A super-efficient cobalt catalyst for electrochemical hydrogen production from neutral water with 80 mV overpotential†
Lin
Chen
a,
Mei
Wang
*a,
Kai
Han
a,
Peili
Zhang
a,
Frederic
Gloaguen
b and
Licheng
Sun
*ac
aState Key Laboratory of Fine Chemicals, DUT-KTH Joint Education and Research Centre on Molecular Devices, Dalian University of Technology (DUT), Dalian, 116024, China. E-mail: symbueno@dlut.edu.cn
bUMR 6521, CNRS, Université de Bretagne Occidentale, CS 93387, 29238 Brest, France
cDepartment of Chemistry, KTH Royal Institute of Technology, Stockholm, 10044, Sweden. E-mail: lichengs@kth.se
First published on 22nd October 2013
Abstract
Self-assembled molecular iron and cobalt catalysts (MP4N2, M = Fe, Co) bearing a multihydroxy-functionalized tetraphosphine ligand electrocatalyze H2 generation from neutral water on a mercury electrode at −1.03 and −0.50 V vs. NHE, respectively. Complex CoP4N2 displays extremely low overpotential (Eonset = 80 mV) while maintaining high activity and good stability. Bulk electrolysis of CoP4N2 in a neutral phosphate buffer solution at −1.0 V vs. NHE produced 9.24 × 104 mol H2 per mol cat. over 20 h, with a Faradaic efficiency close to 100% and without apparent deactivation.
Broader contextHydrogen production by electrolysis of water is an appealing way to store energy if the electric power is obtained from renewable sources of energy, such as photovoltaics. To this aim, catalysts that contain only earth-abundant elements and electrocatalyze hydrogen generation from neutral water at low overpotential with high activity and good durability are ardently desired. Many chemists have devoted themselves to pursue such ideal catalysts. Herein we report a self-assembled molecular cobalt catalyst (CoP4N2) bearing a multihydroxy-functionalized tetraphosphine ligand, which electrocatalyzes hydrogen production from neutral water on a mercury electrode or an amalgamated copper electrode at an onset overpotential of 80 and 130 mV, respectively. Bulk electrolysis of CoP4N2 in a neutral phosphate buffer solution at −1.0 V vs. NHE produced 1490 mol H2 per mol cat. h−1 (cm2 Hg)−1 over 20 h, with a Faradaic efficiency close to 100% and without apparent deactivation. |
Photochemical production of hydrogen from water is the Holy Grail eagerly pursued by many scientists nowadays because it represents an ideal solution to solve the global energy problem.1 In addition to the photolysis of water, hydrogen production by electrolysis of water is also an appealing way to store energy if the electric power is obtained from renewable sources of energy, such as photovoltaics.2 To this aim, one of the crucial scientific challenges facing chemists is development of efficient catalytic systems for electrochemical production of hydrogen. Although platinum and other precious metal-based catalysts are highly efficient for hydrogen production, they are not sustainable materials for large-scale applications due to their limited reserves on earth. To replace these costly metal catalysts, the use of earth-abundant metal complexes as molecular catalysts for the electrochemical hydrogen production has received intensive attention.
In recent years, great progress has been made in this area.3 More than a hundred complexes of non-precious metals have been reported to be catalytically active for electrochemical hydrogen generation. However, most of these molecular catalysts operate in organic solvents or mixtures of organic solvents and water in the presence of different proton sources.4 The number of catalysts known to work efficiently for water reduction to hydrogen in fully aqueous solutions is limited. To date, fewer than thirty molecular catalysts have been found in the literature to be active in fully aqueous solutions,3 and many of them work with low activities and/or large overpotentials. In most cases, acidic aqueous solutions were required.5 Among those catalysts, the complexes, [(PY5Me2)MoO]2+ (PY5Me2 = 2,6-bis(1,1-bis(2-pyridyl)ethyl)pyridine),6 [(PY5Me2)Co(H2O)]2+,7 and [(DPA–Bpy)Co(H2O)]3+ (DPA–Bpy = N,N-bis(2-pyridinylmethyl)–2,2′-bipyridine-6-methanamine),8 are most efficient molecular catalysts for electrochemical hydrogen generation from neutral water on a mercury electrode. With an eventual aim of building up photoelectrochemical cells for total water splitting, it is imperative to develop some electrocatalysts that are cheap in cost, easy to prepare, relying only on earth-abundant elements, and capable of producing hydrogen from neutral water at low overpotential with high activity and good durability.
As an entry to such ideal electrocatalysts, we focused our eyes on the first-row metal complexes containing the hexakis(hydroxymethyl) tetradentate phosphine ligand, hereafter denoted as MP4N2 (M = Fe, Co, Fig. 1). The preparation and structure of the iron complex, FeP4N2, was reported in 2000.9 As MP4N2 complexes are insoluble in organic solvents, electrochemical studies were made in fully aqueous solutions. Complex CoP4N2 displayed a reversible oxidative peak at E1/2 = +0.22 V vs. NHE (normal hydrogen electrode, all potentials mentioned in this paper are versus NHE) at a glassy carbon electrode in 0.1 M KCl aqueous solution (Fig. S1a†), while no reductive peak was observed before the potential of the sharp catalytic current with the onset at −0.94 V. Similarly, FeP4N2 displayed an oxidative peak at E1/2 = −0.12 V (Fig. S1b†). The electrocatalytic properties of FeP4N2, generated in situ from [Fe(NH4)2](SO4)2 and [P(CH2OH)4]2SO4 in aqueous solutions, were initially studied for hydrogen production from neutral water with a mercury pool (∼3.1 cm2) as working electrode in a double-compartment cell. This iron catalyst displays a catalytic current in phosphate buffer at pH 7 (Fig. 2), with the onset at −1.03 V, corresponding to an onset overpotential of 610 mV (the onset overpotential is defined as the difference between the reversible potential of the couple H2/H+, i.e. −420 mV at pH 7, and the potential where electrocatalysis occurs). Up to now, there is no report on electrocatalytic H2 production by iron-based catalysts in fully aqueous solutions without needing any extra acids. To our delight, when [Fe(NH4)2](SO4)2 was replaced by [Co(NH4)2](SO4)2, the in situ-generated CoP4N2, prepared by an identical procedure as that for preparation of FeP4N2,9 displays a sharp catalytic peak at Eonset = −0.50 V during the cathodic scan in a pH 7 phosphate buffer solution, corresponding to an onset overpotential of only 80 mV. Gas bubbles were observed on the surface of the mercury electrode during cathodic scanning at a rate of 100 mV s−1 with the scan stopped at −1.08 V, indicating an electrocatalytic process for water reduction to hydrogen. The buffer solutions containing CoP4N2, made either from [Co(NH4)2](SO4)2 + [P(CH2OH)4]2SO4 or from CoSO4 + (NH4)2SO4 + [P(CH2OH)4]2SO4 in water, give rise to identical features in the cathodic scan for their electrocatalytic performances (Fig. S2†). To make a comparison, the cyclic voltammogram (CV) with a Pt disc (0.2 cm2) as working electrode was measured in the same cell under identical conditions, with a catalytic current arising at −0.43 V. It is impressive to find that CoP4N2 functions as a highly efficient electrocatalyst for production of hydrogen from neutral water with an onset potential comparable to that displayed by a Pt working electrode. The distinct electrocatalytic performances of CoP4N2 and FeP4N2 indicate that the property of the metal centre of the catalyst greatly influences the overpotential in the electrocatalytic water reduction. The onset overpotentials are in the range of 0.52–0.66 V for the electrochemical hydrogen production from neutral water at a mercury electrode in the presence of non-precious metal complexes, including nickel macrocycles,10 cobalt macrocycles,11 [(PY5Me2)MoO]2+,6 [(PY5Me2)Co(H2O)]2+,7a and [(DPA–Bpy)Co(H2O)]3+.8 Utilization of CoP4N2 as an electrocatalyst gives a dramatic decrease in overpotential as compared to the non-precious metal-based molecular catalysts reported to date for the electrochemical hydrogen production from neutral water.
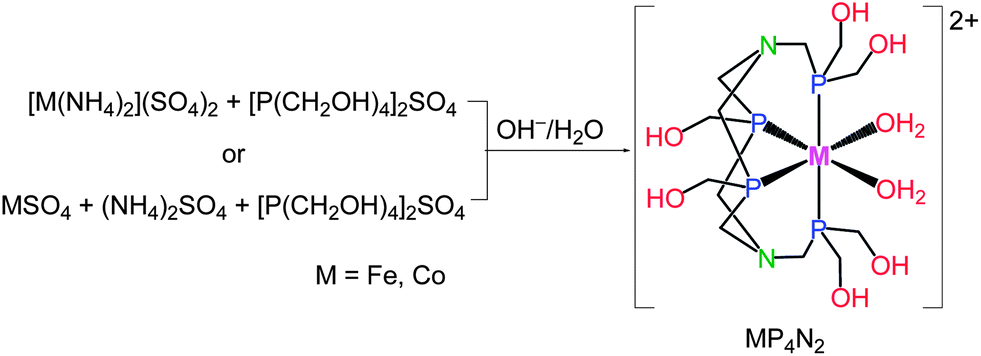 | ||
| Fig. 1 Straightforward preparation of iron and cobalt electrocatalysts. | ||
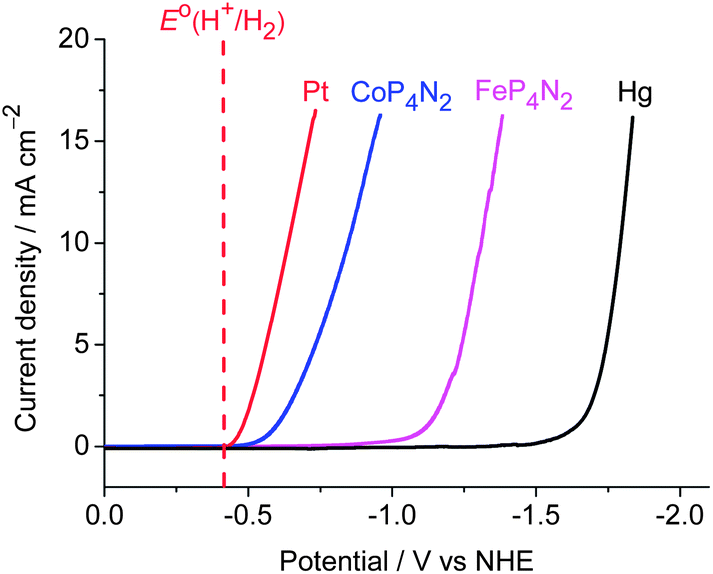 | ||
| Fig. 2 Cathodic scans of the 60 μM solutions of in situ-generated CoP4N2 (blue line) and FeP4N2 (pink line) in 1.0 M phosphate buffer at pH 7. The black and red lines indicate the cathodic scans of buffer solution in the absence of catalyst using a Hg pool and Pt disc as working electrode, respectively, under otherwise identical conditions. | ||
In addition to the extremely low overpotential, CoP4N2 exhibits very high activity in electrochemical water reduction. A current density of 2 mA cm−2 is attained at only 230 mV overpotential for a phosphate buffer solution of CoP4N2 at pH 7. In contrast, the required overpotentials for the [(PY5Me2)Co(H2O)]2+ and [(DPA–Bpy)Co(H2O)]3+ systems to reach 2 mA cm−2 current density are about 830 and 940 mV,7a,8 respectively, under similar conditions. The current density reaches 10 mA cm−2 at an overpotential of 430 mV for the neutral buffer solution of CoP4N2. In the presence of CoP4N2, the decrease of the overpotential by reference to a bare mercury electrode is 890 mV for attaining the same current density under identical conditions. The potential required for attaining the current density of 10 mA cm−2 is 230 mV for a Pt disc working electrode under the same conditions. To our knowledge, none of the reported non-precious metal catalysts display such a high current density at an overpotential as low as 430 mV in neutral water.
A series of control experiments were carried out in phosphate buffer solution at pH 7. The catalytic current was not observed in the CVs of either the solutions of [Co(NH4)2][SO4]2, [P(CH2OH)4]2SO4, CoSO4, (NH4)2SO4, and [P(CH2OH)4]2SO4 alone (Fig. S3†), or the two component solutions of CoSO4 + (NH4)2SO4, (NH4)2SO4 + [P(CH2OH)4]2SO4, and CoSO4 + [P(CH2OH)4]2SO4 (Fig. S4†). In addition, the in situ-generated catalysts from CoX2, (NH4)X and [P(CH2OH)4]X (X = Cl and OAc) exhibit essentially identical cathodic scans to those formed from corresponding sulfate salts. The results of the control experiments indicate that the Co2+, P(CH2OH)3, and NH4+ are all indispensable for the formation of the catalytically active species, which should be a cobalt complex instead of a naked Co2+ ion, while the counter ion in the system is innocent in the electrocatalytic hydrogen production.
The easy formation of the cobalt catalyst was evinced by CV and UV-vis spectroscopic studies. Sulfate salts [Co(NH4)2](SO4)2 and [P(CH2OH)4]2SO4 were successively added to the pH 7 buffer solution. After this solution was stirred for 1 min, the catalytic peak was observed in the cathodic scan and the catalytic current gradually went up when the solution was stirred for an extended interval under open circuit conditions. It reaches a maximum current after the solution is stirred for ∼25 min (Fig. 3(a)). The UV-vis spectroscopic studies show that when [Co(NH4)2](SO4)2 was added to the solution of [P(CH2OH)4]2SO4 in phosphate buffer, two absorption bands appeared at 270 and 440 nm (Fig. 3(b)). The intensities of these bands gradually increase and reach their maxima after the solution is stirred for ∼25 min. In contrast, the characteristic absorptions at 270 and 440 nm are not observed in the UV-vis spectra of [Co(NH4)2](SO4)2, [P(CH2OH)4]2SO4, CoSO4 + [P(CH2OH)4]2SO4, CoSO4 + (NH4)2SO4, and [P(CH2OH)4]2SO4 + (NH4)2SO4 (Fig. S5†). The results obtained from UV-vis spectroscopic studies are consistent with those observed in CVs, indicating that the cobalt catalyst is readily generated in situ from the above-mentioned salts in aqueous solutions. Most reported molecular electrocatalysts for hydrogen production require multistep syntheses of the ligands and the metal complexes. From an application point of view, the convenient availability of this cobalt electrocatalyst by self-assembling from cheap and simple salts in water is a potentially valuable feature of this cobalt-based H2-production system, in addition to its remarkable features of extremely low overpotential and high activity.
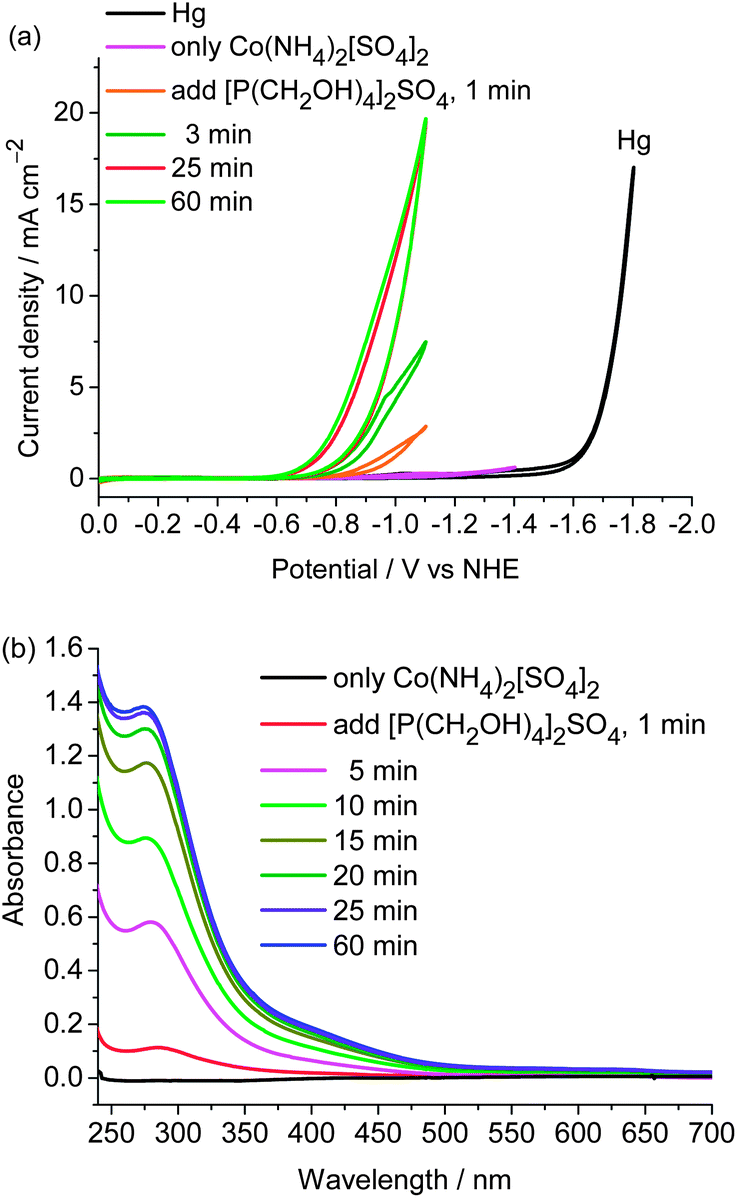 | ||
| Fig. 3 (a) Cyclic voltammograms of the 1.0 mM solution of [Co(NH4)2][SO4]2 in 1.0 M phosphate buffer at pH 7 upon addition of two equiv. of [P(CH2OH)4]2SO4, measured after the mixture was stirred for 1, 3, 25, and 60 min under the open circuit conditions with a mercury electrode at a scan rate of 100 mV s−1; (b) UV-vis spectra of the 1.0 mM solution of [Co(NH4)2][SO4]2 in 1.0 M phosphate buffer at pH 7 upon addition of two equiv. of [P(CH2OH)4]2SO4, measured after the mixture was stirred for 1, 5, 10, 15, 20, 25, and 60 min. | ||
Controlled potential electrolysis (CPE) experiments of a phosphate buffer solution of CoP4N2 at pH 7 were carried out with the applied overpotential varied from 50 to 620 mV over 2 min intervals to assess the rate of hydrogen production at various overpotentials. As shown in Fig. 4, there is no apparent charge consumption at overpotentials below 90 mV, and the charge increases linearly with time during the electrolysis at overpotentials larger than 90 mV. A value of 2.26 C is reached at an overpotential of 620 mV, corresponding to a hydrogen generation rate of 1370 mol H2 per mol cat. h−1 cm−2 (Fig. S6†). The Faradaic efficiency for hydrogen production by CoP4N2 was assessed by CPE experiments (Fig. S7†) to be close to 100%. The activity of hydrogen production for the CoP4N2 system is 1–3 orders of magnitude higher than those displayed by previously reported non-precious metal complexes under similar conditions.6–8,10,11 It is noticeable that the hydrogen-evolving rates given in this paper for all mentioned molecular catalysts are calculated by using the number of catalyst molecules in the entire bulk solution, which provide only lower bounds for their activities.12
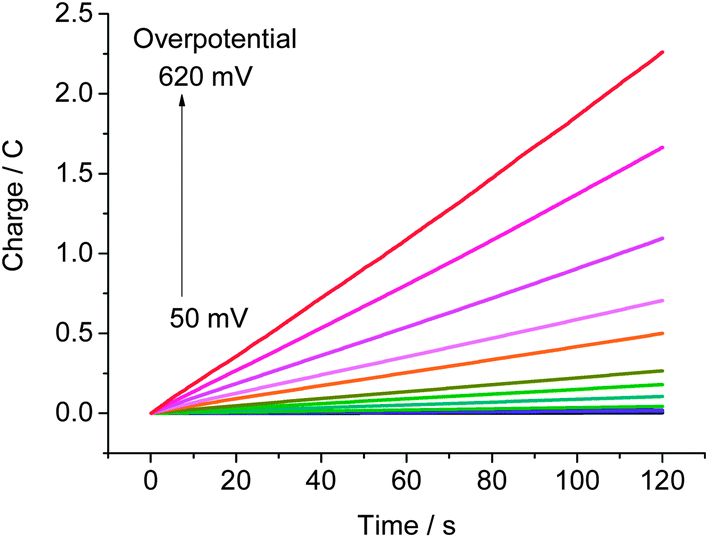 | ||
| Fig. 4 Charge build-up versus time for a 3.3 μM solution of CoP4N2 in 1.0 M phosphate buffer (25 mL) at pH 7 at various overpotentials using a 3.1 cm2 Hg pool electrode. | ||
The stability of CoP4N2 was evaluated by extended CPE experiments at −1.0 V carried out in 2.0 M phosphate buffer at pH 7 (Fig. 5). The pH value of the buffer medium was increased from 7.0 to 7.6 in the water reduction compartment and decreased to 0.6 in the water oxidation compartment after 20 h electrolysis. The significant decrease of the pH value in the water oxidation compartment is possibly the main reason for the slight decrease in the charge accumulation rate in Fig. 5. The turnover number (TON) of hydrogen evolution calculated based on the charges accumulated over the 20 h CPE experiment is 9.24 × 104 mol H2 per mol cat. after subtracting the contribution from the blank experiment, corresponding to an activity of 1490 mol H2 per mol cat. h−1 cm−2 at −1.0 V in pH 7 phosphate buffer, while under identical conditions the phosphate buffer solution of [Co(NH4)2][SO4]2 displays an activity of only ∼7 mol H2 per mol cat. h−1 cm−2. It is unusual that an electrocatalyst exhibits such a high activity at a low applied potential for long-term hydrogen production from neutral water. The performance of CoP4N2 is much better than the most efficient molecular catalysts so far reported in the literature for the electrochemical hydrogen production from neutral water: [(PY5Me2)MoO]2+ that displays an activity of 434 mol H2 per mol cat. h−1 cm−2 at an applied potential of −1.40 V (71 h) in 3.0 M phosphate buffer,6 [(PY5Me2)Co(H2O)]2+ that exhibits a hydrogen-evolving rate of 47 mol H2 per mol cat. h−1 cm−2 at −1.30 V (60 h) in 2.0 M phosphate buffer,7a and [(DPA–Bpy)Co(H2O)]3+ that gives a rate of 62 mol H2 per mol cat. h−1 cm−2 at −1.40 V (1 h) in 1.0 M phosphate buffer.8
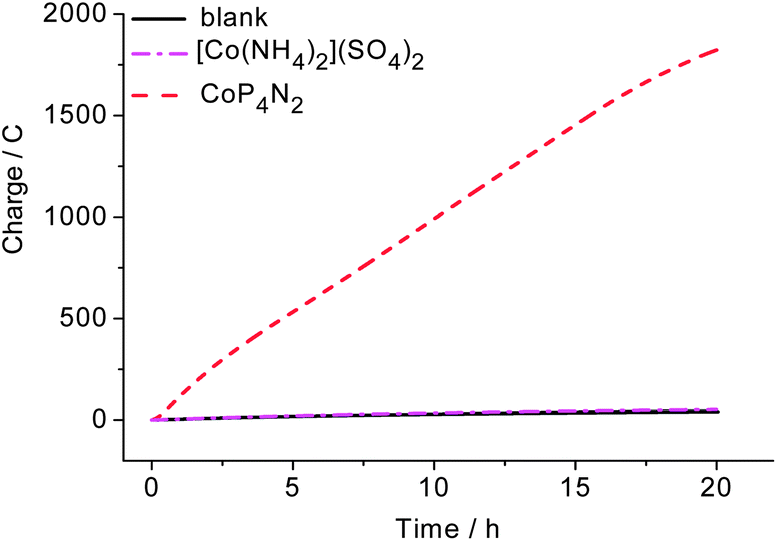 | ||
| Fig. 5 Extended CPE experiments of 4.0 μM CoP4N2 (red line) and [Co(NH4)2](SO4)2 (pink line) in 2.0 M phosphate buffer (25 mL) at pH 7 at −1.0 V, and the blank solution (black) of 2.0 M phosphate buffer; the contribution from the background solution has been subtracted from the red and pink lines. | ||
When an organometallic complex is used as a pre-catalyst for electrochemical H2 production, the actual nature of the active species in the catalytic system is always the most concerned issue in the aspect of reaction mechanism.13,14 We tried to figure out whether the active species in the present cobalt-based catalytic system is a molecular catalyst or electrodeposited metallic particles. Firstly, after the extended CPE experiment of CoP4N2 in a pH 7 phosphate buffer at −1.0 V for 20 h, the mercury electrode remained as shiny as before use. Secondly, after a 20 h CPE experiment of the 2.0 M phosphate buffer solution of CoP4N2 (80 μM) at an applied potential of −1.10 V using an amalgamated copper plate (1.0 cm2) as working electrode, the scanning electronic micrograph (SEM) image and the energy dispersive X-ray (EDX) analysis of the electrode surface have no detectable change compared to that recorded for the electrode before used (Fig. S8†). Accordingly, after the used mercury or amalgamated copper electrode was carefully washed with distilled water, no catalytic activity was observed when it was reused for the electrolysis of phosphate buffer in the absence of catalyst. These results show that the electrodeposition of CoP4N2 in phosphate buffer does not occur at least at an applied potential lower than −1.10 V. As all CPE experiments of CoP4N2 in phosphate buffer were carried out at applied potentials lower than −1.04 V, we postulate that a self-assembled molecular cobalt catalyst, instead of electrodeposited metallic particles, is responsible for the catalytic activity of hydrogen production.
To gain further insight into the actual nature of the active species in the present catalytic system, we recorded CVs of the buffer solution of CoP4N2 using a static mercury dropping electrode with each scan for a new mercury drop, which was kept in the buffer solution for different time intervals (1 to 40 s) before recording the CV. The catalytic current apparently increased as the time interval was extended (Fig. 6(a)). This observation suggests the adsorption of the molecular cobalt catalyst on the surface of the mercury electrode.15 Compared with the diffusion-limited reduction process observed with a glassy carbon electrode (Fig. S9†), the apparent positive shift (∼470 mV) of the catalytic wave with a mercury electrode implicates the stabilization of the molecular cobalt species involved in the electrocatalytic process through adsorption on the mercury electrode.15c Furthermore, the CVs recorded with varying concentrations of CoP4N2 (0.8–9.2 μM) in a pH 7 phosphate buffer reveal that the maximum catalytic current is linearly dependent on the concentration of CoP4N2 when the concentration is lower than ca. 5 μM. As shown in Fig. 6(b), when the concentration of CoP4N2 is higher than 5 μM, the maximum catalytic current deviates from the linear relationship with the concentration of catalyst, presumably due to saturation coverage of the catalyst on the electrode surface. This evidence indicates that the concentration of CoP4N2 in the buffer solution is not a rate determining factor, but that the amount of adsorbed catalyst molecules on the surface of the mercury electrode gives a dominant contribution to the catalytic current. More intensive studies are needed to elucidate the catalytic mechanism of the highly active catalyst CoP4N2 for the electrochemical water reduction to hydrogen.
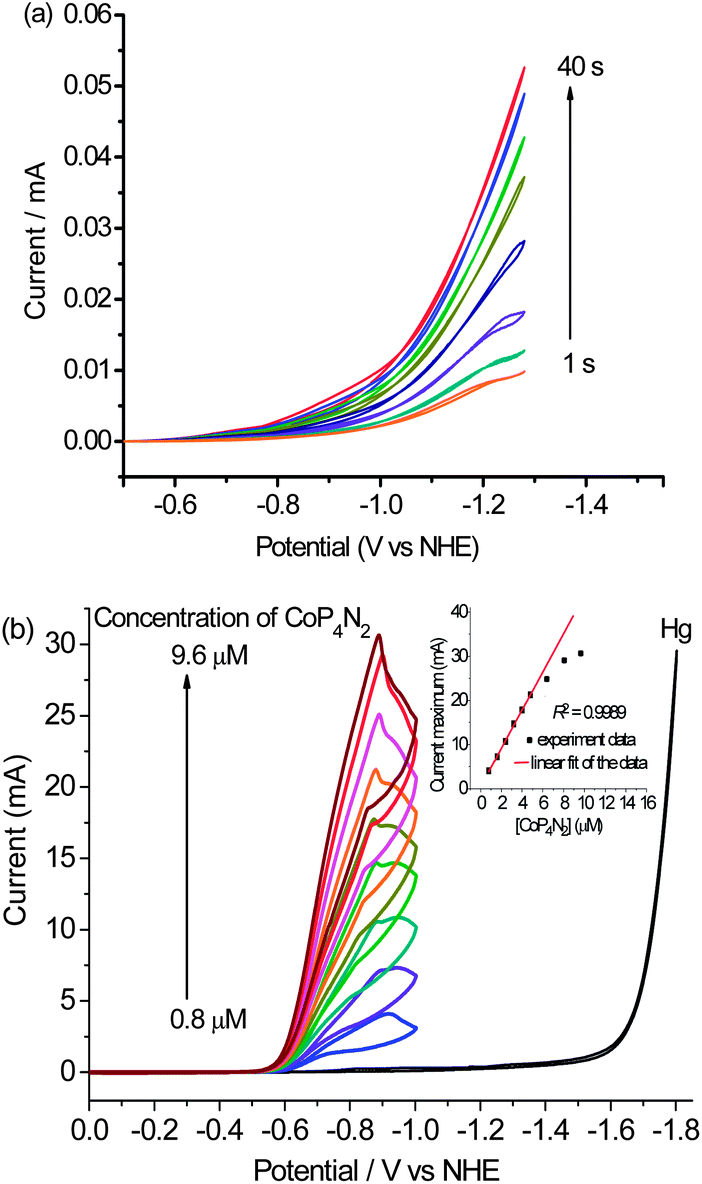 | ||
| Fig. 6 (a) Cyclic voltammograms on a static mercury dropping electrode with each scan for a new mercury drop, which was kept in the 1.0 M phosphate buffer solution of CoP4N2 (4.0 μM) for different time intervals (1 to 40 s) before recording the CV at a scan rate of 200 mV s−1; (b) cyclic voltammograms of 1.0 M phosphate buffer at pH 7 containing different concentrations of CoP4N2, with a mercury pool electrode (3.1 cm2) at a scan rate of 100 mV s−1; (inset of Fig. 6(b)) plot of the maximum catalytic current as a function of the concentration of CoP4N2 in phosphate buffer. | ||
To elucidate the structures of the self-assembled molecular catalysts MP4N2 (M = Fe, Co), solids were isolated as red (FeP4N2) and light-orange (CoP4N2) powder, either from the condensation reaction of [M(NH4)2](SO4)2 and [P(CH2OH)4]2SO4 or from the reaction of MSO4, (NH4)2SO4, and [P(CH2OH)4]2SO4 in water (see the ESI†) according to the protocol in the literature for the preparation of [Fe(H2O)2{RP(CH2N(CH2PR2)CH2)}2PR]SO4 (FeP4N2, R = CH2OH).9 The 1H and 31P NMR as well as the UV-vis spectroscopic data obtained for FeP4N2 are in good agreement with the data reported in the literature. The NMR spectra of CoP4N2 do not give any useful structural information due to its paramagnetic properties. The analysis of the isolated cobalt catalyst by inductively coupled plasma mass spectroscopic analysis (ICP) indicates that the ratio of Co to P is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 and the data of the elemental analysis are in agreement with the composition of the proposed phosphine cobalt complex CoP4N2. The product was also characterized by UV-vis (Fig. S5†), EPR (Fig. S10†), and XPS (Fig. S11†) spectroscopy. The UV-vis spectrum of CoP4N2 shows a broad absorption with the maximum at 270 nm and a broad weak absorption with λmax at 420 nm in an aqueous solution. The EPR spectrum of the CoP4N2 solid measured at room temperature shows a broad signal with a g value at 2.147, implicating the presence of a low-spin cobalt(II) center.16 This is consistent with the results of XPS analysis of the CoP4N2 catalyst. The Co 2p region of the XPS spectrum shows two broad sets of signals with binding energies of 779.6 eV (2p3/2) and 794.9 eV (2p1/2), which are the signature of the Co2+ ion.17 In addition, the P 2p peak centered at 132.0 eV is consistent with metal-bonded phosphorus atoms and the N 1s peak centered at 398.9 eV is in agreement with tertiary amine nitrogen atoms.3a The MP4N2 powder has good solubility in water, but is not soluble in any organic solvent. The electrocatalytic performance of the isolated MP4N2 in phosphate buffer is identical to that displayed by the in situ-generated catalyst (Fig. S1†).
:4 and the data of the elemental analysis are in agreement with the composition of the proposed phosphine cobalt complex CoP4N2. The product was also characterized by UV-vis (Fig. S5†), EPR (Fig. S10†), and XPS (Fig. S11†) spectroscopy. The UV-vis spectrum of CoP4N2 shows a broad absorption with the maximum at 270 nm and a broad weak absorption with λmax at 420 nm in an aqueous solution. The EPR spectrum of the CoP4N2 solid measured at room temperature shows a broad signal with a g value at 2.147, implicating the presence of a low-spin cobalt(II) center.16 This is consistent with the results of XPS analysis of the CoP4N2 catalyst. The Co 2p region of the XPS spectrum shows two broad sets of signals with binding energies of 779.6 eV (2p3/2) and 794.9 eV (2p1/2), which are the signature of the Co2+ ion.17 In addition, the P 2p peak centered at 132.0 eV is consistent with metal-bonded phosphorus atoms and the N 1s peak centered at 398.9 eV is in agreement with tertiary amine nitrogen atoms.3a The MP4N2 powder has good solubility in water, but is not soluble in any organic solvent. The electrocatalytic performance of the isolated MP4N2 in phosphate buffer is identical to that displayed by the in situ-generated catalyst (Fig. S1†).
In conclusion, CoP4N2 proves to be an efficient molecular catalyst for the electrochemical production of hydrogen from neutral water, which displays a low overpotential (Eonset = 80 mV) while maintaining high activity and good durability. It displays an activity of 1490 mol H2 per mol cat. h−1 cm−2 over 20 h CPE experiment at an applied potential of 1.0 V in phosphate buffer at pH 7, without apparent deactivation. The other merit of this catalyst is its self-assembling property from simple salts of earth-abundant elements, which is reminiscent of the self-formation of many metalloenzymes in nature. The results show promise in using earth-abundant metal-based catalysts as surrogates of precious metal-based ones such as Pt to create highly efficient and economical catalyst systems for water reduction to a sustainable fuel, hydrogen gas. To make this electrocatalytic system more practically available, we are curious to know the electrocatalytic properties of CoP4N2 on the solid metal electrodes. Preliminary results show that when an amalgamated copper electrode was used, the onset overpotential of catalytic current is 130 mV and the current density reached 5.2 mA cm−2 per geometric surface area at −0.85 V in a phosphate buffer solution of CoP4N2 at pH 7 (Fig. S12†). The screening of other electrodes made from different materials is under way in our lab.
We are grateful to the Natural Science Foundation of China (Grant no. 21373040, 21120102036, and 91233201), the Basic Research Program of China (Grant no. 2009CB220009), the Swedish Research Council, and the K & A Wallenberg Foundation for financial support of this work.
Notes and references
- (a) A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS; (b) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- (a) J. A. Turner, Science, 1999, 285, 687–689 CrossRef CAS; (b) R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science., 2011, 332, 805–809 CrossRef CAS PubMed.
- (a) G. A. N. Felton, C. A. Mebi, B. J. Petro, A. K. Vannucci, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Organomet. Chem., 2009, 694, 2681–2699 CrossRef CAS PubMed; (b) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed; (c) D. L. DuBois and R. M. Bullock, Eur. J. Inorg. Chem., 2011, 1017–1027 CrossRef CAS; (d) P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC; (e) M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763–6778 RSC; (f) V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- (a) C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817–1824 CrossRef CAS PubMed; (b) X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed; (c) G. M. Jacobsen, J. Y. Yang, B. Twamley, A. D. Wilson, R. M. Bullock, M. R. DuBois and D. L. DuBois, Energy Environ. Sci., 2008, 1, 167–174 RSC; (d) K. D. Welch, W. G. Dougherty, W. S. Kassel, D. L. DuBois and R. M. Bullock, Organometallics, 2010, 29, 4532–4540 CrossRef CAS; (e) U. J. Kilgore, J. A. Roberts, D. H. Pool, A. M. Appel, M. P. Stewart, M. R. DuBois, W. G. Dougherty, W. S. Kassel, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2011, 133, 5861–5872 CrossRef CAS PubMed; (f) M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed; (g) M. J. Rose, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2012, 134, 8310–8313 CrossRef CAS PubMed.
- (a) B. D. Stubbert, J. C. Peters and H. B. Gray, J. Am. Chem. Soc., 2011, 133, 18070–18073 CrossRef CAS PubMed; (b) C. C. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS PubMed; (c) H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed; (d) D. H. Pool, M. P. Stewart, M. O'Hagan, W. J. Shaw, J. A. Roberts, R. M. Bullock and D. L. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15634–15639 CrossRef CAS PubMed; (e) F. Quentel, G. Passard and F. Gloaguen, Energy Environ. Sci., 2012, 5, 7757–7761 RSC.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- (a) Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed; (b) Y. Sun, J. Sun, J. R. Long, P. Yang and C. J. Chang, Chem. Sci., 2013, 4, 118–124 RSC.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS PubMed.
- J. C. Jeffery, B. Odell, N. Stevens and R. E. Talbot, Chem. Commun., 2000, 101–102 RSC.
- J. P. Collin, A. Jouaiti and J. P. Sauvage, Inorg. Chem., 1988, 27, 1986–1990 CrossRef CAS.
- P. V. Bernhardt and L. A. Jones, Inorg. Chem., 1999, 38, 5086–5090 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Saveant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS PubMed.
- (a) R. M. Kellett and T. G. Spiro, Inorg. Chem., 1985, 24, 2373–2377 CrossRef CAS; (b) K. Bujno, R. Bilewicz, L. Siegfried and T. A. Kaden, J. Electroanal. Chem., 1998, 445, 47–53 CrossRef CAS; (c) J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 RSC.
- A. Bakac, M. E. Brynildson and J. H. Espenson, Inorg. Chem., 1986, 25, 4108–4114 CrossRef CAS.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515–2525 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional CVs, UV-vis spectra, SEM images, EDX, EPR and XPS spectra. See DOI: 10.1039/c3ee42194e |
| This journal is © The Royal Society of Chemistry 2014 |