
- M. E. Van Valkenburg, R. L. Vaughn, M. Williams and J. S. Wilkes, Thermochim. Acta, 2005, 425, 181–188 CrossRef CAS.
- C. L. Yaws, Yaw's Handbook of Thermodynamic and Physical Properties of Chemical Compounds, Knovel, Norwich, NY, 2003 Search PubMed.
- T. J. Abraham, D. R. MacFarlane, R. H. Baughman, N. Li, Y. Chen and J. M. Pringle, MRS Online Proc. Libr., 2013, 1575 DOI:10.1557/opl.2013.647.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2013 Search PubMed , in press.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- A. B. McEwen, H. L. Ngo, K. LeCompte and J. L. Goldman, J. Electrochem. Soc., 1999, 146, 1687–1695 CrossRef CAS.
- I. Murayama, N. Yoshimoto, M. Egashira, M. Morita, Y. Kobayashi and M. Ishikawa, Electrochemistry, 2005, 73, 600–602 CAS.
- K. Chiba, T. Ueda and H. Yamamoto, Electrochemistry, 2007, 75, 668–671 CrossRef CAS.
- M. Ue, K. Ida and S. Mori, J. Electrochem. Soc., 1994, 141, 2989–2996 CrossRef CAS.
- A. Krause and A. Balducci, Electrochem. Commun., 2011, 13, 814–817 CrossRef CAS.
- R. Palm, H. Kurig, K. Tonurist, A. Jaenes and E. Lust, Electrochim. Acta, 2012, 85, 139–144 CrossRef CAS.
- A. Balducci, U. Bardi, S. Caporali, M. Mastragostino and F. Soavi, Electrochem. Commun., 2004, 6, 566–570 CrossRef CAS.
- A. Balducci, W. A. Henderson, M. Mastragostino, S. Passerini, P. Simon and F. Soavi, Electrochim. Acta, 2005, 50, 2233–2237 CrossRef CAS.
- A. Balducci, F. Soavi and M. Mastragostino, Appl. Phys. A: Mater. Sci. Process., 2006, 82, 627–632 CrossRef CAS.
- A. Balducci, R. Dugas, P. L. Taberna, P. Simon, D. Plee, M. Mastragostino and S. Passerini, J. Power Sources, 2007, 165, 922–927 CrossRef CAS.
- N. Handa, T. Sugimoto, M. Yamagata, M. Kikuta, M. Kono and M. Ishikawa, J. Power Sources, 2008, 185, 1585–1588 CrossRef CAS.
- C. Largeot, P. L. Taberna, Y. Gogotsi and P. Simon, Electrochem. Solid-State Lett., 2011, 14, A174–A176 CrossRef CAS.
- R. Mysyk, E. Raymundo-Pinero, M. Anouti, D. Lemordant and F. Beguin, Electrochem. Commun., 2010, 12, 414–417 CrossRef CAS.
- L. Timperman, P. Skowron, A. Boisset, H. Galiano, D. Lemordant, E. Frackowiak, F. Beguin and M. Anouti, Phys. Chem. Chem. Phys., 2012, 14, 8199–8207 RSC.
- R. Lin, P.-L. Taberna, S. Fantini, V. Presser, C. R. Perez, F. Malbosc, N. L. Rupesinghe, K. B. K. Teo, Y. Gogotsi and P. Simon, J. Phys. Chem. Lett., 2011, 2, 2396–2401 CrossRef CAS.
- M. Kunze, S. Jeong, E. Paillard, M. Winter and S. Passerini, J. Phys. Chem. C, 2010, 114, 12364–12369 CAS.
- M. Kunze, M. Montanino, G. B. Appetecchi, S. Jeong, M. Schoenhoff, M. Winter and S. Passerini, J. Phys. Chem. A, 2010, 114, 1776–1782 CrossRef CAS PubMed.
- W.-Y. Tsai, R. Lin, S. Murali, L. L. i. Zhang, J. K. McDonough, R. S. Ruoff, P.-L. Taberna, Y. Gogotsi and P. Simon, Nano Energy, 2013, 2, 403–411 CrossRef CAS.
- M. V. Fedorov and A. A. Kornyshev, Electrochim. Acta, 2008, 53, 6835–6840 CrossRef CAS.
- M. Z. Bazant, B. D. Storey and A. A. Kornyshev, Phys. Rev. Lett., 2011, 106, 046102-1–046102-4 CrossRef.
- Y. Shim and H. J. Kim, ACS Nano, 2010, 4, 2345–2355 CrossRef CAS PubMed.
- G. Feng and P. T. Cummings, J. Phys. Chem. Lett., 2011, 2, 2859–2864 CrossRef CAS.
- D.-e. Jiang, Z. Jin and J. Wu, Nano Lett., 2011, 11, 5373–5377 CrossRef CAS PubMed.
- P. Wu, J. Huang, V. Meunier, B. G. Sumpter and R. Qiao, ACS Nano, 2011, 5, 9044–9051 CrossRef CAS PubMed.
- C. Merlet, B. Rotenberg, P. A. Madden, P.-L. Taberna, P. Simon, Y. Gogotsi and M. Salanne, Nat. Mater., 2012, 11, 306–310 CrossRef CAS PubMed.
- A. Foelske-Schmitz and R. Koetz, J. Power Sources, 2013, 225, 84–88 CrossRef.
- B. Scrosati, Applications of electroactive polymers, Chapman & Hall, 1993 Search PubMed.
- Y. Bar-Cohen, Electroactive Polymer (EAP) Actuators as Artificial Muscles: Reality, Potential, and Challenges, Society of Photo Optical, 2004 Search PubMed.
- R. Pelrine, R. Kornbluh, Q. B. Pei and J. Joseph, Science, 2000, 287, 836–839 CrossRef CAS PubMed.
- Y. Osada and A. Matsuda, Nature, 1995, 376, 219 CrossRef CAS PubMed.
- K. Kaneto, M. Kaneko, Y. Min and A. G. MacDiarmid, Synth. Met., 1995, 71, 2211–2212 CrossRef CAS.
- R. H. Baughman, Synth. Met., 1996, 78, 339–353 CrossRef CAS.
- E. Smela, Adv. Mater., 2003, 15, 481–494 CrossRef CAS.
- K. Asaka, K. Oguro, Y. Nishimura, M. Mizuhata and H. Takenaka, Polym. J., 1995, 27, 436–440 CrossRef CAS.
- M. Shahinpoor, Electrochim. Acta, 2003, 48, 2343–2353 CrossRef CAS.
- R. H. Baughman, C. Cui, A. A. Zakhidov, Z. Iqbal, J. N. Barisci, G. M. Spinks, G. G. Wallace, A. Mazzoldi, D. De Rossi, A. G. Rinzler, O. Jaschinski, S. Roth and M. Kertesz, Science, 1999, 284, 1340–1344 CrossRef CAS PubMed.
- American Chemical Society. Division of Polymer, Ionic Liquids In Polymer Systems: Solvents, Additives, And Novel Applications, C. S. Brazel and R. D. Rogers, American Chemical Society [by] Oxford University Press, 2005 Search PubMed.
- M. A. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976–4983 CrossRef CAS PubMed.
- W. Lu, A. G. Fadeev, B. Qi, E. Smela, B. R. Mattes, J. Ding, G. M. Spinks, J. Mazurkiewicz, D. Zhou, G. G. Wallace, D. R. MacFarlane, S. A. Forsyth and M. Forsyth, Science, 2002, 297, 983–987 CrossRef CAS PubMed.
- M. D. Bennett and D. J. Leo, Sens. Actuators, A, 2004, 115, 79–90 CrossRef CAS.
- B. J. Akle, M. D. Bennett and D. J. Leo, Sens. Actuators, A, 2006, 126, 173–181 CrossRef CAS.
- T. Fukushima, K. Asaka, A. Kosaka and T. Aida, Angew. Chem., Int. Ed., 2005, 44, 2410–2413 CrossRef CAS PubMed.
- K. Mukai, K. Asaka, K. Kiyohara, T. Sugino, I. Takeuchi, T. Fukushima and T. Aida, Electrochim. Acta, 2008, 53, 5555–5562 CrossRef CAS.
- M. Watanabe, S.-I. Yamada, K. Sanui and N. Ogata, J. Chem. Soc., Chem. Commun., 1993, 929–931 RSC.
- A. Noda and M. Watanabe, Electrochim. Acta, 2000, 45, 1265–1270 CrossRef CAS.
- S. Seki, M. A. B. H. Susan, T. Kaneko, H. Tokuda, A. Noda and M. Watanabe, J. Phys. Chem. B, 2005, 109, 3886–3892 CrossRef CAS PubMed.
- S. A. M. Noor, P. M. Bayley, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2013, 91, 219–226 CrossRef CAS.
- R. T. Carlin and J. Fuller, Chem. Commun., 1997, 1345–1346 RSC.
- J. Fuller, A. C. Breda and R. T. Carlin, J. Electroanal. Chem., 1998, 459, 29–34 CrossRef CAS.
- Y. He and T. P. Lodge, Macromolecules, 2007, 41, 167–174 CrossRef.
- R. Gao, D. Wang, J. R. Heflin and T. E. Long, J. Mater. Chem., 2012, 22, 13473–13476 RSC.
- M. D. Green, D. Wang, S. T. Hemp, J.-H. Choi, K. I. Winey, J. R. Heflin and T. E. Long, Polymer, 2012, 53, 3677–3686 CrossRef CAS.
- T. Wu, D. Wang, M. Zhang, J. R. Heflin, R. B. Moore and T. E. Long, ACS Appl. Mater. Interfaces, 2012, 4, 6552–6559 CAS.
- H. Gokhan, L. Yang, Z. Ran, Y. Mitra, M. T. Dean, T. Srinivas and Q. M. Zhang, Smart Mater. Struct., 2012, 21, 055015 CrossRef.
- S. Imaizumi, Y. Ohtsuki, T. Yasuda, H. Kokubo and M. Watanabe, ACS Appl. Mater. Interfaces, 2013, 5 , 6307–6315 CAS.
- S. Imaizumi, H. Kokubo and M. Watanabe, Macromolecules, 2011, 45, 401–409 CrossRef.
- S. Imaizumi, Y. Kato, H. Kokubo and M. Watanabe, J. Phys. Chem. B, 2012, 116, 5080–5089 CrossRef CAS PubMed.
- A. Izgorodin, O. Winther-Jensen, B. Winther-Jensen and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2009, 11, 8532–8537 RSC.
- F. L. Zhou, A. Izgorodin, R. K. Hocking, L. Spiccia and D. R. MacFarlane, Adv. Energy Mater., 2012, 2, 1013–1021 CrossRef CAS.
- A. Izgorodin and D. Macfarlane, Patent AU2012905597, 2012.
- A. Chong and et al., 2013, to be published.
- A. Izgorodin, E. Izgorodina and D. R. MacFarlane, Energy Environ. Sci., 2012, 5, 9496–9501 CAS.
- D. R. MacFarlane, R. Vijayaraghavan, H. N. Ha, A. Izgorodin, K. D. Weaver and G. D. Elliott, Chem. Commun., 2010, 46, 7703–7705 RSC.
- K. M. Powell and T. F. Edgar, Chem. Eng. Sci., 2012, 71, 138–145 CrossRef CAS.
- N. Terasawa, S. Tsuzuki, T. Umecky, Y. Saito and H. Matsumoto, Chem. Commun., 2010, 46, 1730–1732 RSC.
- J. Zhu, L. Bai, B. Chen and W. Fei, Chem. Eng. J., 2009, 147, 58–62 CrossRef CAS.
- R. Vijayaraghavan, U. Rana, E. G. D and M. D. R, Energy Technol., 2013 DOI:10.1002/ente.201300101.
- Y. U. Paulechka, J. Phys. Chem. Ref. Data, 2010, 39, 033108–033123 CrossRef.
- R. Ge, C. Hardacre, P. Nancarrow and D. W. Rooney, J. Chem. Eng., 2007, 52, 1819–1823 CAS.
- D. Tomida, S. Kenmochi, T. Tsukada and K. Qiao, Int. J. Thermophys., 2007, 28, 1147–1160 CrossRef CAS.
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- L. Zhang, J. Chen, J. X. Lv, S. F. Wang and Y. Cui, Asian J. Chem., 2013, 25, 2355–2358 CAS.
- X. P. Zhang, X. C. Zhang, H. F. Dong, Z. J. Zhao, S. J. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 CAS.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- J. H. Davis, Abstracts of Papers of the American Chemical Society, 2003, 225, U952–U953 Search PubMed.
- M. D. Soutullo, C. I. Odom, B. F. Wicker, C. N. Henderson, A. C. Stenson and J. H. Davis, Jr., Chem. Mater., 2007, 19, 3581–3583 CrossRef CAS.
- (a) B. F. Goodrich, J. C. de la Fuente, B. E. Gurkan, D. J. Zadigian, E. A. Price, Y. Huang and J. F. Brennecke, Ind. Eng. Chem. Res., 2011, 50, 111–118 CrossRef CAS; (b) B. F. Goodrich, J. C. de la Fuente, B. E. Gurkan, Z. K. Lopez, E. A. Price, Y. Huang and J. F. Brennecke, J. Phys. Chem. B, 2011, 115, 9140–9150 CrossRef CAS PubMed.
- I. Task-specific ILs with amine-functionalized cations have been commercialized for sale by Iolitec GmbH; TSILs with amine-functionalized anions have been commercialized for sale by Frontier Scientific.
- S. Kasahara, E. Kamio, T. Ishigami and H. Matsuyama, J. Membr. Sci., 2012, 415, 168–175 CrossRef.
- D. L. Gin and R. D. Noble, Science, 2011, 332, 674–676 CrossRef CAS PubMed.
- T. K. Carlisle, E. F. Wiesenauer, G. D. Nicodemus, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2013, 52, 1023–1032 CrossRef CAS.
- W. J. Fang, Z. L. Luo and J. W. Jiang, Phys. Chem. Chem. Phys., 2013, 15, 651–658 RSC.
- K. Friess, J. C. Jansen, F. Bazzarelli, P. Izak, V. Jarmarova, M. Kacirkova, J. Schauer, G. Clarizia and P. Bernardo, J. Membr. Sci., 2012, 415, 801–809 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |