
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination frameworks assembled from CuII ions and H2-1,3-bdpb ligands: X-ray and magneto structural investigations, and catalytic activity in the aerobic oxidation of tetralin†
Maciej
Grzywa
a,
Christof
Geßner
b,
Björn
Bredenkötter
a,
Dmytro
Denysenko
a,
Jan
van Leusen
c,
Paul
Kögerler
 c,
Elias
Klemm
b and
Dirk
Volkmer
*a
c,
Elias
Klemm
b and
Dirk
Volkmer
*a
aChair of Solid State and Materials Chemistry, Institute of Physics, Augsburg University, Universitätsstr. 1, 86159 Augsburg, Germany. E-mail: dirk.volkmer@physik.uni-augsburg.de
bInstitute of Chemical Technology, Faculty of Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
cInstitute of Inorganic Chemistry RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
First published on 11th September 2014
Abstract
The syntheses and crystal structures of H2-1,3-bdpb·MeOH, [CuII2(1,3-bdpb)(OCH3)2] (CFA-5) and [CuICl(H2-1,3-bdpb)] (H2-1,3-bdpb = 1,3-bis(3,5-dimethyl-1H-pyrazol-4-yl)benzene) are described. The copper(II) containing metal–organic framework (termed Coordination Framework Augsburg University-5, CFA-5) crystallizes in the trigonal crystal system, within the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (no. 148) and the unit cell parameters are as follows: a = 26.839(3), c = 15.8317(16) Å, V = 9876.2(19) Å3. CFA-5 features a two-fold interpenetrated 3-D microporous framework structure of cross-linked wheel-shaped {CuII(pz)(OMe)}12 fundamental building units, each containing twelve copper(II) ions, μ2-bridging MeO− groups and pyrazolate (pz–) ligands. Replacing copper(II) acetate by copper(II) chloride in the synthesis leads to compound [CuICl(H2-1,3-bdpb)], which crystallizes in the orthorhombic crystal system, within the space group Pnma (no. 62) and the unit cell parameters are as follows: a = 6.1784(8), b = 6.1784(8), c = 6.1784(8) Å, V = 1583.8(4) Å3. In contrast to the former compound, CuCl(H2-1,3-bdpb) is a non-porous compound consisting of CuI–Cl zigzag chains expanding in the direction [100] and H2-1,3-bdpb ligands. CFA-5 is characterized by elemental and thermogravimetric analyses, variable temperature powder X-ray diffraction and IR-spectroscopy; and its porosity and magnetic properties are described in detail. CFA-5 shows a promising catalytic activity in the heterogeneously catalyzed aerobic oxidation of tetralin, which is compared with other catalytically active metal–organic frameworks.
(no. 148) and the unit cell parameters are as follows: a = 26.839(3), c = 15.8317(16) Å, V = 9876.2(19) Å3. CFA-5 features a two-fold interpenetrated 3-D microporous framework structure of cross-linked wheel-shaped {CuII(pz)(OMe)}12 fundamental building units, each containing twelve copper(II) ions, μ2-bridging MeO− groups and pyrazolate (pz–) ligands. Replacing copper(II) acetate by copper(II) chloride in the synthesis leads to compound [CuICl(H2-1,3-bdpb)], which crystallizes in the orthorhombic crystal system, within the space group Pnma (no. 62) and the unit cell parameters are as follows: a = 6.1784(8), b = 6.1784(8), c = 6.1784(8) Å, V = 1583.8(4) Å3. In contrast to the former compound, CuCl(H2-1,3-bdpb) is a non-porous compound consisting of CuI–Cl zigzag chains expanding in the direction [100] and H2-1,3-bdpb ligands. CFA-5 is characterized by elemental and thermogravimetric analyses, variable temperature powder X-ray diffraction and IR-spectroscopy; and its porosity and magnetic properties are described in detail. CFA-5 shows a promising catalytic activity in the heterogeneously catalyzed aerobic oxidation of tetralin, which is compared with other catalytically active metal–organic frameworks.
Introduction
In the last decade the major driving force for the continued interest in metal–organic frameworks (MOFs) has been the promising technical applications of these materials ranging from gas storage and separation,1 drug delivery,2 sensing3 to electrochemistry.4 The large specific surface areas of up to 7140 m2 g−1,5 open pores with apertures of up to 98 Å,6 the high thermal stability and a high volume density of active sites within the framework structures suggest a host of applications in the field of heterogeneous catalysis in particular.1a,7On searching for suitable MOF candidates for catalytic oxidation processes, copper-containing frameworks seem to be particularly interesting owing to the fact that this element is present in the active site of many metalloenzymes, such as copper-containing oxidases or oxygenases.8 Biologically inspired MOF catalysts trying to mimic structural and functional aspects of these enzymes could be applied for wide-ranging synthetic applications in the oxidation of organic intermediates containing non-activated C–H-bonds. However, until now, only a few reports have appeared in the literature where Cu-MOFs were successfully applied as oxidation catalysts. In particular, the Cu-catalyzed hydroxylation of phenol,9 oxidation of trimethylsilyl enolates to α-hydroxy ketones,10 allylic oxidation of cyclohexenes,11 cross-dehydrogenative coupling reaction of ethers with 2-carbonyl-substituted phenols,12 and oxidation of benzene derivatives and benzylic compounds have been described.13
Some years ago we had started the development of Cu-containing MOF catalysts containing pyrazole ligands for the purpose of introducing oxygen functions into the C–H bonds of organic substrates.14 Our previous studies on the reactivity of CFA-2 (a Cu(I)-containing MOF) toward molecular oxygen for instance have shown that this compound is stable during oxidation and reduction of the Cu ions, suggesting its usage in liquid-phase oxidation reactions. The exploration of pyrazolate-based Cu-MOFs follows the simple rational and experimental observation that this ligand forms strong coordinative bonds with Cu(I) and Cu(II), thus leading to robust coordination polymers. Similar to carboxylate-type ligands, pyrazolates are known to form μ2-bridges between metal ions. Unlike the former, the hydrolytic stability of N-heterocyclic ligand-based frameworks containing the latter, i.e. electron-rich transition metal ions, is exceptionally high which has been convincingly demonstrated.15 More speculatively, we might anticipate that pyrazolate heterocycles could mimic the coordination of Cu ions by histidine (= imidazolate) ligands, a coordination motif which is frequently observed in (multi-)copper oxidase enzymes, thus opening routes towards biomimetic catalysis.16
μ2-Bridging pyrazolate ligands have been frequently used to generate trinuclear copper (I/II) [Cu3(μ2-pz)3] metallocycles,17 which later on were introduced as secondary building units (SBUs) into metal–organic frameworks.14,18 Polynuclear metallocyclic coordination units of the type [{CuII(μ-OH)(μ2-pz)}n], n = 6, 8, 9, 12 and 14, have been described,19 which might become SBUs of the as yet undiscovered MOFs. One such example, CFA-5, a novel MOF which contains a dodecanuclear {CuII(pz)(OMe)}12 SBU is presented in this article. CFA-5 was characterized by elemental and thermogravimetric analyses, X-ray diffraction and IR-spectroscopy and its porosity and magnetic properties have been studied in detail. Additionally, the results of catalytic tests of CFA-5 in the aerobic oxidation of tetralin are presented and compared to other Cu-MOFs.
Results and discussion
Syntheses and characterization
H2-1,3-bdpb (1,3-bis(3,5-dimethyl-1H-pyrazol-4-yl)benzene) was synthesized according to the published procedure.20 The compound CFA-5 was obtained as green hexagonal prism crystals (Fig. 1) after heating a MeOH solution of H2-1,3-bdpb and copper(II) acetate hydrate in the presence of 2,6-lutidine. By applying microwave irradiation instead of conventional heating the reaction time was reduced drastically, from 3 d to 25 min. | ||
| Fig. 1 SEM image of CFA-5 (left) and optical micrograph (right). | ||
Replacing copper(II) acetate by copper(II) chloride in the synthesis leads to the compound [CuICl(H2-1,3-bdpb)] (Scheme 1).
 | ||
| Scheme 1 Synthesis routes to CFA-5 [CuII2(1,3-bdpb)(OMe)2] and [CuICl(H2-1,3-bdpb)]; L = C18H22N4. | ||
Single crystal structure analyses
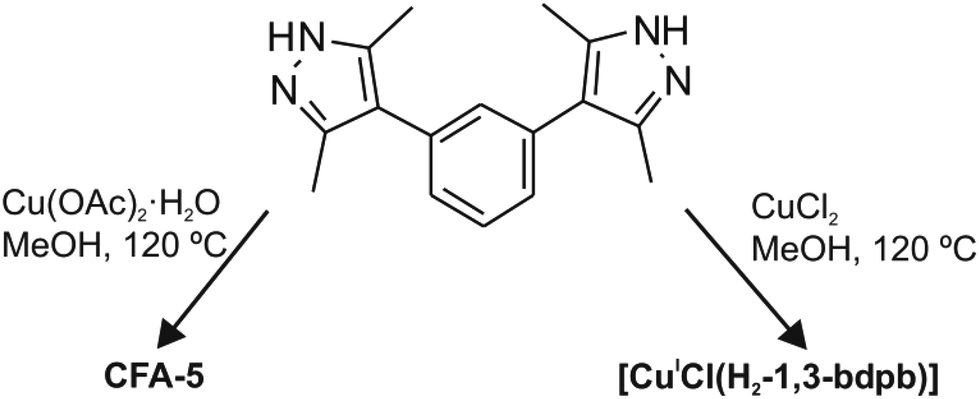
(no. 148). The asymmetric unit consists of eighteen carbon, four nitrogen, two oxygen and two copper atoms. An Ortep style plot of the asymmetric unit of CFA-5 with atom labels is shown in the ESI, Fig. S3.†CFA-5 features a 3-D two-fold interpenetrated microporous structure. The twelve metal atoms are connected by twelve μ2-bridging MeO− and by twelve pyrazolate moieties belonging to 1,3-bdpb2− ligands, respectively, creating dodecanuclear copper(II) rings, as shown in Fig. 2a. In the literature several examples of copper pyrazolate structures created from four up to tetradecanuclear copper rings are known.19 To the best of our knowledge, CFA-5 represents the first example of a copper-containing MOF in which a dodecanuclear copper(II) SBU was structurally characterized. In these SBUs, each Cu(II) center exhibits an unusual distorted tetrahedral coordination geometry (as shown in Fig. 2b). The coordination geometry of each Cu(II) (N,N′,O,O′) subset lies between that of the tetrahedrally coordinated Cu(I) ions and that of Cu(II) complexes featuring a square planar coordination environment. The Cu–N distances range between 1.924(5) and 1.963(5) Å, whereas the Cu–O distances range between 1.904(4) and 1.929(5) Å (see Table 1). These values are in good agreement with those found in structurally related copper compounds.19
 | ||
| Fig. 2 (a) Wheel-shaped SBU of CFA-5, a framework featuring dodecanuclear Cu(II) coordination units showing cross-sectional non-bonding CuII⋯CuII distances. (Symmetry transformations used to generate equivalent Cu atoms: (i) –x + y, −x, z; (ii) −y, x − y, z; (iii) −y + 2/3, x − y + 1, 3z + 1/3.) (b) Selected part of the dodecanuclear SBU in CFA-5 showing the μ2-bridging modes of the coordinated pyrazolate and methoxide ligands within a dodecanuclear {CuII(pz)(OMe)}12 ring. (Symmetry transformations used to generate equivalent Cu atoms: y − 1/3,−x + y + 1/3,−z + 1/3.) (c) Crystal packing diagram of CFA-5 including the positions of open channels running along the c direction of the crystal lattice. (d) View perpendicular to the channel axis onto a packing model of CFA-5, including subsets of the two symmetry-related interpenetrating frameworks which are shown in black and red color. (e) Packing diagram of CFA-5 showing the two-fold interpenetrated porous 3D framework (methyl groups from 1,3-bdpb2− ligands were omitted for clarity). | ||
| Atoms | Bond length (Å) |
|---|---|
| a Symmetry transformations used to generate equivalent atoms: #1 x − y + 2/3,x + 1/3,−z + 1/3. | |
| Cu(1)–O(1) | 1.923(4) |
| Cu(1)–N(4) | 1.924(5) |
| Cu(1)–N(1) | 1.925(5) |
| Cu(1)–O(2) | 1.929(5) |
| Cu(2)–O(1) | 1.904(4) |
| Cu(2)–O(2)#1 | 1.921(4) |
| Cu(2)–N(3) | 1.942(5) |
| Cu(2)–N(2) | 1.963(5) |
Placing a least-squares plane through the twelve Cu(II) ions of each SBU, six 1,3-bdpb2− ligands (blue and purple ligands in Fig. 2a) are found above and the six remaining ones (black and green) are placed below this plane. Among the twelve coordinating pyrazolate moieties six are almost perpendicular to the plane passing through the Cu centers (blue and green ligands; the angle between the Cu12 plane and a least-squares plane passing through the pyrazole ring is equal to 79.8(1)°). Pyrazole moieties of the other six 1,3-bdpb2− ligands (black and purple) each include a more flatter angle of 46.71(9)° compared with the {Cu12} plane. The μ2-bridging MeO− ligand is always found opposite to the 1,3-bdpb2− ligand that bridges the same pair of Cu(II) ions. Of the twelve methoxides, six are pointing their methyl groups towards the center of the dodecanuclear SBU, thus leading to a hydrophobic internal channel surface.
Defining the wheel-shaped {CuII(pz)(OMe)}12 fragment as SBU of CFA-5, these SBUs are connected by the meta-substituted benzene ring of the 1,3-bis(3,5-dimethyl-1H-pyrazol-4-yl)benzene ligand, thus creating one-dimensional channels expanding in the c-direction of the crystal lattice (see Fig. 2c and d). Taking the van der Waals radii of hydrogen atoms (1.2 Å) into account, the narrowest channel diameter calculated between the hydrogen atoms of the MeO− groups is 4.5 Å, whereas the pore width, calculated between carbon atoms of the methyl groups and pyrazolate rings, is 5.86 Å (see Fig. S4, ESI†). An estimation with the program SQUEEZE reveals21 that the initial solvent's accessible void volume is 1213.3 Å3, which is 12.3% of the unit cell volume (9876.2(19) Å3) for a probe radius of 1.68 Å, corresponding to the approximate van der Waals radius of argon.22 For MeOH molecules with an approximate van der Waals radius of 1.97 Å, the value of 1091.8 Å3 (11.1% of the unit cell volume) is calculated. The CFA-5 coordination network can be described as a twofold interpenetrated 3D (see Fig. 2e) uninodal six-connected net of the pcu type (alpha-Po primitive cubic), with a point (Schläfli) symbol (412 × 63), with the Cu12 rings as six-connected nodes and the 1,3-bdpb2− ligands as spacers expanding 8.78(7) Å in length.23 The atomic coordinates and isotropic thermal parameters, selected bond lengths and angles are presented in Tables S3 and S4 (ESI†), respectively.
In the dodecanuclear coordination units found in [{CuII(μ2-OH)(μ2-pz)}12] compounds, [PPN+][Cl−{[Cu(μ-OH)(μ2-pz)]6+12}2], [Bu4N+][Cl−{[Cu(μ-OH)(μ2-pz)]6+12}2], six pyrazoles are in-plane and six out-of-plane created by Cu(II) ions. In comparison, in the dodecanuclear {CuII(pz)(OMe)}12 coordination unit of CFA-5 six pyrazoles of 1,3-bdpb2− ligands are above and six below the plane. The cross-sectional non-bonding CuII⋯CuII distances in the dodecanuclear ring of CFA-5 (see Fig. 2a) are slightly smaller (12.2 Å) than in the above-mentioned compounds (12.7 Å). The Cu–O distances from the OH groups in the molecular {CuII(OH)(pz)}12 compounds are slightly larger (1.909(6)–1.957(6)) than the Cu–O distances of the OMe− groups (CFA-5) 1.904(4)–1.929(5). Apart from the {Cu(μ-pz)}12 compounds, two additional literature reports on dodecanuclear metallomacrocycles appeared, containing Zn or Co(III) ions, namely ([(μ2-5-methyl-3-phenylpyrazole)2Zn2(μ3-OCH2CH2S)]6)24 and ([PPN]2[Co12(μ2-OH)12(μ2-4-NO2-pz)12(μ2-3,5-Me2-pz)12](NO2)2).25
TGA and XRPD studies
The phase purity of CFA-5 was confirmed by XRPD measurement under ambient conditions. The experimental XRPD pattern is consistent with the simulated one as gleaned from the single crystal X-ray diffraction data, as shown in Fig. 3. Differences in peak intensities are due to occluded solvent molecules.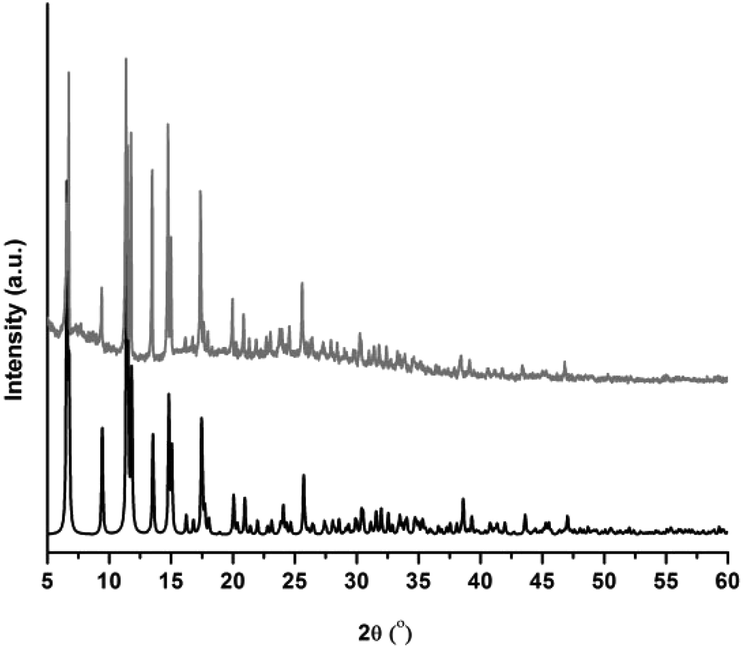 | ||
| Fig. 3 Calculated and measured X-ray powder patterns for CFA-5. | ||
In addition, the thermal stability of CFA-5 was determined by thermogravimetric (TG) and VTXRPD measurements. Prior to TG measurement, the sample was heated at 100 °C under vacuum for 2 h in order to remove the occluded solvent molecules (MeOH). As shown in Fig. 4, the thermogravimetric profile of CFA-5 under nitrogen exhibits a weight loss of 6.8% between 200 and 300 °C corresponding to the loss of one MeOH molecule per formula unit. The next degradation step (−37.5%), in which a fraction of the organic ligand is lost, is observed between 300 and 600 °C. According to the XRPD data presented in Fig. 5, the sample is stable up to ca. 150 °C. At 200 °C characteristic peaks at 6.60–6.76° 2θ (corresponding to the (2![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and (101) planes), 9.44 (20), 11.42 ((300),(3
0) and (101) planes), 9.44 (20), 11.42 ((300),(3![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) )), 11.80 (10), 13.54 (202), 14.82 (41), 15.05 (32), 17.47 (50), 17.58 (5) become weaker and the peaks >20° in 2θ disappear, which indicates framework damage. Subsequent heating of the sample leads to amorphization of the material.
)), 11.80 (10), 13.54 (202), 14.82 (41), 15.05 (32), 17.47 (50), 17.58 (5) become weaker and the peaks >20° in 2θ disappear, which indicates framework damage. Subsequent heating of the sample leads to amorphization of the material.
 | ||
| Fig. 4 Temperature dependent weight loss of CFA-5 under flowing nitrogen gas. | ||
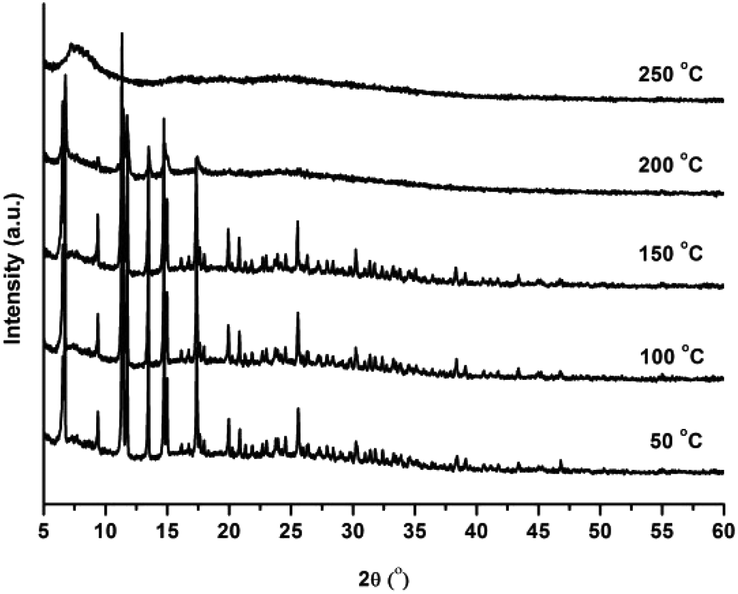 | ||
| Fig. 5 VTXRPD plots of CFA-5 kept in air, sampled in a temperature range of 50–250 °C. | ||
Physisorption results
CFA-5 exhibits permanent porosity, which is confirmed by argon gas sorption. Prior to sorption measurement, the crystalline sample was heated at 100 °C under vacuum for 1 h. The argon adsorption isotherm follows type I behaviour, typical for microporous solids. The maximum uptake achieved at 77 K and p/p0 = 0.99 is 252 cm3 g−1 (Fig. 6). The micropore volume obtained from the sorption isotherm is 0.2 cm3 g−1 on applying the NLDFT method.27 The same value was obtained using the de Boer t-method micropore analysis.28 The adsorption data were fitted to the BET equation to give a surface area of 460 m2 g−1, the Langmuir surface area is equal to 510 m2 g−1. To evaluate the pore size distribution, the argon sorption isotherms sampled at 77 K were analyzed using non-local density functional theory (NLDFT) implementing a carbon equilibrium transition kernel for argon adsorption at 77 K based on a slit-pore model.29 The distribution calculated by fitting the sorption data reveals micropores with a diameter of ca. 6.6 Å (Fig. 7, which is slightly higher than the data from the crystal structure (5.86 Å)).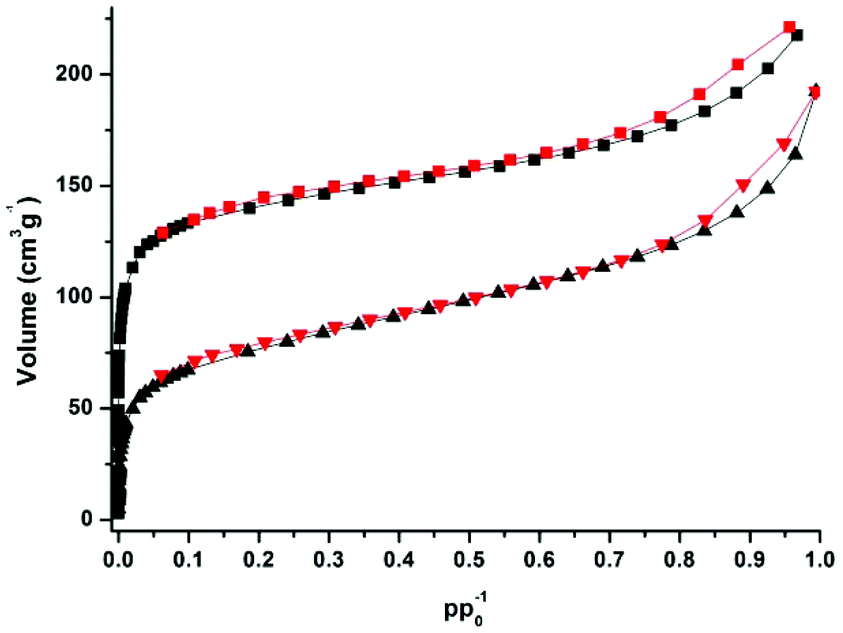 | ||
| Fig. 6 Argon adsorption (black squares, triangles) and desorption (red squares, triangles) isotherms measured for samples of CFA-5, desolvated at 100 °C (top) and 250 °C (bottom curves), respectively. | ||
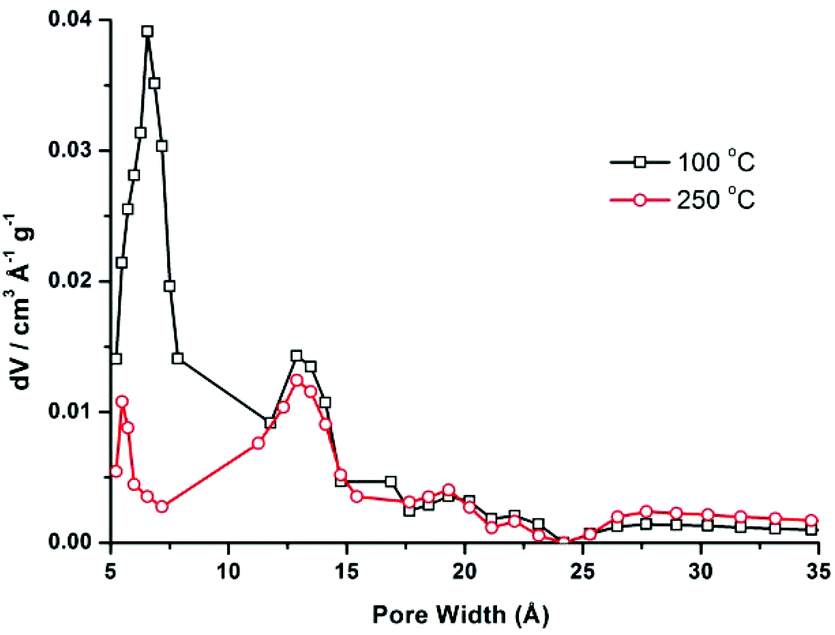 | ||
| Fig. 7 Pore size distribution for CFA-5 desolvated at 100 °C (black line) and 250 °C (red line) calculated by fitting the NLDFT model to the argon sorption data. | ||
CFA-5 becomes amorphous after prolonged heating at T > 250 °C, as indicated by VTXRPD data. However, the Ar gas sorption measurement, performed for the sample heated at 250 °C under vacuum for 1 h, shows that the material remains porous. The maximum uptake achieved at 77 K and p/p0 = 0.99 is 192 cm3 g−1 (Fig. 6). The micropore volume obtained from the sorption isotherm is 0.1 cm3 g−1 applying the NLDFT method.27 The adsorption data were fitted to the BET equation to give a surface area of 241 m2 g−1, the Langmuir surface area is equal to 278 m2 g−1. The pore size distribution calculated by fitting the sorption data shows that the volume of small pores, corresponding to the channels as described in the crystallographic section (<10 Å), is strongly reduced and the maximum is shifted to a lower value (5.5 Å), whereas the volume of larger pores remains nearly the same (Fig. 7). These larger pores are probably related to crystal defects or intercrystalline cavities.
Magnetic studies
The magnetic measurements of CFA-5 are presented in Fig. 8 as the temperature dependence of the effective magnetic moment μeff per asymmetric unit at 0.1 and 1.0 tesla. At 290 K, the effective moment of 1.37μB (0.1 T; 1.35μB at 1.0 T) reveals dominant strong antiferromagnetic exchange interactions within the compound since these moments are considerably smaller than the values expected for the two uncoupled Cu(II) centers (2.40–3.11μB).30 Upon cooling, the effective moment steadily decreases to 0.34μB (0.1 T; 0.32μB at 1.0 T) at 2.0 K, further indicating antiferromagnetic exchange interactions. The μeff curve exhibits a change in the curvature at approximately 45 K with a less steep slope at further decreasing temperatures indicating paramagnetic impurities or/and minor ferromagnetic exchange interactions within the compound. A rapid increase of χm (not shown) by decreasing the temperature in this interval is in agreement with this observation. Because of the high symmetry of the compound and the known tendency of compounds consisting of multiple coupled Cu(II) centers to contain paramagnetic impurities,31 the aforementioned explanation of this behaviour due to ferromagnetic interactions seems to be less probable.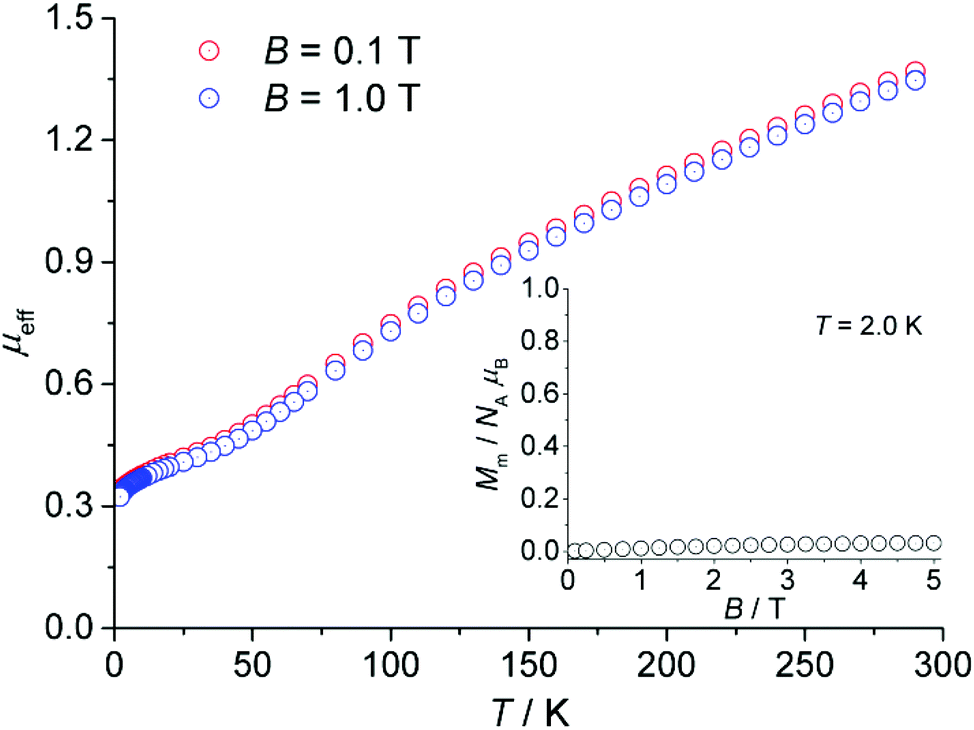 | ||
| Fig. 8 Temperature dependence of the effective magnetic moment μeff per asymmetric unit of CFA-5 (cf. Fig. 3b) at 0.1 and 1.0 tesla; inset: field dependence of the molar magnetization Mm at 2.0 K. | ||
The dependence of the molar magnetization Mm on the magnetic field B up to 5 T at 2.0 K is depicted in the inset of Fig. 8. In this region, the magnetization is almost independent of the applied field and retains a value close to zero. Therefore, the ground state of the compound corresponds to an S = 0 state, i.e. singlet spin ground state.
Catalytic studies
Cu-MOFs mimicking copper-containing enzymes like pMMO should be able to catalyse direct oxidation of methane to methanol with molecular oxygen. However, since the binding energy of the C–H bond of methane is quite high (421 kJ mol−1), a catalyst is indispensable to open reaction pathways with low activation energies comparable to the enzymatic oxidation with pMMO. Currently, solid catalysts for selective oxidation of methane with molecular oxygen in the gas phase need high reaction temperatures of more than 200 °C. These high temperatures favour total oxidation of methanol, which can be oxidised more easily than methane. Thus, for preliminary catalytic studies it is advisable to choose a substrate with an activated C–H bond like benzylic compounds,13ce.g. oxidation of tetralin gives tetralol and tetralon as products (see Scheme 2). As shown in Scheme 2, there are two major mechanisms. One is the radical autoxidation mechanism proceeding via tetralin hydroperoxide as an intermediate which is decomposed into the products. The latter step is by far not as simple as shown in Scheme 2, because two different propagation cycles consuming tetralin hydroperoxide in bimolecular elementary steps are postulated.32 The radical autoxidation can be enhanced by a catalyst through radical formation (initiation) and hydroperoxide decomposition (Haber–Weiss cycle).33 The other mechanism is analogous to the enzymatic mono oxygenation through a proton driven formation of a high-valent metal-oxo species from molecular oxygen which oxygenates C–H bonds forming the corresponding alcohols.34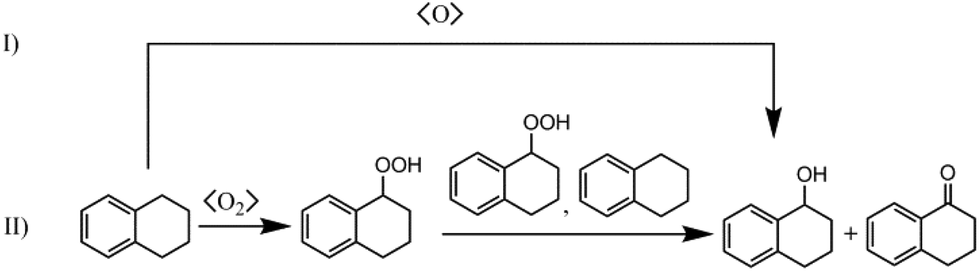 | ||
| Scheme 2 Reaction scheme of the oxidation of tetralin: (I) mono oxygenation according to nature (s-MMO, CP 450), (II) radical autoxidation mechanism. | ||
In order to benchmark catalytic results, not only CFA-5, but also [Cu(2-pymo)2] and [Cu3(BTC)2] (HKUST-1) were tested. [Cu(2-pymo)2] had already been used in the aerobic oxidation of tetralin by Xamena et al.13b They observed that [Cu(2-pymo)2] significantly enhances the tetralin hydroperoxide formation according to the radical autoxidation mechanism. Ryan et al. investigated both [Cu(2-pymo)2] and [Cu3(BTC)2] by DFT calculations and came to the conclusion that on both Cu-MOFs the decomposition of the tetralin hydroperoxide can only occur at the outer surface.35 In the case of [Cu(2-pymo)2] the pore window size is 5.04 Å and, thus, is smaller than the kinetic diameter of tetralin which is expected to be larger than 5.85 Å.35 Since the narrowest channel diameter of CFA-5 is 4.5 Å, tetralin and tetralin hydroperoxide are also not expected to access the inner surface of the MOF. In the case of [Cu3(BTC)2] the pore size is large enough (10.7 Å according to Schlichte et al.)36 and the coordination at the free coordination site of the Cu paddlewheel structural motif indeed occur, but the calculated activation energy was not lower than the one for the gas phase.35 Therefore, the inner surface of [Cu3(BTC)2] should not act as a catalyst. However, the DFT calculations showed that undercoordinated Cu clusters at the outer surface of Cu-MOFs should decompose tetralin hydroperoxide.35
In Fig. 9, the measured time dependence of the conversion of tetralin over time is shown for the three catalysts CFA-5, [Cu(2-pymo)2] and [Cu3(BTC)2]. All the three catalysts exhibit an induction period which is typical for the radical autoxidation mechanism (see Scheme 2, mechanism II). CFA-5 shows an activity very similar to that of [Cu(2-pymo)2], but both catalysts display a significantly shorter induction period than [Cu3(BTC)2]. Since we verified experimentally that no uncatalysed initiation of the reaction occurred, Cu at the outer surface of the MOFs seems to be an active site in the initial formation of tetralin radicals. Xamena et al. excluded leaching of copper.13b By an optimized ICP-MS measurement procedure we could detect only 3.5 ppm Cu in the solution after 24 hours which corresponds to leaching of approx. 1.5% of Cu from the original Cu-MOF. Since Xamena et al. have shown that copper acetate corresponding to an assumed copper leaching of 10% gives a much slower increase of conversion over time,13b we also assume that copper leaching is negligible. However, this will be investigated more deeply in future work.
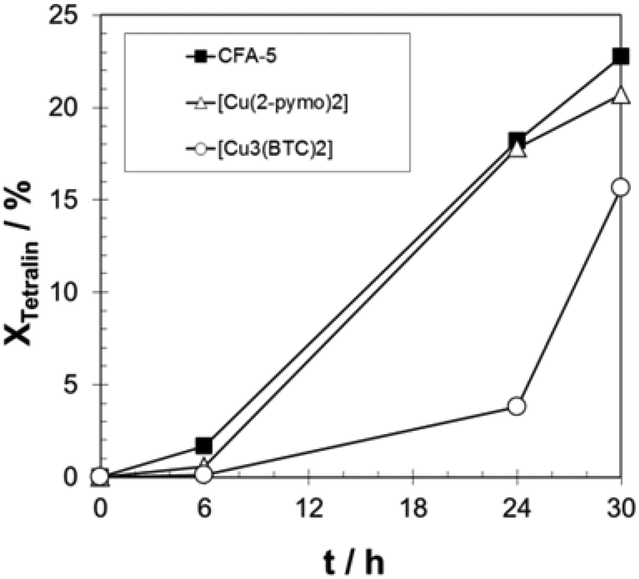 | ||
| Fig. 9 Progress of tetralin conversion over time for CFA-5, [Cu(2-pymo)2], and [Cu3(BTC)2]. T = 90 °C; 20 g tetralin, tetralin/Cu molar ratio: 2000/1. | ||
The observed induction period (see Fig. 9) is an indication for the radical autoxidation mechanism proceeding via tetralin hydroperoxide (THP) as an intermediate. As expected, the measured selectivities of THP are significant and reach more than 80% in the case of [Cu3(BTC)2] (see Fig. 10). Surprisingly CFA-5 and [Cu(2-pymo)2] show a different course of THP selectivity, because for these Cu-MOFs THP selectivity is not increasing, but decreasing with progressing time. Thus, the most likely explanation might be that CFA-5 and [Cu(2-pymo)2] catalyse the decomposition of THP, whereas [Cu3(BTC)2] is not able to do this. For further verification of this conclusion, a hot filtration test was performed after 6 h reaction time. As can be seen from Fig. 11 the reaction proceeds even after removal of the catalyst due to the radical autoxidation mechanism with an increasing selectivity to THP. This is a clear evidence that CFA-5 and [Cu(2-pymo)2] catalyse the decomposition of THP whereas [Cu3(BTC)2] does not. Since the course of conversion over time after 6 h is almost the same, irrespective of whether or not the catalyst is present (not shown), this allows the conclusion that all three catalysts do not catalyse the THP formation, if the initiation has been completed (which is evidently the case after 6 h).
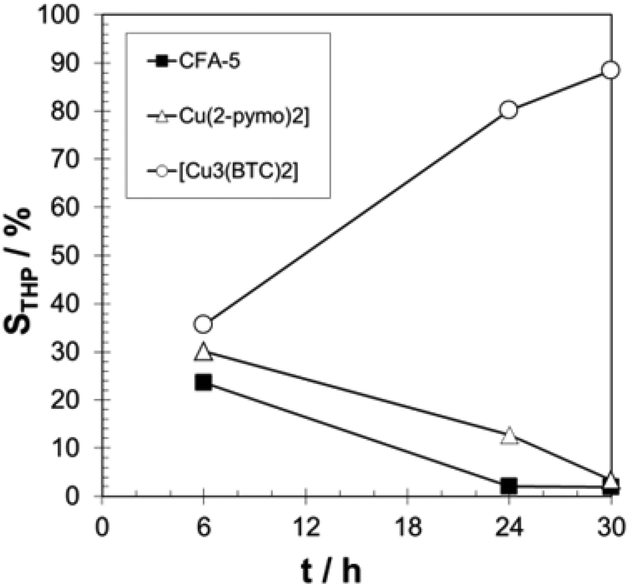 | ||
| Fig. 10 Progress of THP selectivity over time for CFA-5, [Cu(2-pymo)2], and [Cu3(BTC)2]. T = 90 °C; 20 g tetralin, tetralin/Cu molar ratio: 2000/1. | ||
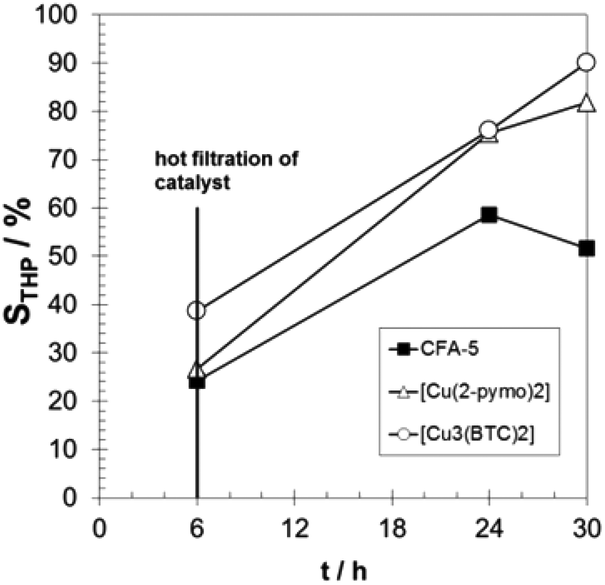 | ||
| Fig. 11 Progress of THP selectivity over time with hot filtration of the catalyst after 6 h for CFA-5, [Cu(2-pymo)2], and [Cu3(BTC)2]. T = 90 °C; 20 g tetralin, tetralin/Cu molar ratio: 2000/1. | ||
To sum up, based on our catalytic studies and on the published knowledge13b,35 a reaction mechanism can be proposed as shown in Fig. 12:
• Initiation of radical THP formation by all the investigated Cu-MOFs.
• Heterogeneously catalyzed THP decomposition by CFA-5 and [Cu(2-pymo)2].
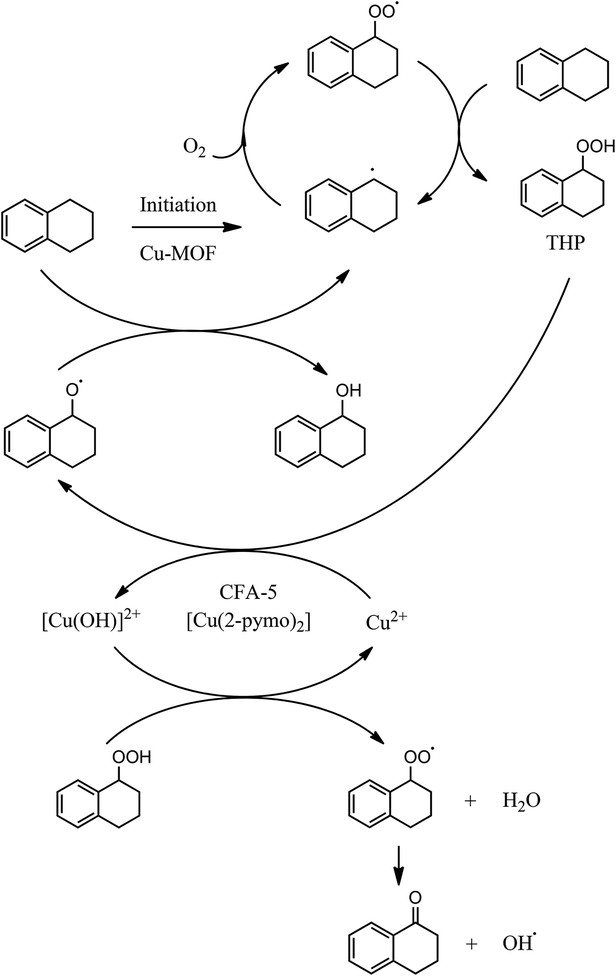 | ||
| Fig. 12 Proposed reaction mechanism as a combination of homogeneous radical propagation cycles and heterogeneously catalyzed steps (initiation, THP decomposition).35 | ||
Finally, due to the proposed mechanism as shown in Fig. 12, it can be assumed that CFA-5 will be also an active catalyst for the oxidants H2O2 or TBHO since it catalyzes the decomposition of hydroperoxide species. This is already known from other Cu-MOFs.13a,c
Conclusions
The work reported here focuses on the synthesis and characterization of a novel metal–organic framework constructed from wheel-shaped dodecanuclear {CuII(pz)(OMe)}12 coordination units, created by twelve copper(II) ions connected by twelve μ2-bridging MeO− groups and pyrazolate (pz−) ligands. The compound remains porous after solvent removal, as confirmed by TG, VTXRPD and gas sorption. The magnetic measurements reveal dominant strong antiferromagnetic exchange interactions within the compound. Catalytic tests of CFA-5 in the aerobic oxidation of tetralin show a similar performance as with [Cu(2-pymo)2]. Both Cu-MOFs catalyse the decomposition of tetralin hydroperoxide after 6 h reaction time and initiate its formation within the first 6 h reaction time. For both Cu-MOFs it can be suggested that low coordinated Cu centers at the outer surface are responsible for the observed activity since in both cases the substrate cannot access the pore system. Both Cu MOFs contain diazaheterocyclic linkers which might be relevant for their catalytic activity. Under the applied test conditions [Cu3(BTC)2], with its carboxylate linkers, does not seem to form catalytically active Cu centers, neither at the inner nor at the outer surface.Experimental
Materials and general methods
Commercially available reagents of analytical grade were used as received without further purification. [Cu(2-pymo)2] was synthesized according to the published procedure,37 [Cu3(BTC)2] was purchased from Sigma-Aldrich (Basolite C-300).Solvothermal method. A mixture of Cu(OAc)2·H2O (11 mg, 0.055 mmol) and H2-1,3-bdpb (15 mg, 0.056 mmol) was dissolved in MeOH (4 mL). 2,6-Dimethylpyridine (2,6-lutidine) (0.05 mL) was added and the solution was placed in a glass tube (10 mL). The tube was closed with a cap and heated at 120 °C for 3 d and subsequently cooled to room temperature. The green crystals were filtered off by suction and washed thoroughly with MeOH. The synthesis can be similarly performed at larger quantities (upscale factor: 50). Yield: 12 mg, 96% (based on Cu(OAc)2·H2O). [Cu2(C16H16N4)(OCH3)2] calcd: C: 47.67, H: 4.89, N: 12.35; found C: 47.44, H: 4.83, N: 12.28. IR: (cm−1) 1605 w, 1584 w, 1539 w, 1492 w, 1424 m, 1377 w, 1331 m, 1301 w, 1244 m, 1167 w, 1130 w, 1063 w, 1034 w, 900 w, 816 m, 776 s, 709 s, 700 m, 662 w, 547 s, 447 s. The IR and UV/Vis spectra of CFA-5 are shown in Figs S7 and S8,† respectively.
Microwave irradiation method. A mixture of Cu(OAc)2·H2O (11 mg, 0.055 mmol) and H2-1,3-bdpb (15 mg, 0.056 mmol) was dissolved in MeOH (3 mL). 2,6-Dimethylpyridine (2,6-lutidine) (0.05 mL) was added and the solution was placed in a Pyrex sample tube (10 mL). The tube was closed with a cap and placed in a microwave synthesizer (CEM, Discover S). The resulting mixture was heated to 150 °C at 300 W for 25 min and cooled to room temperature. The green microcrystalline material was filtered off and washed with MeOH. Yield: 12 mg, 96% (based on Cu(OAc)2·H2O). This material exhibited the same analytical results as the ones obtained by the solvothermal method.
Microwave irradiation method. A mixture of CuCl2 (9 mg, 0.067 mmol) and H2-1,3-bdpb (15 mg, 0.056 mmol) was dissolved in MeOH (2 mL). 2,6-Dimethylpyridine (2,6-lutidine) (0.05 mL) was added and the solution was placed in a Pyrex sample tube (10 mL). The tube was closed with a cap and placed in a microwave synthesizer (CEM, Discover S). The resulting mixture was heated to 140 °C at 100 W for 30 min and cooled to room temperature. The colorless microcrystalline material was filtered off and washed with MeOH. Yield: 11 mg, 54% (based on H2-1,3-bdpb). This material exhibited the same analytical results as the ones obtained by the solvothermal method.
Single-crystal X-ray diffraction
X-ray data for the single crystal structure determinations of H2-1,3-bdpb·MeOH, CFA-5 and CuCl(H2-1,3-bdpb) were collected on a Bruker D8 Venture diffractometer. Intensity measurements were performed using monochromated (doubly curved silicon crystal) MoKα radiation (0.71073 Å) from a sealed microfocus tube. Generator settings were 50 kV, 1 mA. Data collection temperature was −173 °C. APEX2 software was used for preliminary determination of the unit cell.38 Determination of integrated intensities and unit cell refinement were performed using SAINT.39 The structures were solved and refined using the Bruker SHELXTL Software Package.40 Selected crystal data and details of structure refinements for H2-1,3-bdpb·MeOH, CFA-5 and CuCl(H2-1,3-bdpb) are provided in Table 2. Complete crystallographic data for the structures H2-1,3-bdpb·MeOH, CFA-5 and CuCl(H2-1,3-bdpb) reported in this paper have been deposited in the CIF format with the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB21EZ, UK as supplementary publication no. CCDC 1006742-1006744.| Compound | H2-1,3-bdpb·MeOH | CFA-5 | CuCl(H2-1,3-bdpb) |
|---|---|---|---|
| a R 1 = ∑||F0| − |Fc||/∑|F0|. b wR2 = ∑[w(F02 − Fc2)2]/∑[w(F02)2]1/2. | |||
| Empirical formula | C17H22N4O | C18H22Cu2N4O2 | C16H18N4ClCu |
| Formula | C16H18N4, CH3OH | [Cu2(C16H16N4)(OCH3)2] | C16H18N4ClCu |
| M r/g mol−1 | 298.39 | 453.48 | 365.33 |
| T/K | 100(2) | 100(2) | 100(2) |
| Wavelength/Å | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Trigonal | Orthorhombic |
| Space group | P21/m (no. 11) |
R (no. 148) |
Pnma (no. 62) |
| a/Å | 4.7416(3) | 26.839(3) | 6.1784(8) |
| b/Å | 12.1325(7) | 26.839(3) | 12.9041(17) |
| c/Å | 14.0001(8) | 15.8317(16) | 19.866(3) |
| α/° | 90 | 90 | 90 |
| β/° | 95.107(2) | 90 | 90 |
| γ/° | 90 | 120 | 90 |
| V/Å3 | 802.19(8) | 9876.2(19) | 1583.8(4) |
| Z | 2 | 18 | 4 |
| D c/g cm−3 | 1.235 | 1.372 | 1.532 |
| μ/mm−1 | 0.080 | 1.955 | 1.549 |
| F(000) | 320 | 4176 | 752 |
| θ Range/° | 2.92 to 26.36 | 2.65 to 25.00 | 3.16 to 25.00 |
| Refls. collected | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 984 984 |
19551 |
12815 |
| Refls. unique | 1705 | 3874 | 1460 |
| R(int) | 0.0634 | 0.1439 | 0.0391 |
| GooF | 1.129 | 1.036 | 1.140 |
| R 1 [I > 2σ(I)]a | 0.0521 | 0.0679 | 0.0440 |
| wR2 (all data)b | 0.1049 | 0.1515 | 0.1243 |
| Largest diff. peak and hole/Å−3 | 0.244 and −0.219 | 0.763 and −0.925 | 0.448 and −0.441 |
Physical methods
Fourier transform infrared (FTIR) spectra were recorded with ATR in the range 4000–400 cm−1 on a Bruker Equinox 55 FT-IR spectrometer. The following indications are used to characterize absorption bands: very strong (vs), strong (s), medium (m), weak (w). Elemental analyses (C, H, N) were carried out on a Perkin-Elmer 2400 Elemental Analyzer. Thermogravimetric analysis (TGA) was performed with a TGA Q500 analyser in the temperature range of 25–800 °C under flowing nitrogen at a heating rate of 10 K min−1. The argon sorption isotherms at −196 °C up to p/p0 = 1 were measured using a Quantachrome Autosorb-I ASI-CP-8 instrument. High purity gas was used for the adsorption experiments (argon 99.999%). Ambient temperature X-ray powder diffraction (XRPD) patterns were measured with a Seifert XRD 3003 TT diffractometer equipped with a Meteor 1D detector operated at 40 kV, 40 mA, CuKα (λ = 1.54247 Å) with a scan speed of 10 s per step and a step size of 0.02° in 2θ. The variable temperature XRPD experiment data were collected in the 2θ range of 5–60° with 0.02° steps, with a Bruker D8 Advance diffractometer equipped with a Lynxeye linear position-sensitive detector, an MRI TCPU1 oven, in transmission geometry. The sample was loaded into a capillary (Hilgenberg) made from special glass no. 10, 0.5 mm diameter and 0.01 mm wall thickness. The patterns were recorded in a temperature range from 30 to 250 °C, in the 5–60° 2θ range, with one step per 1 s, and an angular step width of 0.02° in 2θ. Temperature program between measurements: heating rate (0.5 °C s−1), then 10 min isothermal. Magnetic susceptibility data of CFA-5 were recorded using a Quantum Design MPMS-5XL SQUID magnetometer. The polycrystalline samples were compacted and immobilized into cylindrical PTFE capsules. The susceptibility data were acquired as a function of the field (0.1–5.0 T) and temperature (2.0–290 K). All data were corrected for the contribution of the sample holder and the diamagnetic contributions of CFA-5 calculated from tabulated values (χdia(CFA-5) = –2.8 × 10−9 m3 mol−1 per asymmetric unit).Catalytic investigations
The method used to determine the catalytic activity is based on the work of Xamena et al.13b Catalytic tests were carried out in a three-necked flask equipped with a reflux condenser and a gas inlet tube. Before each experiment, the glassware was passivated for a period of 12 h with 1 M aqueous sodium hydroxide solution, subsequently neutralized by rinsing with 1 M hydrochloric acid for 30 min and finally washed with demineralized water and dried at 80 °C in an oven. With this passivation procedure, uncatalysed initiation of the reaction could be avoided. The reaction temperature was set to 90 °C and the applied molar ratio of tetralin/metal was always 2000:1. 20 g of tetralin were used both as a solvent and a reactant. The required amount of catalyst was calculated from the previously mentioned ratio. Synthetic air at a flow rate of 0.5 mL s−1 was adjusted by a needle valve. A HP 6890N GC with AutoSampler was used to analyze the reaction mixture. All samples were measured five times and the results were averaged. Peroxide concentration was determined by the TPP method.41 In time-resolved experiments, small samples of the reaction solution were taken after 6, 24 and 30 hours.
Conversion (X) of tetralin and selectivity (S) to tetralin hydroperoxide were calculated according to the following formulas:
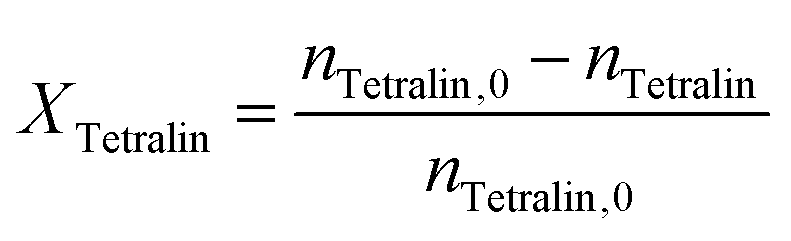
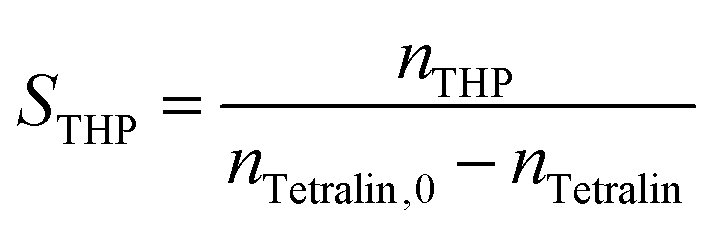
The sum of selectivities to THP, tetralol and tetralone was always larger than 95%. Thus, the consecutive oxidation of tetralol and tetralone was negligible and the carbon balance fulfilled.
Acknowledgements
This work was supported by a grant from the Ministry of Science, Research and Art Baden-Württemberg and by the German Research Foundation (DFG) within Priority Program “Porous Metal–Organic Frameworks” (SPP 1362, MOFs).Notes and references
- (a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed; (b) J. Teufel, H. Oh, M. Hirscher, M. Wahiduzzaman, L. Zhechkov, A. Kuc, T. Heine, D. Denysenko and D. Volkmer, Adv. Mater., 2013, 4, 635 CrossRef PubMed; (c) J. Sculley, D. Yuan and H.-C. Zhou, Energy Environ. Sci., 2011, 4, 2721 RSC; (d) D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44(2), 123 CrossRef CAS PubMed.
- (a) P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 5974 CrossRef CAS PubMed; (b) J. Y. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376 CrossRef CAS PubMed; (c) P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, J. Am. Chem. Soc., 2008, 130, 6774 CrossRef CAS PubMed; (d) R. C. Huxford, J. D. Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14(2), 262 CrossRef CAS PubMed.
- (a) S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249 CrossRef CAS PubMed; (b) O. Shekhah, J. Liu, R. A. Fischer and Ch. Wöll, Chem. Soc. Rev., 2011, 40, 1081 RSC.
- A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269 CAS.
- O. K. Farha, C. E. Wilmer, I. Eryazici, B. G. Hauser, P. A. Parilla, K. O'Neill, A. A. Sarjeant, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 9860 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 CrossRef CAS PubMed.
- (a) Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112 CrossRef CAS PubMed; (b) L. Ma and W. Lin, Top. Curr. Chem., 2010, 293, 175 CrossRef CAS PubMed; (c) M. Tonigold, Y. Lu, A. Mavrandonakis, A. Puls, R. Staudt, J. Möllmer, J. Sauer and D. Volkmer, Chem. – Eur. J., 2011, 17, 8671 CrossRef CAS PubMed; (d) A. Dhakshinamoorthy, M. Alvaro and H. García, Catal. Sci. Technol., 2011, 1, 856 RSC; (e) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333 CrossRef CAS PubMed.
- L. Jian, C. Chen, F. Lan, S. Deng, W. Xiao and N. Zhang, Solid State Sci., 2011, 13, 1127 CrossRef CAS.
- D. Jiang, T. Mallat, F. Krumeich and A. Baiker, J. Catal., 2008, 275, 390 CrossRef.
- D. Jiang, T. Mallat, D. M. Meier, A. Urakawa and A. Baiker, J. Catal., 2010, 270, 26 CrossRef CAS.
- N. T. S. Phan, P. H. L. Vu and T. T. Nguyen, J. Catal., 2013, 306, 38 CrossRef CAS.
- (a) S. Marx, W. Kleist and A. Baiker, J. Catal., 2011, 281, 76 CrossRef CAS; (b) F. X. Llabres i Xamena, O. Casanova, R. Galiasso Tailleur, H. Garcia and A. Corma, J. Catal., 2008, 255, 220 CrossRef CAS; (c) S. Wang, L. Li, J. Zhang, X. Yuan and C.-Y. Su, J. Mater. Chem., 2011, 21, 7098 RSC.
- M. Grzywa, C. Geßner, D. Denysenko, B. Bredenkötter, F. Gschwind, K. Fromm, W. Nitek, E. Klemm and D. Volkmer, Dalton Trans., 2013, 42, 6909 RSC.
- (a) M. Tonigold, Y. Lu, A. Mavrandonakis, A. Puls, R. Staudt, J. Möllmer, J. Sauer and D. Volkmer, Chem. – Eur. J., 2011, 17, 8671 CrossRef CAS PubMed; (b) V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchic and J. R. Long, Chem. Sci., 2011, 2, 1311 RSC.
- (a) L. Que and W. B. Tolman, Nature, 2008, 455, 333 CrossRef CAS PubMed; (b) J. T. Rubino and K. J. Franz, J. Inorg. Biochem., 2012, 107, 129 CrossRef CAS PubMed.
- (a) A. A. Mohamed, Coord. Chem. Rev., 2010, 254, 1918 CrossRef CAS; (b) H. V. R. Dias, H. V. K. Diyabalanage, M. G. Eldabaja, O. Elbjeirami, M. A. Rawashdeh-Omary and M. A. Omary, J. Am. Chem. Soc., 2005, 127, 7489 CrossRef CAS PubMed; (c) H. V. R. Dias and H. V. K. Diyabalanage, Polyhedron, 2006, 25, 1655 CrossRef CAS; (d) M. Casarin, C. Corvaja, C. di Nicola, D. Falcomer, L. Franco, M. Monari, L. Pandolfo, C. Pettinari, F. Piccinelli and P. Tagliatesta, Inorg. Chem., 2004, 43, 5865 CrossRef CAS PubMed; (e) S. M. Tekarli, T. R. Cundari and M. A. Omary, J. Am. Chem. Soc., 2008, 130, 1669 CrossRef CAS PubMed.
- (a) J. P. Zhang and S. Kitagawa, J. Am. Chem. Soc., 2008, 130, 907 CrossRef CAS PubMed; (b) J. P. Zhang, S. Horike and S. Kitagawa, Angew. Chem., Int. Ed., 2007, 46, 889 CrossRef CAS PubMed; (c) J. He, Y.-G. Yin, T. Wu, D. Li and X.-C. Huang, Chem. Commun., 2006, 2845 RSC.
- (a) G. Mezei, P. Braun and R. G. Raptis, Angew. Chem., Int. Ed., 2004, 43, 574 CrossRef CAS PubMed; (b) R. W. M. Hoedt, F. B. Hulsbergen, G. C. Verschoor and J. Reedijk, Inorg. Chem., 1982, 21, 2369 CrossRef; (c) G. Mezei, P. Baran and R. G. Raptis, Angew. Chem., Int. Ed., 2004, 43, 574 CrossRef CAS PubMed; (d) G. A. Ardizzoia, M. A. Angaroni, G. La Monica, F. Cariati, M. Moret and N. Masciocchi, J. Chem. Soc., Chem. Commun., 1990, 1021 RSC; (e) G. A. Ardizzoia, M. A. Angaroni, G. L. Monica, F. Cariati, S. Cenini, M. Moret and N. Masciocchi, Inorg. Chem., 1991, 30, 4347 CrossRef CAS.
- (a) S.-H. Li, H.-P. Huang, S.-Y. Yu and X.-P. Li, Chin. J. Chem., 2006, 24, 1225 CrossRef CAS; (b) M. Rancan, A. Dolmella, R. Seraglia, S. Orlandi, S. Quici, L. Sorace, D. Gatteschi and L. Armelao, Inorg. Chem., 2012, 51, 5409 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- Quantachrome Autosorb, Version 1.56, 2009 Search PubMed.
- V. A. Blatov, Struct. Chem., 2012, 23, 955 CrossRef CAS.
- D. T. Puerta and S. M. Cohen, Chem. Commun., 2003, 1278 RSC.
- H. N. Miras, I. Chakraborty and R. G. Raptis, Chem. Commun., 2010, 46, 2569 RSC.
- (a) G. Mezei and R. G. Raptis, Inorg. Chim. Acta, 2004, 357, 3279 CrossRef CAS; (b) M. A. Angaroni, G. A. Ardizzoia, T. Beringhelli, G. D'Alfonso, G. La Monica, N. Masciocchi and M. Moret, J. Organomet. Chem., 1989, 363, 409 CrossRef CAS.
- (a) P. I. Ravikovitch and A. V. Neimark, Colloids Surf., 2001, 187, 11 CrossRef; (b) S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, Academic Press, London, UK, 1982, p. 42 Search PubMed.
- F. Rouquerol, J. Rouquerol and K. Sing, in Adsorption by Powders & Porous Solids, Academic Press, San Diego, 1999 Search PubMed.
- J. Jagiello and M. Thommes, Carbon, 2004, 42, 1227 CrossRef CAS.
- H. Lueken, Magnetochemie, Stuttgart, Teubner, Germany, 1999 Search PubMed.
- See e.g. (a) A. P. Ginsberg, Inorg. Chim. Acta Rev., 1971, 5, 45 CrossRef; (b) E. Sinn, Inorg. Chem., 1970, 9, 2370 CrossRef; (c) A. Harada, M. Tsuchimoto, S. Ohba, K. Iwasawa and T. Tokii, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 654 Search PubMed; (d) F. P. W. Agterberg, H. A. J. Provo Kluit, W. L. Driessen, H. Oevering, W. Buijs, M. T. Lakin, A. L. Spek and J. Reedijk, Inorg. Chem., 1997, 36, 4321 CrossRef CAS PubMed.
- I. Hermans, T. L. Nguyen, P. A. Jacobs and J. Peeters, ChemPhysChem, 2005, 6, 637 CrossRef CAS PubMed.
- F. Haber and J. Weiss, Proc. R. Soc., 1934, 147, 332 CrossRef CAS.
- C. E. Tinberg and S. J. Lippard, Biochemistry, 2010, 49, 7902 CrossRef CAS PubMed.
- P. Ryan, I. Konstantinov, R. Snurr and L. Broadbelt, J. Catal., 2012, 286, 95 CrossRef CAS.
- K. Schlichte, T. Katzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81 CrossRef CAS.
- L. C. Tabares, J. A. R. Navarro and J. M. Salas, J. Am. Chem. Soc., 2001, 123, 383 CrossRef CAS.
- APEX2 Version 2011.6, Bruker AXS Inc Search PubMed.
- SAINT Version 8.32B, Bruker AXS Inc., 2013 Search PubMed.
- XL Version 2013/3 CrossRef CAS PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. M. Fares, M. El-Khateeb and K. J. Asali, J. Inorg. Organomet. Polym., 2003, 13, 143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Atomic coordinates, bond lengths and angles, packing diagrams, IR and UV/Vis spectra. CCDC 1006742–1006744. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01880j |
| This journal is © The Royal Society of Chemistry 2014 |