
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcium carbonate crystal growth beneath Langmuir monolayers of acidic β-hairpin peptides†
Haofei
Gong
a,
Yi
Yang
b,
Manuela
Pluntke
c,
Othmar
Marti
c,
Zsuzsa
Majer
d,
Norbert
Sewald
b and
Dirk
Volkmer
*a
aInstitute of Physics, Chair of Solid State and Materials Chemistry, Augsburg University, Universitätsstrasse 1, D-86159 Augsburg, Germany. E-mail: dirk.volkmer@physik.uni-augsburg.de
bDepartment of Chemistry, Organic and Bioorganic Chemistry, Bielefeld University, PO Box 100131, D-33501 Bielefeld, Germany
cInstitute of Experimental Physics, Ulm University, Albert-Einstein-Allee 11, D-89081 Ulm, Germany
dInstitute of Chemistry, Eötvös Loránd University, PO Box 32, H-1518 Budapest 112, Hungary
First published on 22nd September 2014
Abstract
Four amphiphilic peptides with designed hairpin structure were synthesized and their monolayers were employed as model systems to study biologically inspired calcium carbonate crystallization. Langmuir monolayers of hairpin peptides were investigated by surface pressure area isotherms, surface potential isotherms, Brewster angle microscopy (BAM), atomic force microscopy (AFM) and Fourier transform infrared (FTIR) spectroscopy. A β-hairpin conformation was found for all peptides at the air–water interface although their packing arrangements seem to be different. Crystallization of calcium carbonate under these peptide monolayers was investigated at different surface pressures and growth times both by in situ optical microscopy, BAM and ex situ investigations such as scanning electron microscopy (SEM) and transmission electron microscopy (TEM). An amorphous calcium carbonate precursor was found at the initial crystallization stage. The crystallization process occurred in three stages. It starts from the nucleation of amorphous particles being a kinetically controlled process. Crystal nuclei subsequently aggregate to large particles and vaterite crystals start to form inside the amorphous layer, with the monolayer fluidity exerting an important role. The third process includes the re-crystallization of vaterite to calcite, which is thermodynamically controlled by monolayer structural factors including the monolayer flexibility and packing arrangement of the polar headgroups. Thus, the kinetic factors, monolayer fluidity and flexibility as well as structure factors govern the crystal morphology and polymorph distribution simultaneously and synergistically.
Introduction
Living organisms are capable to build intricate shells and casings from calcium carbonate which are widely known as biominerals.1,2 Most of these biominerals are structurally well-organized composites of inorganic and organic components. Among the most frequently studied biominerals, nacre, the lustrous inner part of many mollusc shells, consists of alternating layers of aragonite crystal tablets that are interspersed with thin sheets of an organic matrix, the latter consisting of a complex mixture of proteins.3 These protein layers inside the mineral phase not only act as a structure component but also have been recognized to be a template or modifier of calcium carbonate mineralization.4,5 Such an unusual “brick-and-mortar” structure makes nacreous biominerals exceptionally tough and resistant.6 Some proteins isolated from the nacreous parts of mollusc biominerals are particularly rich in acidic amino acid residues, e.g. aspartic or glutamic acid.7In vitro experiments provide strong evidence for the hypothesis that these acidic peptides are directly involved in the mineralization process and play a crucial role in modulating the mineral crystallization process and thus determine the final morphology or polymorph of the minerals.3,8–15 Moreover, it was proposed that the uniform growth direction of calcium carbonate crystals induced by the biological organic matrix might relate to structurally well-organized packing motifs of carboxylate residues emanating from β-sheet domains of the acidic proteins.16 However, a direct structural relationship, in the sense of an epitaxial match between these self-organizing peptides and the juxtaposed CaCO3 crystal face has been subject of a controversial debate.17Interfacial properties of polypeptides in living organisms seem to play a key role in modifying calcium carbonate crystal growth since it is assumed that the initial crystal nucleation takes place at peptide-modified surfaces or interfaces. However, only few investigations employed artificial peptides as CaCO3 crystal growth modifiers, which included water-soluble peptides,18–20 and water-insoluble Langmuir monolayers.21,22 Insoluble monolayers of peptides in particular might form an organized supramolecular template matrix with a well-defined two-dimensional structure, thus providing a potential means to control the nucleation and crystallization of inorganic crystals. From an experimentalist's point of view it is straight-forward to adjust monolayer properties such as molecular conformation and/or charge density by changing the experimental conditions (i.e. monolayer surface pressure, pH and ion conc. of the aqueous subphase). The crystallization beneath peptide monolayers can be initiated at different conditions and in a controlled manner. In addition, the two-dimensional monolayer matrix structure can be characterized structurally, both by in situ methods and by ex situ characterization of the transferred Langmuir–Blodgett films, finally revealing the key factors which determine the surface-initiated crystallization process. Langmuir monolayers of peptides thus constitute reductionistic model systems for peptide-induced biomineralisation, i.e. to investigate crystal growth at self-organized organic matrices.
However, although monolayer-controlled crystal growth might seems to be a simplistic model system, diverse and partly contradictory hypotheses can be found in literature about calcium carbonate crystallization beneath monolayers of amphiphilic molecules covering a wide range of different molecular structures. In seminal studies, Mann and Heywood et al. attributed the oriented nucleation to the geometrical and/or stereochemical match between the monolayer packing arrangement and the oriented mineral surface.23–26 Later, a multistep assembly process from an amorphous to a crystalline phase was suggested by Xu et al.27 Other groups demonstrated that the CO2 evaporation rate effectively influences crystal growth beneath monolayers, which may add to or even overlay the template effect of the monolayer.28In situ synchrotron X-ray diffraction studies have demonstrated that additives such as poly(sodium acrylate) and Mg2+ ions dissolved in the aqueous subphase lead to changes in the kinetics of the crystallization process.29 Employing a vast range of structurally different, tailored amphiphiles it was concluded by Volkmer et al. that non-specific factors such as the monolayer's surface charge density or its effective dipole moment seems to be key factors that determine the morphology and polymorph of the crystals grown underneath the monolayer.30–33 In addition, monolayer flexibility and its corresponding dynamic structural changes during crystallization were shown to be crucial factors in the mineralization process.34
Continuing these model studies we here report on CaCO3 crystallization underneath monolayers of amphiphilic, structurally well-defined β-hairpin peptides which were chosen for reasons of structural analogy with natural polyacidic peptides often found in biominerals. These hairpin peptides are composed of alternating hydrophobic and hydrophilic amino acid residues that spontaneously fold into a D-Pro-Gly-induced β-hairpin conformation (Scheme 1) at the air–water interface not only by interfacial assembly but also by inter-strand hydrogen bonding interactions.35–37 Artificial amphiphilic hairpin peptides represent a novel class of functional model peptides that form insoluble monolayers. The film-forming properties of these molecules as yet have not been studied in depth with respect to different in-film molecular packing arrangements. The possibility to adjust the monolayer's surface charge density in conjunction with different molecular packing arrangements provides additional degrees of experimental freedom if compared to single strand model peptides comprised of alternating hydrophobic/hydrophilic residues. From an experimental point of view, the latter peptides, although structurally more closely related to natural proteins, are very difficult to prepare by standard solid-phase synthesis owing to their high propensity to form insoluble aggregates, which furthermore renders them difficult to spread as monolayer films in a controlled and reproducible fashion. Calcium carbonate crystallization beneath β-hairpin peptide monolayers having different amino acid sequences will be described in the following. Although these peptides have similar primary structure, it is found that CaCO3 nucleation and crystallization beneath each monolayer they form follow distinct pathways, which might be ascribed to the different peptide packing arrangements at the air–water interface.
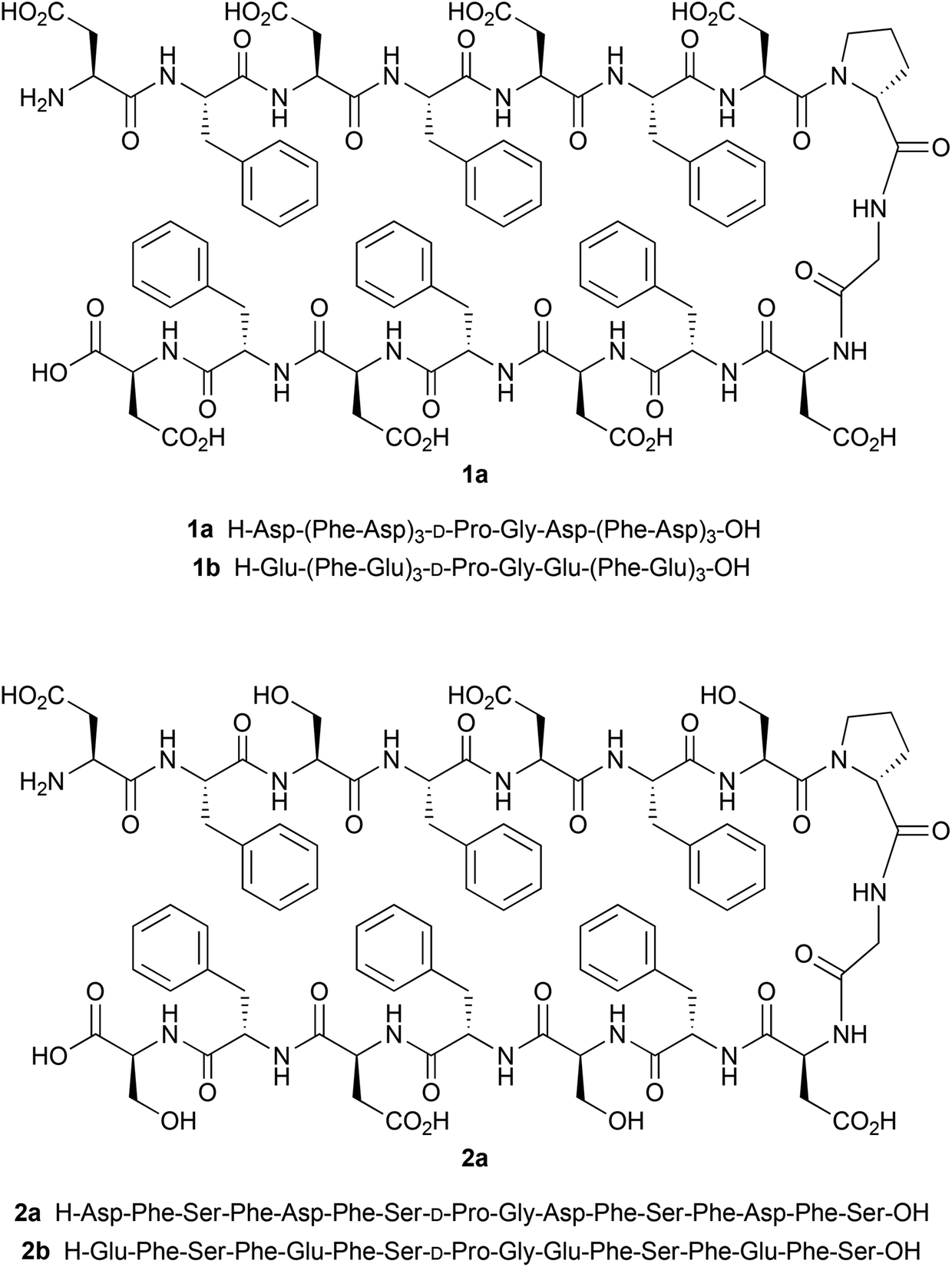 | ||
| Scheme 1 Molecular structures and primary sequences of the hairpin peptides. | ||
Experimental section
General
All chemicals were purchased from Sigma Aldrich (Hamburg, Germany), Acros (Geel, Belgium), Alfa Aesar (Ward Hill, USA) and VWR (Darmstadt, Germany) and were employed without additional purification. Dimethylformamide was distilled in vacuum from ninhydrin. 2-Chlorotrityl resin (0.6 mmol g−1), coupling reagents and all Fmoc-amino acids were purchased from Iris Biotech (Marktredwitz, Germany) and Orpegen (Heidelberg, Germany). Peptide synthesis was performed manually. Preparative RP-HPLC was done on a Thermo Separation Products system equipped with a Vydac 218 TP 1022 efficiency column (22 × 250 μm, 10 μm) with water–acetonitrile gradients as the eluent and UV detection at 220 nm. Analytical RP-HPLC was performed on a Thermo Separation Products system equipped with a Phenomenex Jupiter 4 μ Proteo 90 Å column (250 × 4.6 mm, 4 μm) with water–acetonitrile gradients as the eluent and UV detection at 220 nm.MALDI-ToF mass spectra were measured on a Voyager DE Instrument (PE Biosystems, Weiterstadt, Germany) mounted with a 1.2 m flight tube. 2,5-Dihydroxybenzoic acid was used as the matrix. Depending on the mass range the ions were accelerated at 15 to 25 kV with the option of detecting positive or negative ions. The instrument default calibration was used for calibrating the mass axis.
Manual solid phase peptide synthesis – general procedure
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:96, v/v) for 10 min at room temperature to cleave the Fmoc group. The resin was washed with CH2Cl2 (4×), DMF (4×), CH2Cl2 (4×), CH3OH (4×) and dried under vacuum overnight.
:2:96, v/v) for 10 min at room temperature to cleave the Fmoc group. The resin was washed with CH2Cl2 (4×), DMF (4×), CH2Cl2 (4×), CH3OH (4×) and dried under vacuum overnight.
For peptide elongation the corresponding Fmoc-amino acid (3–4 equiv. with respect to resin loading) was dissolved in a minimum volume of DMF (1 mmol amino acid mL−1) and pre-activated for up to 5 min with 3–4 equiv. of TBTU, dissolved in a minimum volume of DMF, and 6 equiv. DIPEA were added to the reaction mixture. The pre-activated Fmoc-amino acid was then added under argon to the resin loaded with the first amino acid or peptide and agitated for 45 minutes at room temperature. The reaction progress result was monitored by MALDI-ToF MS or Kaiser test.
For capping of unreacted amino groups 3 equiv. Ac2O (based on resin loading) and 3 equiv. Et3N, dissolved in DMF, were added and the mixture was agitated at room temperature for 30 min. Then the resin was washed with CH2Cl2 (4×), DMF (4×), CH2Cl2 (4×).
After assembly of the protected peptide on the resin, it was cleaved by adding 5 mL TFA in CH2Cl2 (1%, v/v). After 5 min the resin was filtered and the filtrate was neutralized with pyridine. The cleavage was repeated once and the combined filtrates were evaporated to dryness under reduced pressure. The crude fully protected peptides were lyophilized.
Deprotection of the protected peptides was achieved by treatment with the cleavage cocktail TFA–H2O–TIS (95:2.5:2.5) for several hours at ambient temperature. The crude products were purified by preparative HPLC using acetonitrile–water–TFA gradients. The pure peptides were obtained in low yields because of their high tendency towards aggregation and precipitation.
MS (MALDI-ToF): m/z = 1978.37 [M + H]+, 1999.42 [M + Na]+, 2015.59 [M + K]+.
Calculated Mass (monoisotopic): [M + H]+ = 1977.73, [M + Na]+ = 1999.71, [M + K]+ = 2015.69.
MS (MALDI-ToF): m/z = 2086.92 [M + H]+, 2108.32 [M + Na]+, 2124.35 [M + K]+.
Calculated Mass (monoisotopic): [M + H]+ = 2087.84, [M + Na]+ = 2109.83, [M + K]+ = 2125.80.
MS (MALDI-ToF): m/z = 1886.83 [M + Na]+; calculated mass (monoisotopic): [M + Na]+ = 1885.72.
MS (MALDI-ToF): m/z = 1941.36 [M + Na]+, 1957.61 [M + K]+; calculated mass (monoisotopic): [M + Na]+ = 1941.78, [M + K]+ = 1957.76.
CD spectroscopy
CD spectra were obtained on a J-810 spectrometer equipped with a CDF-4265 Peltier unit for temperature control (Jasco, Groß-Umstadt, Germany). The spectra were recorded using a 0.2 mm path length quartz cell in the range of 185–300 nm at a scanning rate of 50 nm min−1 with three accumulations, a data pitch of 0.2 nm, a spectral band width of 1 nm and a response time of 1 s. Molar ellipticity per amino acid residue [θ]mrw was calculated as follows: [θ]mr = θ/(10·N·c·l). θ represents the ellipticity in millidegrees, N the number of amino acid residues, c the molar concentration in mol L−1 and l the cell path length in cm. The CD spectra were smoothened using the means movement algorithm.Langmuir monolayer investigations
Measurements of surface pressure area (π-A) isotherms and film depositions were carried out on a double barrier. Langmuir balance (NIMA 702 BAM, Nima Technology, UK). The Teflon trough has large dimensions (72 cm long × 10 cm wide × 0.5 cm deep). The volume of the subphase is about 360 mL in all experiments.The surface pressure of the monolayers was measured using a filter paper Wilhelmy plate. The Langmuir film was formed by spreading a chloroform solution of peptides (0.2 mg mL−1, with 10% DMF) onto Milli-Q purified water (ρ > 18 MΩ cm). After evaporation of the solvent (approx. 15 min), π-A isotherms of the spread films were recorded at room temperature (approx. 22 °C) by compressing or expanding the Langmuir film at a barrier speed of 15 cm2 min−1. Surface potential measurements were performed with a Trek electrometer. A vibrating electrode was placed about 2 mm above the air–water interface and a stainless steel plate as the reference electrode was immersed in the subphase. The potential solubilisation of peptides in the aqueous subphase was tested by measuring at least 3 repeated compression/decompression cycles of all monolayer samples (pure water or 9 mM CaCl2). In no case any shift of the limiting area up to a maximum compression of 20 mN m−1 was found.
Monolayers and multilayers of peptides were transferred vertically at 15 mN m−1 on freshly cleaved mica, CaF2, and silicon substrates for AFM, FT-IR and XPS measurements, respectively. The transfer speed was 2 mm min−1 for both upwards and downwards strokes.
Crystal growth experiments
Solutions of calcium bicarbonate were prepared by bubbling carbon dioxide gas through a stirred aqueous solution of Ca(HCO3)2 (9 mM) for a period of 2 h. The monolayer was formed by adding 0.2 mg mL−1 of peptide solution (chloroform–DMF, 90%/10% vol) drop-wise onto the surface of freshly prepared calcium bicarbonate solution followed by compression to a pressure of 0.1 mN m−1. Crystal growth underneath the monolayer was studied at several time intervals either by in situ optical microscopy, Brewster angle microscopy or by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). For SEM the crystals floating at the air–water interface were transferred onto glass cover slips laid on the monolayer. TEM samples were prepared by carefully contacting the Cu grids with the monolayer and subsequent lift-off.Analytical characterization techniques
Infrared spectra were obtained using a Bruker IFS113v spectrometer. Raman spectroscopy was performed with a Horiba Jobin Yvon spectrometer. Brewster angle microscopy (BAM) was carried out with a NIMA Langmuir trough (NIMA 702 BAM) using a BAM-2 (I-Elli2000 supplied by NFT, Nanofilm Technologie, Göttingen, Germany). Images of the films were recorded with a lateral resolution of 2 μm. Crystal morphologies were transferred onto cover slides for SEM observation. Scanning electron microscopy (SEM) was carried out on Hitachi Tabletop 1000. Atomic force microscopy (AFM) was performed with the Agilent 5500 AFM and standard scanning probes from Nanosensors (PPP-FMR) with radii typically smaller than 7 nm (nominal force constant k = 2.8 N m−1). Transmission electron microscope (TEM) investigations were carried out on a Philips CM20 operating at 200 kV. X-ray photoelectron spectra (XPS) were acquired with a Physical Electronics PHI 5800 Multi ESCA system with monochromic Al Kα radiation.Results and discussion
Conformational analysis of the hairpin peptides by CD spectroscopy
The solution conformation of peptides 1a and 1b (Scheme 1), respectively, was investigated in hexafluoroisopropanol (HFIP) and HFIP–H2O solutions. Halogenated alcohols such as TFE and HFIP are widely applied solvents for peptides in CD spectroscopy because of their excellent solubilizing properties for some otherwise hardly soluble peptides such as β-sheet derivatives, and their inclination to stabilize ordered structures, e.g. α-helical conformations in peptides. Structures containing turns, β-hairpins, β-sheets, and hydrophobic clusters are also observable in the presence of TFE or HFIP. HFIP is a better hydrogen bond donor but a poorer hydrogen bond acceptor compared to water.38 HFIP preferentially serves as a hydrogen bond donor interacting with the main chains of the peptide. This solvation preference of HFIP leads to enhanced intra-chain amide to carbonyl hydrogen bonding.The peptides were designed as β-hairpins and their folded conformation is based on three stabilizing factors: (i) hydrogen bonds between backbone amide moieties across the strands, (ii) side chain interactions across two β-strands, and (iii) the presence of a reverse turn (β-turn). In particular, the interaction between aromatic side chains forming hydrophobic clusters as a stabilizing effect has been shown for a number of β-hairpin peptides and it is apparent as an aromatic contribution in the CD curve.39
The CD spectrum of 1a in HFIP indicates the predominance of β-turns, e.g. βI–III type with random coil contributions, characterized by negative bands at 195 nm, a broader one centralized at 212 nm (see Fig. 1a) and a weak one at 230 nm. Solvents like TFE or HFIP in principle promote sheet or helix formation by reducing the water activity.40 However, the overall influence depends on the structure propensity of the sequence. Fluorinated alcohols like TFE or HFIP have been found to attenuate hydrophobic interactions between residues that are distant in the peptide sequence, like e.g. across strands in some β-sheets, and, therefore, are able to disrupt these structures.38 In contrast to peptide 1a, the CD spectrum of 1b in HFIP indicates the predominance of a β-hairpin, composed of β-sheet and β-turn conformations, with the typical positive band at 194 nm and the negative band at 221 nm, as shown in Fig. 1b. The positive shoulder at 202 nm that increased with concentration (data not shown) is characteristic of the β-turn present in the β-hairpin conformation.
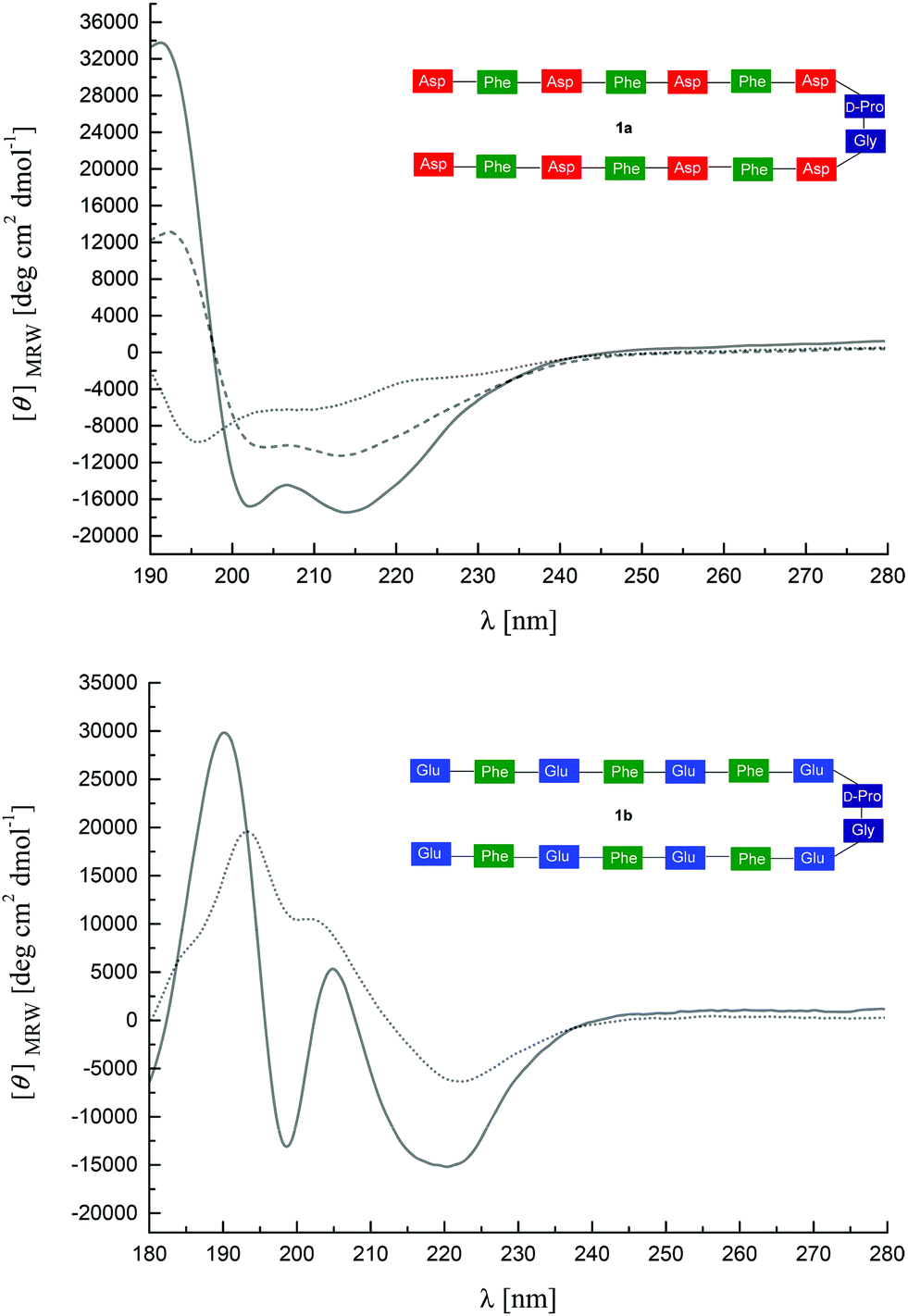 | ||
| Fig. 1 (a) CD spectra of 1a (0.5 mg mL−1 in HFIP, dotted line; 0.5 mg mL−1 in HFIP–H2O 1:1, dashed line; 0.33 mg mL−1 in HFIP–H2O 1:2, solid line), (b) CD spectra of 1b (0.3 mg mL−1 in HFIP, dotted line; 0.15 mg mL−1 in HFIP–H2O 1:1, solid line). | ||
A conformational change was observed for both peptides upon addition of water to the HFIP solutions. The CD spectrum of the Asp-containing peptide 1a indicated increasingly ordered structural units, e.g. with β-hairpin/β-turn (mainly βI–III) characteristics as well as some aromatic contributions of the Phe residues (positive band at 194 nm, increase in negative intensity at 215 nm) with increasing water contents (Fig. 1a). Likewise, the CD-spectrum of the Glu-containing peptide 1b showed a positive band at 190 nm and a negative band at 220 nm, also indicating β-hairpin conformation (Fig. 1b). However, for both peptides 1a and 1b the positive CD effects at 190–194 nm are accompanied by negative bands at 200–203 nm and additional positive bands at 207–208 nm. This unusual pattern in aqueous solutions can be assigned to an interaction of aromatic chromophores. Such effects on hairpin stability have in particular been described for specific Trp/Trp, Trp/Tyr, and Tyr/Tyr interactions.39 It has been suggested that they efficiently stabilize the secondary structure and can be employed to monitor the cross-strand interaction between the aromatic side chains. Moreover, such orthogonal arrangements of aromatic rings have been observed in proteins and have been suggested as a source of tertiary structure stabilization.41 This shows that the increase of the water concentration stabilizes the β-hairpin conformation especially of peptide 1b, presumably by hydrophobic interactions between the phenylalanine side chains.
Monolayer properties of hairpin peptides
Fig. 2 shows the π-A isotherms of all peptides on water and CaCl2 subphase at room temperature (ca. 22 °C). The isotherms of the peptides 1a and 1b on the water subphase show two liquid expanded stages separated by a phase transition. The limiting area of the peptides on the water subphase is 225 Å2 for 1a, and 250 Å2 for 1b, respectively.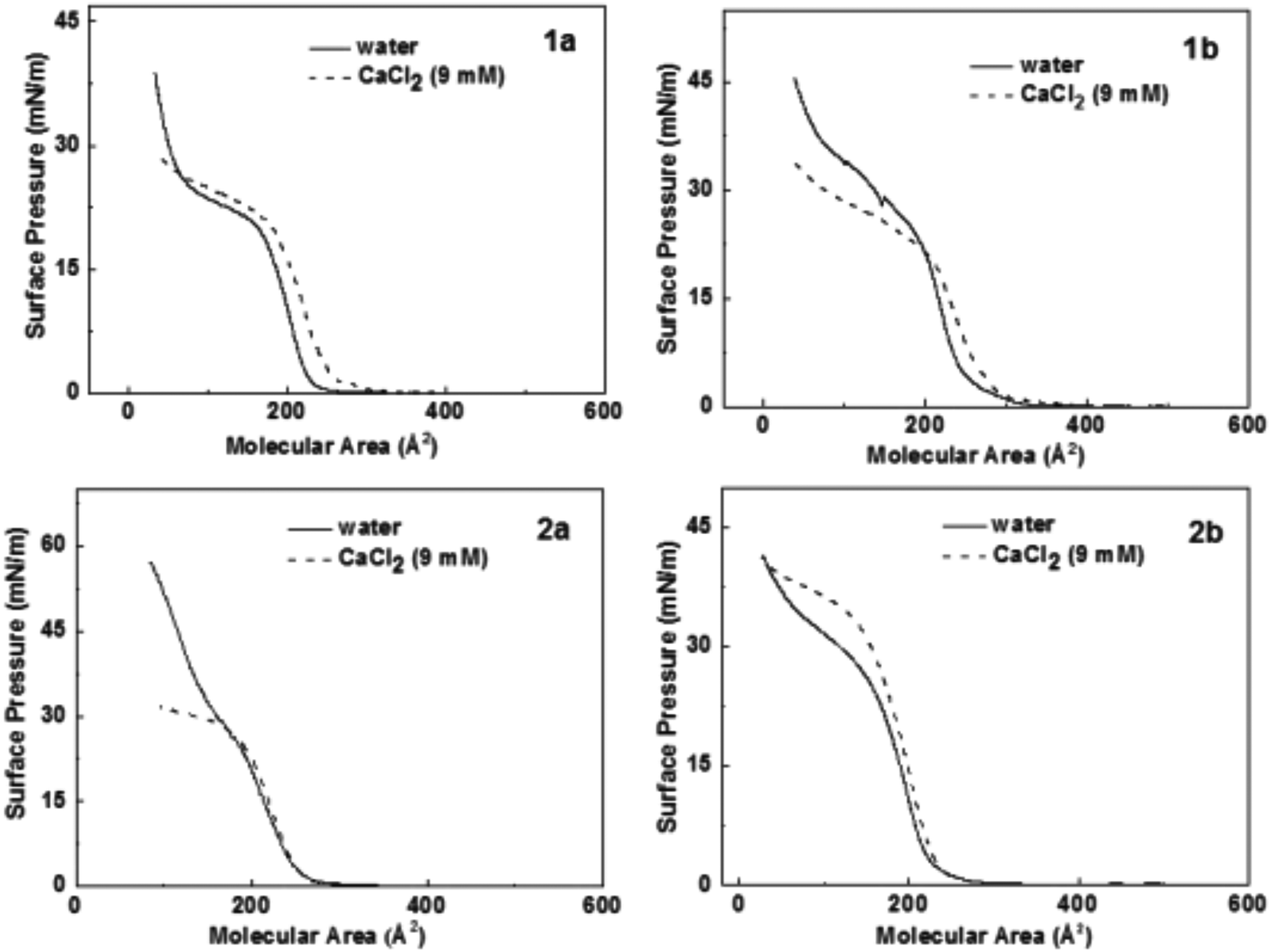 | ||
| Fig. 2 Surface pressure area isotherms of all hairpin peptides at 22 °C. The solid lines indicate the isotherms on pure water surface. The dashed lines indicate the isotherms on aqueous CaCl2 (9 mM) subphase. | ||
Molecular modelling of hairpin peptides assuming an ordered head-to-head β-hairpin packing arrangement42 suggests an occupied area of 259 Å2 for one peptide (Scheme 2), which closely matches the limiting area of peptide 1b on the water subphase. This result indicates that 1b organizes in a head-to-head packing arrangement on the water surface as shown in Scheme 2, while a relatively smaller limiting area of 1a suggests a less ordered peptide packing arrangement on the water surface.
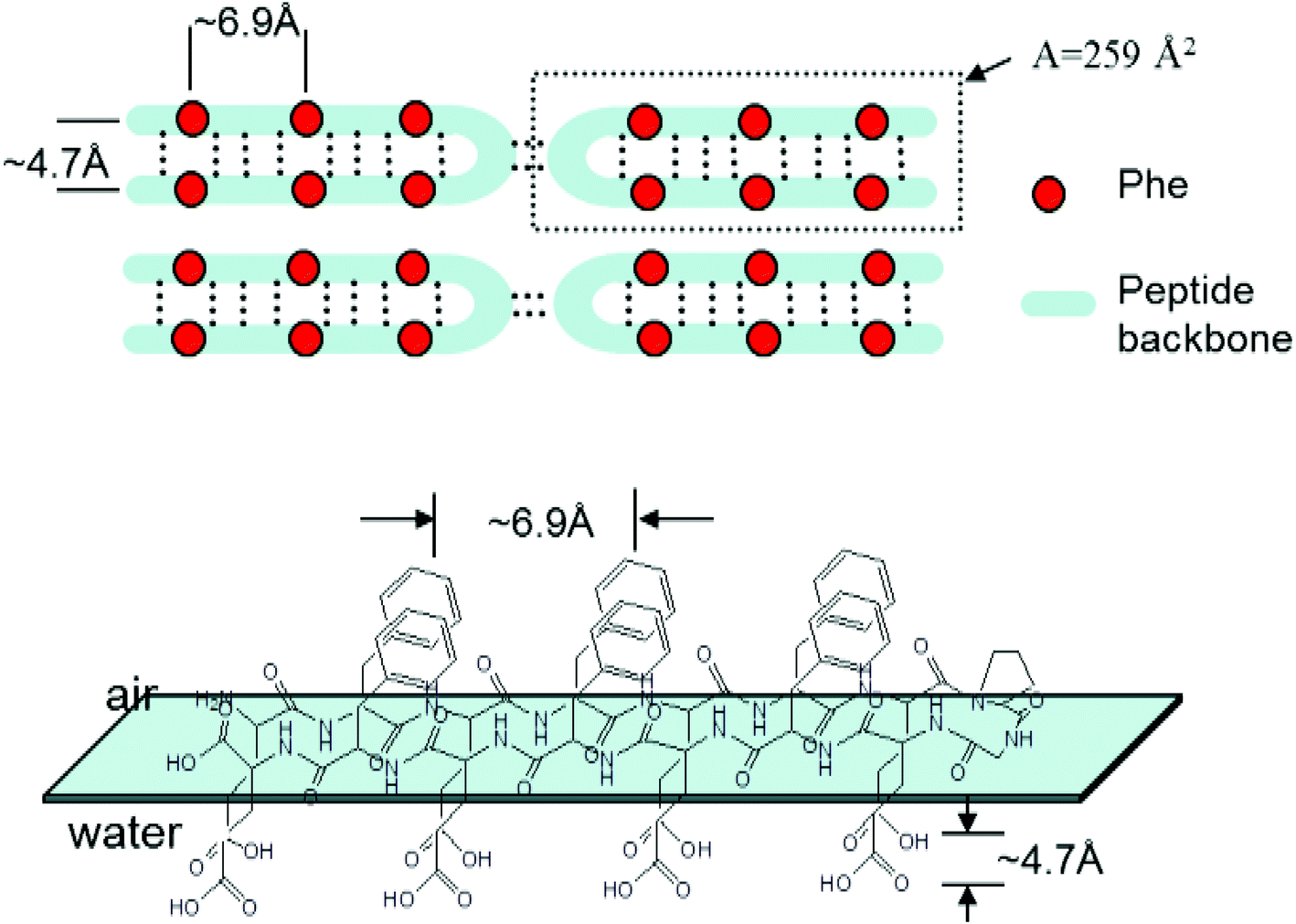 | ||
| Scheme 2 β-Hairpin packing arrangement of the peptides at air–water interface. The interstrand distance and the distance between two adjacent phenyl group was taken from ref. 42. | ||
The surface pressure increases after the phase transition of both peptides 1a and 1b on the water surface, and no obvious collapse pressure was observed within the range of the compression, demonstrating a strong flexibility of the peptide monolayers, i.e. the monolayers are highly compressible at very low surface area. In addition, the limiting area of the second liquid condensed phase is about half the area of the first liquid expanded limiting area for peptide 1a and 1b on the water surface, which indicates that the peptides overlap to form a double layer during the phase transition. For peptide 2a, a significantly higher surface pressure was observed compared to 1a or 1b at the same molecular area, suggesting higher flexibility of peptide 2a on the water surface. The isotherms of peptide 2b have a shape similar to the isotherms of 1b except for a smaller limiting area, which points at a tighter packing of 2b on the water surface than 1b.
On the subphase of CaCl2, the isotherms of the peptides 1a and 1b both show significant shifts towards larger molecular area. The limiting areas both are about 30 Å2 larger than the ones on pure water surface, while the limiting areas for the peptides 2a and 2b change to a smaller value. The results indicate that the packing arrangement of the peptides on the Ca2+ ion subphase is closely related with the density of charged carboxyl groups.
The ordered head to head packing arrangement of peptide 1b is considered to be a result of the subtle balance between hydrophobic interactions and carboxyl group interactions. However, on a subphase with calcium ions, this interaction balance is broken due to Ca2+ ion binding. Ion binding weakens intermolecular hydrogen bonding interactions and thus alters the inter- and intra-peptide distances. This explains the increase of the limiting areas of the peptides on Ca2+ ion subphase. Calculations that imply the modification of the distances between adjacent β-strands revealed that an interstrand distance change from 4.7 Å to 5.1 Å can explain the limiting area change of peptide 1b on a CaCl2 subphase.43
BAM investigations of the peptides spread on water and on an aqueous CaCl2 subphase were performed in order to elucidate monolayer properties in depth. Typical BAM images are shown for 1b and 2b in Fig. 3, because the monolayer morphologies of 1a and 1b as well as the structures of 2a and 2b are similar both on water and CaCl2 subphase. The BAM images of the peptides 1a and 1b on water show large domains with small holes inside (Fig. 3a). Upon further compression, a uniform film was observed within the resolution of BAM (not shown). However, on the subphase of CaCl2, the monolayer consists of many small domains at the onset pressure (Fig. 3b). This monolayer morphology change suggests that the intermolecular interactions change in the monolayer after binding with Ca2+ ions, which is consistent with the results of surface pressure-area isotherms.
 | ||
| Fig. 3 Brewster angle microscopy (BAM) images recorded for a monolayer of 1b and 2b spread on pure water and aqueous CaCl2 (9 mM) subphase at the onset point of surface pressure. (a) 1b on water subphase. (b) 1b on CaCl2 subphase. (c) 2b on water subphase. (d) 2b on CaCl2 subphase. (The size of each BAM image corresponds to a monolayer area of 430 μm in width.) | ||
Peptides 2a and 2b form larger domains on the water subphase at the onset of the surface pressure, while the film splits into many small domains on the subphase of CaCl2. These domains merge and form a homogeneous monolayer at high pressure. The morphology difference of 2a or 2b on water and CaCl2 subphase also suggests a strong binding interaction between Ca2+ ion and peptide monolayers.
Characterizations of peptide Langmuir–Blodgett films
The morphologies of all peptide Langmuir–Blodgett films (1a, 1b, 2a and 2b) transferred from water to mica were imaged with AFM in tapping mode (Fig. 4 and ESI†). All four peptides form dense films on hydrophilic surfaces, each with a distinct morphology. Both peptides 1b and 2b organize in Langmuir films to give fibrous structures, while peptides 1a and 2a form connected clusters with random shapes. | ||
| Fig. 4 AFM images of peptide 1b monolayer on mica transferred from a pure water subphase at π = 15 mN m−1. (a) scanning area 1 μm × 1 μm. (b) scanning area 0.2 μm × 0.2 μm. | ||
For the LB films with peptide 1b, a distinctive arrangement was observed (Fig. 4a). The molecules were seen to form a highly regular stripe pattern with no preferential orientation, indicating lack of correlation between the peptide arrangement and the crystalline orientation of mica. Thus it can be excluded that the substrate itself has an impact on the structure of the peptide film. The measured film height is about 1 nm, confirming that only one monolayer has been transferred to the mica substrate. Phase images of the LB films (providing higher lateral resolution than the topographies) are shown in Fig. 4b and reveal a double stripe pattern with a separation of 3.1 nm ± 0.1 nm. A few single strands are also observed. As the phase of an oscillating AFM tip depends on the specimen properties (such as elasticity and adhesion), the strong contrast in the AFM phase images may result from the tip interaction with hydrophobic and hydrophilic areas of the sample, or more specifically, with the peptide molecules (hydrophobic sidechains and hydrophilic termini) and mica (in the film gaps) or partially adsorbed water molecules. The observed stripe pattern therefore confirms the assumption that the hairpins form β-sheets containing opposing rows of equally oriented molecules (each with a width of about 3 nm).
AFM scans were repeated on monolayers formed from different subphases from peptide 1b. The LB films transferred from CaCl2 (0.01 M) subphase exhibit shapes similar to the ones discussed above. The film height is 1.00 nm ± 0.03 nm, indicating that the subphase ions have not altered the dimensionality of the peptide film.
The secondary structures of the peptides were analyzed by FT-IR spectroscopy. Multilayers (i.e. 40 layers) of the peptides 1a and 1b were transferred onto CaF2 substrates and analysed, as shown in Fig. 5. Transfer ratios were recorded during multilayer transfer on CaF2 substrates and they were found to be close to unity during all cycles and for all peptides being transferred. The IR absorption band between 1600–1700 cm−1 is assigned to the amide I stretching band, while the peak position in this range is characteristic for the secondary structure of peptides. The amide I vibrational band around 1630 cm−1 and 1696 cm−1 existing in all peptide spectra indicate the presence of similar β-pleated sheet structure motifs of all peptides at the air–water interface.44 FT-IR spectra were also recorded for peptide 1b on a CaCl2 subphase, as shown in Fig. 5b. While the amide I band at 1630 cm−1 is still visible, the relative absorption intensity was significantly reduced due to the interactions with calcium ion stemming from the aqueous subphase.
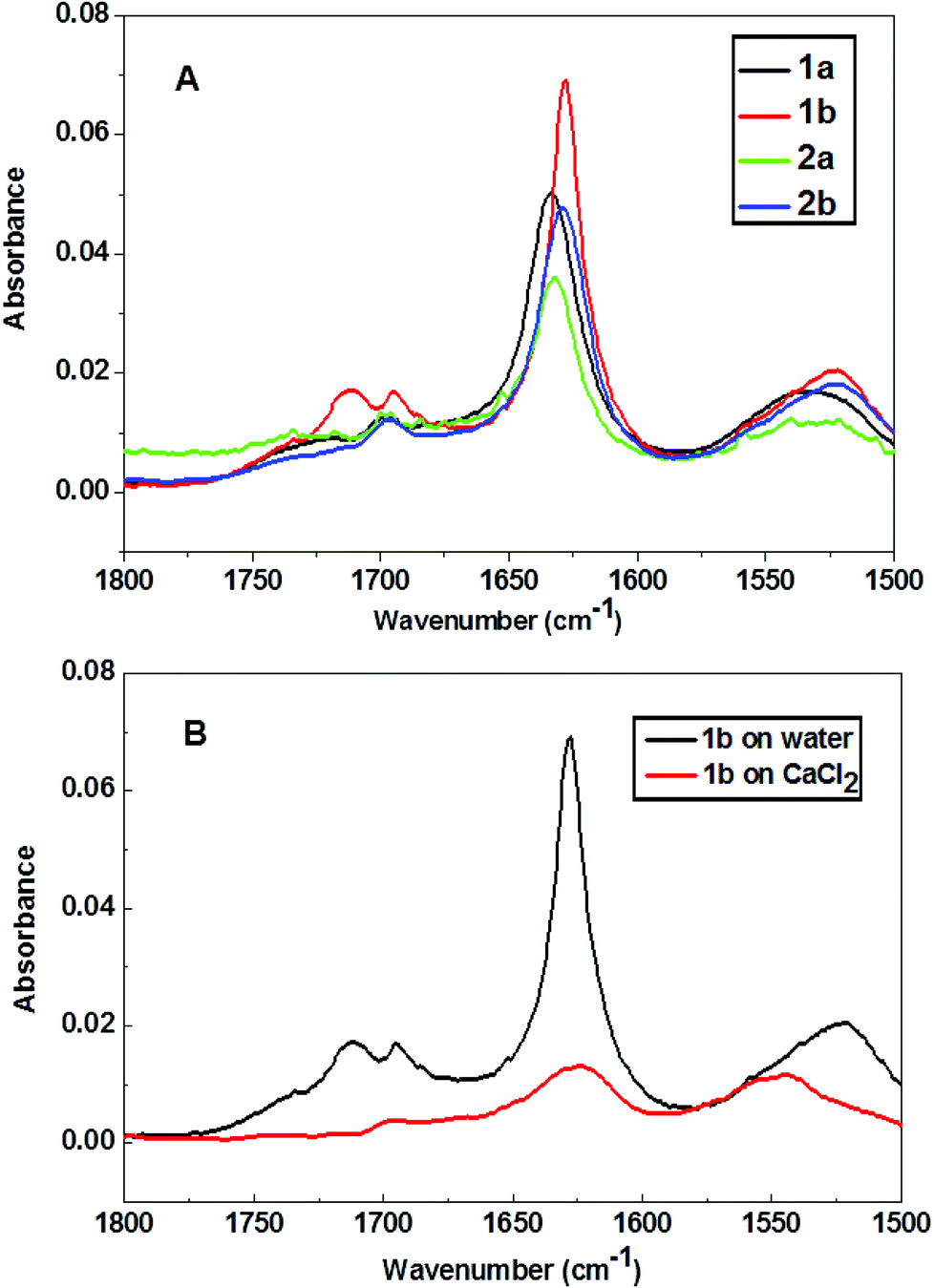 | ||
| Fig. 5 (A) FT-IR spectra of peptide LB multilayers transferred from water subphase at 15 mN m−1. (B) FT-IR spectra of peptide 1b multilayers transferred from water and CaCl2 subphase (9 mM) at 15 mN m−1. | ||
Calcium carbonate crystallization beneath peptide monolayers
Fig. 6 shows calcium carbonate crystals isolated from beneath a monolayer of peptide 1a after 10 min to 4 hours growth time. In the first 10 minutes, olive-shaped particles with the size of 300–500 nm were observed by TEM (Fig. 6a). They were very thin and could be easily decomposed by the high energy electron beam of the microscope. High Resolution TEM (HR-TEM) images showed that the particles are composed of amorphous calcium carbonate particles and contained single crystal domains in their interior (Fig. 6c). After 1 hour of growth, the crystals dominating the surface were pancake-shaped (Fig. 6b). Their Raman spectra showed a peak at 752 cm−1, characteristic of vaterite polymorphs (see Fig. S3, ESI†). After another 3 hours, two types of calcite crystals were isolated at the interface, as shown in Fig. 6d and e. They were oriented with the {10.0} and {12.2} crystal faces pointing towards the monolayer. Remarkably, these orientations were only observed beneath 1a peptide monolayers. | ||
| Fig. 6 Crystallization of CaCO3 beneath peptide 1a monolayer at 0.1 mN m−1. (a) TEM image of crystals separated after 10 min growth time. (b) SEM images of crystals separated after 1 hour. (c) High resolution TEM image of crystals separated after 10 min growth time. (d) and (e) SEM images of crystals separated after 4 hours. | ||
Calcite crystals with a <01.2> orientation were dominating the surface for 1b peptide monolayers after 4 hours of growth time (Fig. 7d). The immature crystals isolated after 10 minutes growth time showed a triangular morphology (Fig. 7a). Some particles had a rectangular morphology (Fig. 7b) with inhomogeneous electron density distribution, indicating that these particles are composed of even smaller particles. HR-TEM showed that they are composed of amorphous calcium carbonate (ACC) with crystalline interior domains (Fig. 7c). This indicates that the crystals were formed via transformation from an amorphous phase into a crystalline one (Fig. 7c).
 | ||
| Fig. 7 Crystallization of CaCO3 beneath peptide 1a monolayer at 0.1 mN m−1. (a–c) TEM images of crystals separated after 10 min growth time. (d) SEM image of crystals separated after 4 hours. | ||
Crystallization experiments beneath peptide 2a and 2b monolayers resulted in each case in {10.4} rhombohedral crystals with a concave pyramidal-shaped face. These crystals were predominant beneath the monolayer of peptide 2a, but were less prominent underneath peptide 2b. Here calcite crystals with <01.2> orientation could be additionally found (Fig. 8).
 | ||
| Fig. 8 SEM images of CaCO3 crystals grown beneath peptide 2a (a, b) and 2b (c, d) monolayer at 0.1 mN m−1. Crystals were separated after 4 hours (a, b) and 16 hours (c, d). | ||
In situ observations of the crystallization process
Observations of the crystallization were performed in situ employing optical and Brewster angle microscopy to further reveal the crystal growth process under peptide monolayers. These methods provide a direct and non-interfering way to observe crystal growth processes. Optical microscopy shows directly the crystal morphology and its distribution at the interface as well as the nucleation density, while BAM observations reveal monolayer domain changes during crystallization and CaCO3 nucleation sites in the monolayer.Immediately after spreading the 1b peptide monolayer on Ca(HCO3)2 solution, domains with holes were observed with BAM (Fig. 9). Bright spots appeared simultaneously, indicating calcium carbonate nucleation under these domains. The nucleation sites multiplied and grew with time, while the peptide domains became more and more tightly packed until they appeared completely homogenous (after approximately 7 h). Similar monolayer domain changes were also observed for other peptides during CaCO3 crystallization (see Fig. S4 and S5 in the ESI†). As far as it is possible to judge from BAM images the growth of calcium carbonate seems to take place mainly at the edges of the peptide monolayer domains. Typical images of the crystallization process beneath 2b monolayers are shown in Fig. 10. Peptide 2b forms irregular monolayer domains on an aqueous Ca(HCO3)2 solution right after spreading (Fig. 10a). After a few minutes, some CaCO3 particles could be seen at the edge of the domains (Fig. 10b). Within 1 hour their number and size increased considerably. Simultaneously, the peptide domains were pushed together and formed a tightly packed monolayer within few hours of crystallization, as shown in Fig. 10c and d.
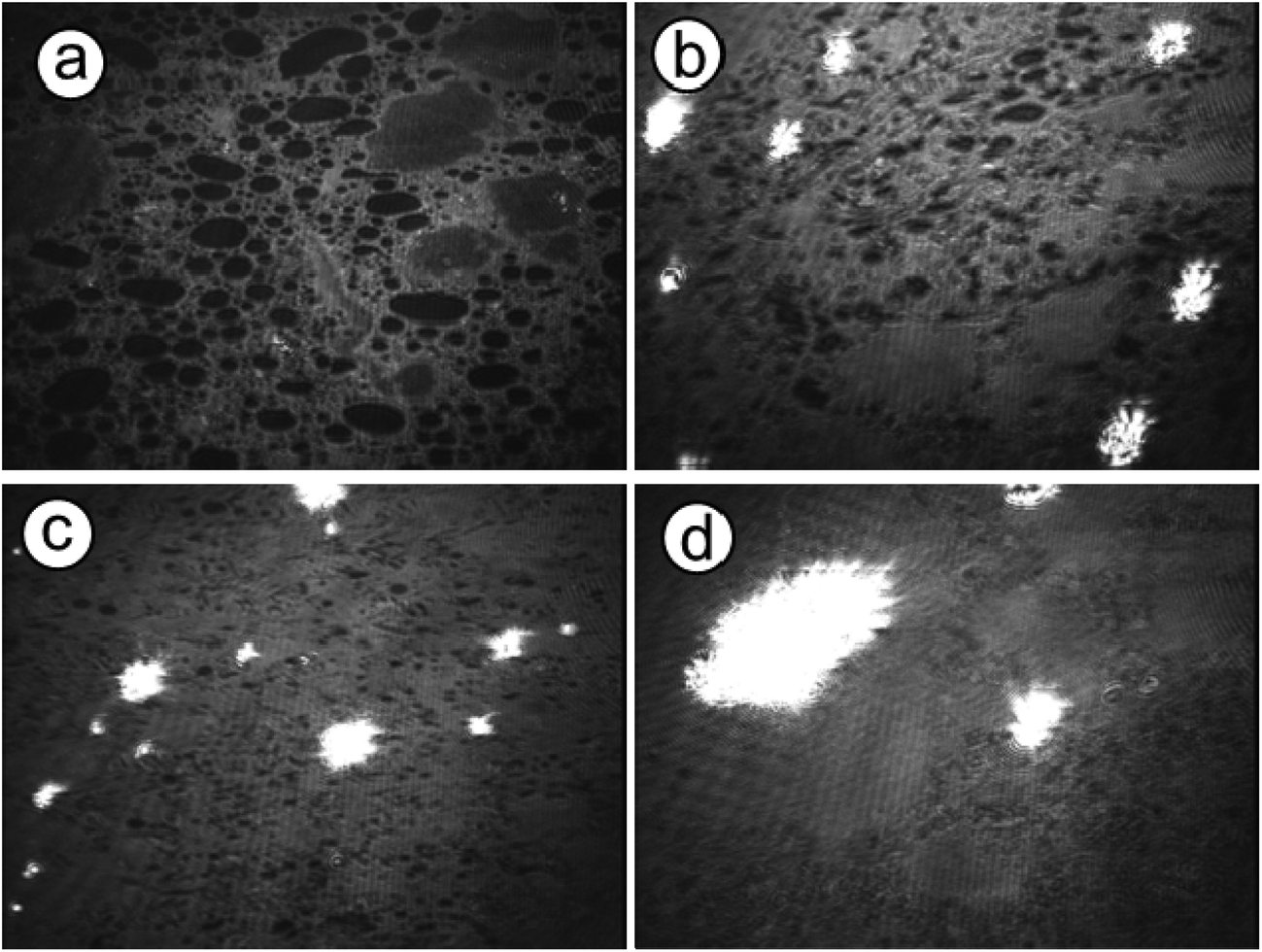 | ||
| Fig. 9 BAM images of CaCO3 crystallization beneath peptide 1b for 0, 1, 2, and 7 hours. The images were recorded at the onset point of surface pressure. (The size of each BAM image corresponds to a monolayer area of 430 μm in width.) | ||
 | ||
| Fig. 10 BAM images of CaCO3 crystallization beneath peptide 2b for 0, 1, 2, and 3 hours. The images was taken at the onset point of surface pressure. (The size of each BAM image corresponds to a monolayer area of 430 μm in width.) | ||
Discussion
Although Langmuir monolayers acting as CaCO3 crystallization modifiers have been systematically investigated for over 20 years, many open questions still remain. For example, the hypothesis of a specific structure and/or geometric match between monolayers and certain crystal faces stands in contrast to the frequent occurrence of calcite crystals having their {01.2} crystal face oriented towards the monolayer, which is an experimental observation for a vast variety of structurally different monolayers.45–49 The latter observation suggests that factors which are non-specific or characteristic of the monolayer structure govern CaCO3 crystal growth and polymorph selection. In a few cases, the template model of selective monolayer-directed CaCO3 crystallization seems to apply, which is the case for calcite crystals growing beneath amphiphilic monolayers of aliphatic sulfonates.50 In this particular case the {00.1} crystal face of calcite seems to match the spatial arrangement of polar head groups in the monolayer.51In the present study, different CaCO3 crystal morphologies and crystal orientations were found to evolve underneath different hairpin peptide monolayers, suggesting that structural or geometrical factors of the monolayers might have some influence at different stages of crystal formation. As a feature common to all peptide monolayers we observed formation of an ACC precursor within the first minutes, thus pointing to a general crystallization pathway being operative under Langmuir monolayers at the early stage of crystal nucleation.
Scheme 3 highlights the likely succession of events accompanying the formation of CaCO3 crystals beneath peptide monolayers. First, ACC primary particles (small black circles) form preferentially at the boundary of separate monolayer domains (blue patches), as indicated by time-resolved in situ BAM investigations. These nucleation sites might serve two different purposes: the edges of the monolayer are structurally flexible, being able to co-adapt to virtually any spatial arrangement of ions constituting the nascent ACC primary particles. Moreover, the non-covered water surface allows for efficient CO2 escape from the aqueous subphase, leading to a local supersaturation of CaCO3 at close proximity to the edges of the freely floating monolayer domains. The LC phase of the monolayer itself is characterized by a dense packing arrangement of amphiphilic molecules, and thus by a high surface charge density. The cumulative electrostatic effects should lead to preferential accumulation of Ca2+ and HCO3– ions underneath patches of the monolayer. However, the evaporation of CO2 and thus the transformation of bicarbonate into carbonate should preferentially occur at the edges of monolayer domains, which might serve to rationalize the preferred formation of CaCO3 particles at this location.
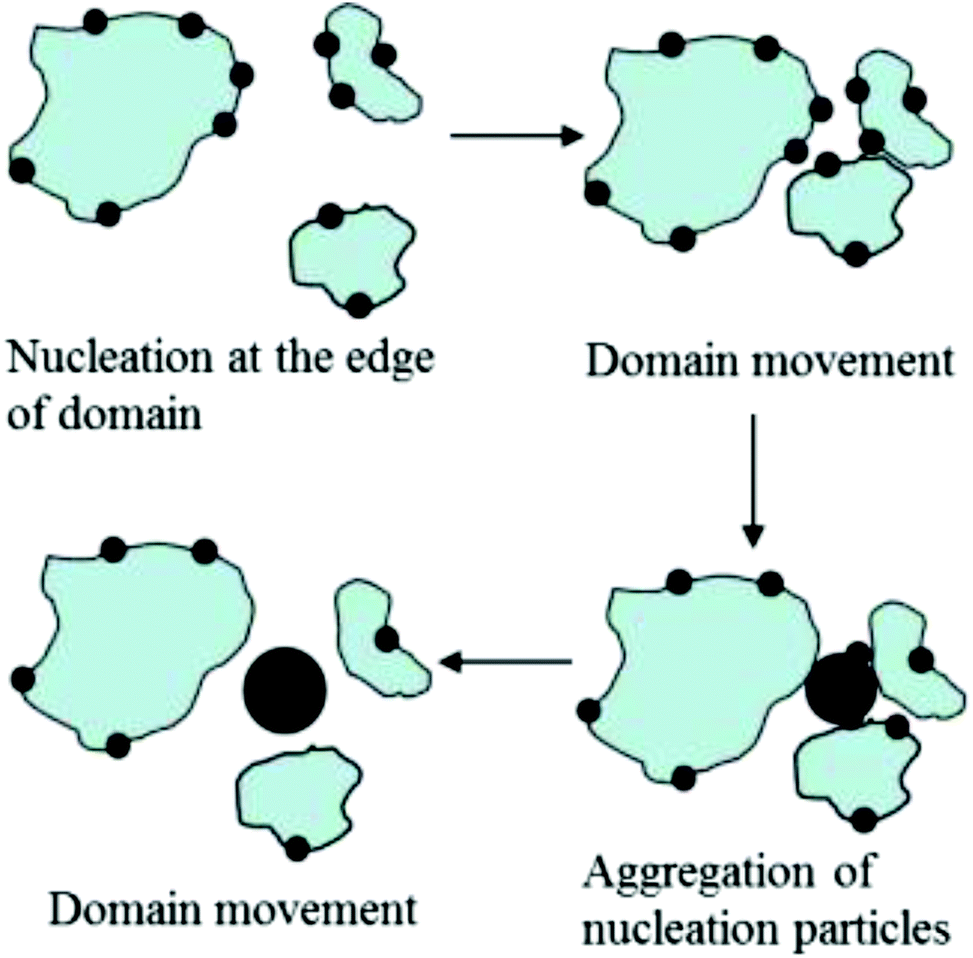 | ||
| Scheme 3 Subsequent stages of calcium carbonate nucleation and aggregation, and changes of peptide monolayer domains during crystallization. | ||
Where monolayer domains approach each other, larger aggregates of ACC particles can thus form via fusion of smaller particles being attached to edges of the monolayer domains (Scheme 3). According to results from TEM investigations CaCO3 crystals start to grow from the inside of larger ACC particles, giving rise to CaCO3 single crystals. This statement gleans (indirect) support from the fact that orientations, sizes and shapes of CaCO3 crystals observed in the present and similar model systems often show broad distributions, which lack a specific and direct correlation with regard to the structure of the templating monolayer.
ACC phases have been observed in natural nacre52 and artificial model systems.53–59 It is reported that these ACC phases are mostly stabilized by polyanions such as poly(aspartic acid)60 or poly(acrylic acid)61 in solution. It is thus reasonable to assume that a thin layer of ACC can also form beneath monolayers of acidic peptides owing to their polyanionic character. Hence, CaCO3 crystallization starts subsequent to the accumulation of amorphous particles, and there is no experimental evidence for a direct epitaxy of CaCO3 crystals emanating from a two-dimensional patch of the peptide monolayer. Our findings seem to support recent studies of multistep crystallization which in a colloidal model system demonstrated the formation of crystal nuclei of subcritical size inside an amorphous precursor phase and their subsequent fusion and transformation into single crystals.62
Subsequent to the formation of ACC particles, vaterite crystals have been observed in the early stage of all crystallization experiments performed with peptide monolayers. After 4 hours the proportion of vaterite crystals decreased significantly for peptide 1a monolayers, and the number of calcite crystals increased at the same time. We conclude that vaterite is a transient polymorph which transforms into calcite during the later stage of the crystallization process. The relatively slow transformation of vaterite into calcite suggests, however, that this process might be influenced by the monolayer. However, for peptide 1b monolayers, vaterite and calcite crystals occurred simultaneously and their relative ratio did not change significantly within the experimental period. Since a less ordered β-hairpin packing arrangement for peptide 1a is indicated by surface pressure-area isotherms, FTIR and AFM investigations, we might tentatively suggest that the more flexible nature of peptide 1a compared with peptide 1b, leads to rapid formation of vaterite nuclei under 1a monolayers and their subsequent transformation into <10.0> oriented calcite crystals. Similar results have been suggested by Kuther et al.63 for a self-assembled monolayer showing a low degree of structural organisation, which promoted the formation of aragonite over calcite under experimental conditions different to ours.
Various orientations and morphologies of calcite crystals were observed for the different peptide monolayers at the late stages of crystallization, i.e. after a few hours. For peptides 1a and 1b, XPS results show that individual peptide molecules on average bind about 4 calcium ions if being spread on an aqueous subphase containing calcium acetate (ESI S6†). Therefore, the different orientations of calcite crystals grown under 1a and 1b peptide monolayers cannot be attributed to different monolayer charge densities. AFM investigations indicate that 1b assumes a highly ordered head-to-head packing arrangement (cf. Scheme 2), while 1a self-assembles into a less well-organized packing arrangement. Based on these results, we conclude that monolayer structure flexibility, i.e. the ability of the monolayer to co-adapt to structural changes imposed by the aggregating and transforming CaCO3 particles is a crucial factor in controlling the orientations and shapes of the mature calcite single crystals.
Conclusions
The monolayers of four acidic model hairpin peptides were investigated for templating effects with respect to CaCO3 crystallization. Unlike in solution, all peptides adopt a β-hairpin conformation at the air–water interface, which results from the sequence of alternating hydrophilic and hydrophobic amino acid residues. Peptide 1b forms a highly ordered head-to-head packing arrangement in the monolayer, whereas peptide 1a is structurally more flexible. These slight structural differences cause a significant variation of CaCO3 crystals that grow underneath the monolayers. The crystals were characterized by SEM, TEM, and in situ optical and BAM. An amorphous calcium carbonate precursor was found during the initial stage of crystallization. TEM images indicate that this amorphous precursor contains nanosized crystals which form preferentially inside the amorphous phase, indicating that the crystallization process starts subsequent to aggregation and merging of amorphous precursor particles. The formation of amorphous calcium carbonate particles mainly occurs at the boundary of LC domains or in the LE phase of the peptide monolayers, the movement of monolayer domains being crucial for the lateral diffusion or aggregation of ACC particles. CaCO3 crystals preferentially form within fused patches of ACC particles. It should be noted that in our hands all attempts have failed to provide experimental evidence for a direct and two-dimensional contact between the CaCO3 crystals and the peptide monolayer domains. This statement stands in sharp contrast to the original notion of a templating monolayer and the mechanistic concept of heteroepitaxial crystal growth in such model systems. The significance of ACC precursor phases yet remains an open question. It seems evident that the ACC phase serves as nutrient solution for the maturing CaCO3 crystals. If and how the ACC phase influences the growth characteristics of single crystals emanating from its surrounding precursor currently remains an open question, as does the functional role of the monolayer during this stage of crystal growth. Addressing such fundamental aspects of biologically inspired crystallisation will require experimental set-ups different to those employed in the present investigations.Acknowledgements
We are grateful to Dr P. Reuter, Dr J. Biskupek for their support with SEM, TEM measurements, respectively. Financial support provided by the Deutsche Forschungsgemeinschaft (DFG Priority Program 1117, “Principles of Biomineralization”, DFG grants VO 829/3 and SE 609/6-1) and the Hungarian Scientific Research Fund (OTKA, grant no. K100720 to Zs.M.) is gratefully acknowledged.References
- K. Simkiss and K. M. Wilbur, Biomineralization: Cell Biology and Mineral Deposition, Academic Press Inc., San Diego, 1989 Search PubMed.
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, New York, Oxford, 1989 Search PubMed.
- L. Addadi and S. Weiner, Interactions between acidic proteins and crystals: stereochemical requirements in biomineralization, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4110–4114 CrossRef CAS.
- A. M. Belcher, X. H. Wu, R. J. Christensen, P. K. Hansma, G. D. Stucky and D. E. Morse, Control of crystal phase switching and orientation by soluble mollusc-shell proteins, Nature, 1996, 381, 56–58 CrossRef CAS.
- Y. Levi-Kalisman, G. Falini, L. Addadi and S. Weiner, Structure of the nacreous organic matrix of a bivalve mollusk shell examined in the hydrated state using cryo-TEM, J. Struct. Biol., 2001, 135, 8–17 CrossRef CAS PubMed.
- J. Y. Sun and B. Bhushan, Hierarchical structure and mechanical properties of nacre: a review, RSC Adv., 2012, 2, 7617–7632 RSC.
- M. Suzuki, K. Saruwatari, T. Kogure, Y. Yamamoto, T. Nishimura, T. Kato and H. Nagasawa, An Acidic Matrix Protein, Pif, Is a Key Macromolecule for Nacre Formation, Science, 2009, 325, 1388–1390 CrossRef CAS PubMed.
- Y. Levi, S. Albeck, A. Brack, S. Weiner and L. Addadi, Control Over Aragonite Crystal Nucleation and Growth: An In Vitro Study of Biomineralization, Chem. – Eur. J., 1998, 4, 389–396 CrossRef CAS.
- D. B. DeOliveira and R. A. Laursen, Control of Calcite Crystal Morphology by a Peptide Designed To Bind to a Specific Surface, J. Am. Chem. Soc., 1997, 119, 10627–10631 CrossRef CAS.
- C. Li, G. D. Botsaris and D. L. Kaplan, Selective in Vitro Effect of Peptides on Calcium Carbonate Crystallization, Cryst. Growth Des., 2002, 2, 387–393 CAS.
- L. A. Estroff, C. D. Incarvito and A. D. Hamilton, Design of a synthetic foldamer that modifies the growth of calcite crystals, J. Am. Chem. Soc., 2004, 126, 2–3 CrossRef CAS PubMed.
- S. Collino and J. S. Evans, Structural Features That Distinguish Kinetically Distinct Biomineralization Polypeptides, Biomacromolecules, 2007, 8, 1686–1694 CrossRef CAS PubMed.
- G. Falini, S. Albeck, S. Weiner and L. Addadi, Control of Aragonite or Calcite Polymorphism by Mollusk Shell Macromolecules, Science, 1996, 271, 67–69 Search PubMed.
- I. W. Kim, S. Collino, D. E. Morse and J. S. Evans, A Crystal Modulating Protein from Molluscan Nacre That Limits the Growth of Calcite in Vitro, Cryst. Growth Des., 2006, 6, 1078–1082 CAS.
- G. Fu, S. Valiyaveettil, B. Wopenka and D. E. Morse, CaCO3 Biomineralization: Acidic 8-kDa Proteins Isolated from Aragonitic Abalone Shell Nacre Can Specifically Modify Calcite Crystal Morphology, Biomacromolecules, 2005, 6, 1289–1298 CrossRef CAS PubMed.
- S. Mann, Biomineralization, Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, Oxford, 2001 Search PubMed.
- J. S. Evans, ‘Apples’ and ‘oranges’: comparing the structural aspects of biomineral- and ice-interaction proteins, Curr. Opin. Colloid Interface Sci., 2003, 8, 48–54 CrossRef CAS.
- D. Volkmer, M. Fricke, T. Huber and N. Sewald, Acidic peptides acting as growth modifiers of calcite crystals, Chem. Commun., 2004, 1872–1873 RSC.
- M. Ndao, E. Keene, F. F. Amos, G. Rewari, C. B. Ponce, L. Estroff and J. S. Evans, Intrinsically Disordered Mollusk Shell Prismatic Protein That Modulates Calcium Carbonate Crystal Growth, Biomacromolecules, 2010, 11, 2539–2544 CrossRef CAS PubMed.
- R. A. Metzler, J. S. Evans, C. E. Killian, D. Zhou, T. H. Churchill, N. P. Appathurai, S. N. Coppersmith and P. U. P. A. Gilbert, Nacre Protein Fragment Templates Lamellar Aragonite Growth, J. Am. Chem. Soc., 2010, 132, 6329–6334 CrossRef CAS PubMed.
- S. Cavalli, D. C. Popescu, E. E. Tellers, M. R. J. Vos, B. P. Pichon, M. Overhand, H. Rapaport, N. A. J. M. Sommerdijk and A. Kros, Self-Organizing β-Sheet Lipopeptide Monolayers as Template for the Mineralization of CaCO3, Angew. Chem., 2006, 118, 753–758 ( Angew. Chem., Int. Ed. , 2006 , 45 , 739–744 ) CrossRef.
- N. R. Chevalier, C. Chevallard, M. Goldmann, G. Brezesinski and P. Guenoun, CaCO3 Mineralization under β-Sheet Forming Peptide Monolayers, Cryst. Growth Des., 2012, 12, 2299–2305 CAS.
- S. Mann, Molecular recognition in biomineralization, Nature, 1988, 332, 119–124 CrossRef CAS.
- B. R. Heywood and S. Mann, Template-directed nucleation and growth of inorganic materials, Adv. Mater., 1994, 6, 9–20 CrossRef CAS.
- B. R. Heywood and S. Mann, Crystal recognition at inorganic–organic interfaces: Nucleation and growth of oriented BaSO4 under Compressed Langmuir Monolayers, Adv. Mater., 1992, 4, 278–281 CrossRef CAS.
- B. R. Heywood and S. Mann, Organic template-directed inorganic crystallization: oriented nucleation of barium sulfate under compressed Langmuir monolayers, J. Am. Chem. Soc., 1992, 114, 4681–4685 CrossRef CAS.
- G. Xu, N. Yao, I. A. Aksay and J. T. Groves, Biomimetic Synthesis of Macroscopic-Scale Calcium Carbonate Thin Films. Evidence for a Multistep Assembly Process, J. Am. Chem. Soc., 1998, 120, 11977–11985 CrossRef CAS.
- E. DiMasi, M. J. Olszta, V. M. Patel and L. B. Gower, When is template directed mineralization really template directed?, CrystEngComm, 2003, 5, 346–350 RSC.
- E. DiMasi, S. Y. Kwak, F. F. Amos, M. J. Olszta, D. Lush and L. B. Gower, Complementary Control by Additives of the Kinetics of Amorphous CaCO3 Mineralization at an Organic Interface: In situ Synchrotron X-Ray Observations, Phys. Rev. Lett., 2006, 97, 045503 CrossRef.
- D. Volkmer, M. Fricke, C. Agena and J. Mattay, Interfacial electrostatics guiding the crystallization of CaCO3 underneath monolayers of calixarenes and resorcarenes, J. Mater. Chem., 2004, 14, 2249–2259 RSC.
- D. Volkmer, M. Fricke, M. Gleiche and L. Chi, Elucidating the role of charge density on the growth of CaCO3 crystals underneath Calix[4]arene monolayers, Mater. Sci. Eng., C, 2005, 25, 161–167 CrossRef PubMed.
- M. Fricke, D. Volkmer, C. E. Krill III, M. Kellermann and A. Hirsch, Vaterite Polymorph Switching Controlled by Surface Charge Density of an Amphiphilic Dendron-calix[4]arene, Cryst. Growth Des., 2006, 6, 1120–1123 CAS.
- M. Fricke and D. Volkmer, Crystallization of Calcium Carbonate Beneath Insoluble Monolayers: Suitable Models of Mineral–Matrix Interactions in Biomineralization?, Top. Curr. Chem., 2007, 270, 1–41 CrossRef CAS.
- D. C. Popescu, M. M. J. Smulders, B. P. Pichon, N. Chebotareva, S.-Y. Kwak, O. L. J. van Asselen, R. P. Sijbesma, E. DiMasi and N. A. J. M. Sommerdijk, Template Adaptability Is Key in the Oriented Crystallization of CaCO3, J. Am. Chem. Soc., 2007, 129, 14058–14067 CrossRef CAS PubMed.
- G. Xu, W. Wang, J. T. Groves and M. H. Hecht, Self-assembled monolayers from a designed combinatorial library of de novo beta-sheet proteins, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3652–3657 CrossRef CAS PubMed.
- H. Isenberg, K. Kjaer and H. Rapaport, Elasticity of Crystalline β-Sheet Monolayers, J. Am. Chem. Soc., 2006, 128, 12468–12472 CrossRef CAS PubMed.
- H. Rapaport, K. Kjaer, T. R. Jensen, L. Leiserowitz and D. A. Tirrell, Two-Dimensional Order in β-Sheet Peptide Monolayers, J. Am. Chem. Soc., 2000, 122, 12523–12529 CrossRef CAS.
- M. Buck, Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins, Q. Rev. Biophys., 1998, 31, 297–355 CrossRef CAS.
- L. Wu, D. McElheny, T. Takekiyo and T. A. Keiderling, Geometry and Efficacy of Cross-Strand Trp/Trp, Trp/Tyr, and Tyr/Tyr Aromatic Interaction in a β-Hairpin Peptide, Biochemistry, 2010, 49, 4705–4714 CrossRef CAS PubMed.
- J. Hilario, J. Kubelka and T. A. Keiderling, Optical Spectroscopic Investigations of Model β-Sheet Hairpins in Aqueous Solution, J. Am. Chem. Soc., 2003, 125, 7562–7574 CrossRef CAS PubMed.
- S. Aravinda, N. Shamala, R. Rajkishore, H. N. Gopi and P. Balaram, A Crystalline β-Hairpin Peptide Nucleated by a Type I′ Aib-D-Ala β-Turn: Evidence for Cross-Strand Aromatic Interactions, Angew. Chem., 2002, 114, 4019–4021 ( Angew. Chem., Int. Ed. , 2002 , 41 , 3863–3865 ) CrossRef.
- E. T. Powers, S. I. Yang, C. M. Lieber and J. W. Kelly, Ordered Langmuir–Blodgett Films of Amphiphilic β-Hairpin Peptides Imaged by Atomic Force Microscopy, Angew. Chem., 2002, 114, 135–138 ( Angew. Chem., Int. Ed. , 2002 , 41 , 127–130 ) CrossRef.
- S. Cavalli, J.-W. Handgraaf, E. E. Tellers, D. C. Popescu, M. Overhand, K. Kjaer, V. Vaiser, N. A. J. M. Sommerdijk, H. Rapaport and A. Kros, Two-Dimensional Ordered β-Sheet Lipopeptide Monolayers, J. Am. Chem. Soc., 2006, 128, 13959–13966 CrossRef CAS PubMed.
- S. Krimm and J. Bandikar, Vibrational Spectroscopy and Conformation of Peptides, Polypeptides, and Proteins, Adv. Protein Chem., 1986, 38, 181–364 CrossRef CAS.
- H. Y. Li and L. A. Estroff, Hydrogels Coupled with Self-Assembled Monolayers: An in Vitro Matrix to Study Calcite Biomineralization, J. Am. Chem. Soc., 2007, 129, 5480–5483 CrossRef CAS PubMed.
- A. M. Travaille, E. G. A. Steijven, H. Meekes and H. van Kempen, Thermodynamics of Epitaxial Calcite Nucleation on Self-Assembled Monolayers, J. Phys. Chem. B, 2005, 109, 5618–5626 CrossRef CAS PubMed.
- A. M. Travaille, L. Kaptijn, P. Verwer, B. Hulsken, J. A. A. W. Elemans, R. J. M. Nolte and H. van Kempen, Highly Oriented Self-Assembled Monolayers as Templates for Epitaxial Calcite Growth, J. Am. Chem. Soc., 2003, 125, 11571–11577 CrossRef CAS PubMed.
- Y.-J. Han and J. Aizenberg, Effect of Magnesium Ions on Oriented Growth of Calcite on Carboxylic Acid Functionalized Self-Assembled Monolayer, J. Am. Chem. Soc., 2003, 125, 4032–4033 CrossRef CAS PubMed.
- D. J. Ahn, A. Berman and D. Charych, Probing the Dynamics of Template-Directed Calcite Crystallization with in Situ FTIR, J. Phys. Chem., 1996, 100, 12455–12461 CrossRef CAS.
- D. Volkmer, N. Mayr and M. Fricke, Crystal structure analysis of [Ca(O3SC18H37)2(DMSO)2], a lamellar coordination polymer and its relevance for model studies in biomineralization, Dalton Trans., 2006, 4889–4895 RSC.
- S. Kewalramani, K. Kim, B. Stripe, G. Evmenenko, G. H. B. Dommett and P. Dutta, Observation of an Organic–Inorganic Lattice Match during Biomimetic Growth of (001)-Oriented Calcite Crystals under Floating Sulfate Monolayers, Langmuir, 2008, 24, 10579–10582 CrossRef CAS PubMed.
- N. Nassif, N. Pinna, N. Gehrke, M. Antonietti, C. Jäger and H. Cölfen, Amorphous layer around aragonite platelets in nacre, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12653–12655 CrossRef CAS PubMed.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Sea urchin spine calcite forms via a transient amorphous calcium carbonate phase, Science, 2004, 306, 1161–1164 CrossRef CAS PubMed.
- Y. Politi, Y. Levi-Kalisman, S. Raz, F. Wilt, L. Addadi, S. Weiner and I. Sagi, Structural Characterization of the Transient Amorphous Calcium Carbonate Precursor Phase in Sea Urchin Embryos, Adv. Funct. Mater., 2006, 16, 1289–1298 CrossRef CAS.
- S. Weiner, I. Sagi and L. Addadi, Choosing the crystallization path less traveled, Science, 2005, 309, 1027–1028 CrossRef CAS PubMed.
- J. R. I. Lee, T. Y.-J. Han, T. M. Willey, D. Wang, R. W. Meulenberg, J. Nilsson, P. M. Dove, L. J. Terminello, T. van Buuren and J. J. De Yoreo, Structural Development of Mercaptophenol Self-Assembled Monolayers and the Overlying Mineral Phase during Templated CaCO3 Crystallization from a Transient Amorphous Film, J. Am. Chem. Soc., 2007, 129, 10370–10381 CrossRef CAS PubMed.
- J. Bolze, B. Peng, N. Dingenouts, P. Panine, T. Narayanan and M. Ballauff, Formation and Growth of Amorphous Colloidal CaCO3 Precursor Particles as Detected by Time-Resolved SAXS, Langmuir, 2002, 18, 8364–8369 CrossRef CAS.
- H. Cölfen and S. Mann, Higher-Order Organization by Mesoscale Self-Assembly and Transformation of Hybrid Nanostructures, Angew. Chem., 2003, 115, 2452–2468 ( Angew. Chem., Int. Ed. , 2003 , 42 , 2350–2365 ) CrossRef.
- H. A. Lowenstam, Minerals formed by organisms, Science, 1981, 211, 1126–1131 CAS.
- X. Cheng and L. B. Gower, Molding mineral within microporous hydrogels by a polymer-induced liquid-precursor (PILP) process, Biotechnol. Prog., 2006, 22, 141–149 CrossRef CAS PubMed.
- M. J. Olszta, S. Gajjeraman, M. Kaufman and L. B. Gower, Nanofibrous Calcite Synthesized via a Solution–Precursor–Solid Mechanism, Chem. Mater., 2004, 16, 2355–2362 CrossRef CAS.
- T. H. Zhang and X. Y. Liu, How Does a Transient Amorphous Precursor Template Crystallization?, J. Am. Chem. Soc., 2007, 129, 13520–13526 CrossRef CAS PubMed.
- J. Kuther, R. Seshadri, G. Nelles, W. Assenmacher, H. J. Butt, W. Mader and W. Tremel, Mercaptophenol-Protected Gold Colloids as Nuclei for the Crystallization of Inorganic Minerals: Templated Crystallization on Curved Surfaces, Chem. Mater., 1999, 11, 1317–1325 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional AFM topography images of Langmuir–Blodgett films of peptide monolayer transferred from water subphase, BAM images of CaCO3 crystallization under monolayers of peptide 1a and 2a. Raman spectrum of crystals grown under the monolayer of peptide 1a. See DOI: 10.1039/c4dt01154f |
| This journal is © The Royal Society of Chemistry 2014 |