 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of high π-density on the coordination properties of π-excess aromatic neutral σ2P ligands – P(π)-donor bonds to Ag+ and HgCl2†
Mohammed
Ghalib
a,
László
Könczöl
b,
László
Nyulászi
*b,
Peter G.
Jones
*c,
Gottfried J.
Palm
d and
Joachim W.
Heinicke
*a
aInstitut für Biochemie (Anorganische Chemie), E.-M.-A.-Universität Greifswald, 17487 Greifswald, Germany. E-mail: heinicke@uni-greifswald.de; Fax: +49-3834-864377; Tel: +49-3834-864318
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Hungary
cInstitut für Anorganische und Analytische Chemie, Techn. Universität Braunschweig, D-38023 Braunschweig, Germany
dInstitut für Biochemie (Molekulare Strukturbiologie), E.-M.-A.-Universität Greifswald, 17487 Greifswald, Germany
First published on 22nd October 2013
Abstract
Unprecedented coordination types of non-zerovalent d10 transition metals (AgX, HgCl2) by π-excess aromatic P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ligands involving P(π)-donor bond contributions were detected by structural and quantum chemical studies.
C ligands involving P(π)-donor bond contributions were detected by structural and quantum chemical studies.
Trivalent phosphorus compounds are widely used ligands in coordination chemistry and homogenous late transition metal catalysis, but studies on ligands with dicoordinated (σ2P) phosphorus are restricted almost completely to phosphabenzenes1 and some phosphaalkenes.2 Complexes with π-excess-aromatic σ2P-azaphosphole ligands, apart from metal(0) complexes,3,4 are extremely sparsely documented,5,6 and only one, a 1,2-azaphosphole allylnickel chloride complex,6 has been characterized by crystal structure analysis. Complexes of the π-rich 1,3-azaphospholes with phosphorus in conjugation to nitrogen (N–C
P ↔ N+C–P−) are still unknown for non-zerovalent transition metals. We report here the first examples with d10 metal cations; these 1,3-benzazaphosphole Ag(I) and HgCl2 complexes display a hitherto unprecedented coordination pattern. Structurally characterized σ2P–Ag(I) complexes are very rare,7,8 and we are not aware of any Hg(II) complexes with σ2P-ligands.
Addition of toluene and NH3 to an equimolar mixture of 19 and AgCl at −60 °C and warming to r.t. led to the formation of an inversion-symmetric benzazaphosphole silver chloride complex with a central Ag2Cl2 ring (Scheme 1). Colourless crystals of 2 were obtained on overlayering a concentrated solution in THF with hexane. The crystal structure analysis (Fig. 1) displays distorted tetrahedral coordination of Ag+ with an interplanar angle of 80.8° between the Ag2Cl2 and the P3–Ag–P3′ planes. The P–Ag bonds do not lie within the extended ligand ring plane, but are tilted away by 32.4 and 12.7°. The bond lengths and angles within the ligand are only marginally changed (P-C3a slightly shortened) compared to 1.9 A cationic silver complex of a different coordination type was formed in THF at room temperature from a mixture of 1 and AgSbF6. Crystals of 3 were grown by slow concentration of a solution of the pale brown crude product in CD3OD. They proved to be less stable than 2 and decomposed within a few days, but crystallographic analysis of freshly prepared crystals of 3 clearly revealed two [Ag4(1)4THF4(SbF6)2]2+ clusters with μ-P coordination and four uncoordinated SbF6− counter-ions per unit cell (in space group P21/c). Full refinement was not possible, with the THF molecules and SbF6− ions being disordered (for details see ESI†). The sharp singlets lacking coupling with 107/109Ag in the CD3OD solution of silver complexes 2 and 3 indicate a rapid ligand exchange and thus kinetic lability. Complex formation is indicated mainly by upfield coordinated 31P chemical shifts, much stronger for the μ-P bridged 3 (Δδ = −76.7 ppm) than the terminally coordinated 2 (Δδ = −18.9 ppm). Characteristic features in the 13C NMR spectra are decreasing 1JPC coupling constants of C2 and C3a and downfield shifts of the C-2 signals, whereas proton NMR data are scarcely altered.
 | ||
| Scheme 1 Formation of silver complexes of 1 (Np = neopentyl) and 3 (schematic structure presentation of 3). | ||
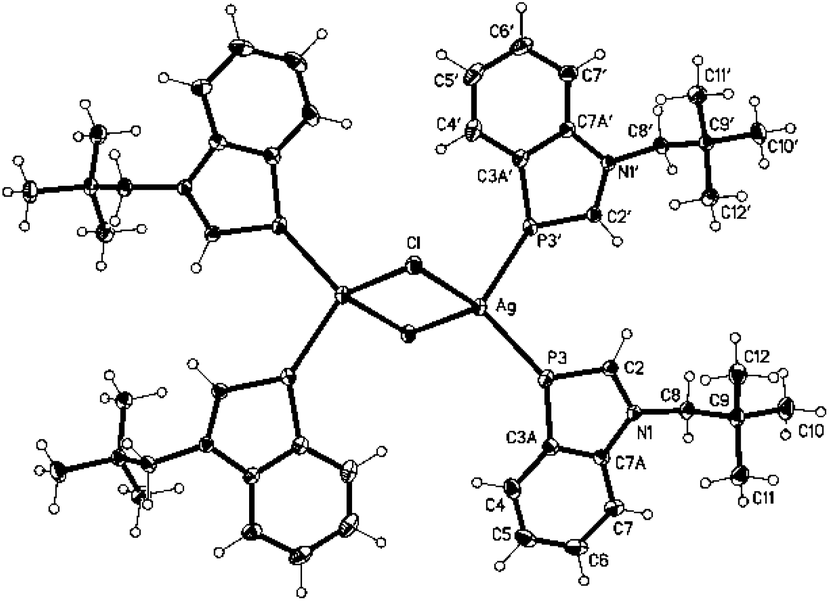 | ||
| Fig. 1 Structure of 2 in the crystal (ellipsoids with 50% probability). | ||
To find out if divalent mercury is also able to form complexes with 1,3-benzazaphospholes, the reaction of 1 with HgCl2 in THF was explored. Oxidation of phosphorus to a λ5P heterocycle as reported for the reaction of 2,4,6-triphenylphosphabenzene with Hg(OAc)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 was not observed. The NMR spectra of the yellow solid, with a marked upfield 31P coordination chemical shift in C6D6 (Δδ31 = −84.8 ppm), indicated complex formation, but attempts to grow single crystals for a detailed structural characterization failed. A crystalline 1,3-benzazaphosphole–HgCl2 complex was finally obtained from the recently reported σ3P,σ2P-hybrid benzazaphosphole 411 and HgCl2 in THF (Scheme 2). XRD analysis of the yellow single crystals of 5 (Fig. 2), grown from concentrated THF solution, showed a P,P′-chelate structure as expected. However, whereas in the recently reported Mo(0)(CO)4(P,P′)-chelate complex with 4 the zero-valent metal is only slightly (by 14.4°) tilted out of the ring plane with shorter ring-P–Mo than Ph2P–Mo(0) distances and marginal changes within the ligand,11 the HgCl2 complex 5 coordinates mercury almost perpendicular to the benzazaphosphole ring plane with much longer Hg–P3 (2.6978(7) Å) than Hg–P1 bonds (2.4219(5) Å) and altered P–C and C–N bond lengths within the ring. The angle of the Hg–P vector to the ring plane of 80.3° (deviation from plane 2.62 Å) and the angles C2–P–Hg (86.40(8)) and C3a–P–Hg (98.92(8)), average 92.7°, hint at a π-donor bond from the ring-P atom to Hg(II). In combination with the elongated C2–P and shortened N–C2 bond (1.770(3) and 1.344(3) Å compared to 1.7173(17) and 1.369(2) Å in the Mo(0) complex11) this implies a high weight for a zwitterionic P-coordinated phosphidomethylenimmonium “resonance structure” (cf. Scheme 2). The increased NH acidity favours N–H⋯Cl hydrogen bonds (H01⋯Cl(2)#1 2.42(3), N1⋯Cl(2)#1 3.238(2) Å, N–H⋯Cl 169(3)°) in the crystal packing and in solution, the latter indicated by a strongly downfield shifted NH proton signal (Δδ = 3.0 ppm). The coordination of HgCl2 at the two P-atoms is indicated in solution by a downfield shift for the phosphanyl group Δδ = 31.0 ppm, similar to that of (Ph3P)2HgCl2 (Δδ = 39 ppm12), and upfield shift for the ring-P atom, which is however much smaller than for the μ-bridging coordination in 3 and even smaller than for the two terminal benzazaphosphole ligands in 2. The coordination at the ring-P atom is labile, displaying a rather sharp doublet by two-bond P–P-coupling but no 1J(119Hg31P) satellites, whereas the coordinated phosphanyl group exhibits a broad signal associated with the dynamic behaviour at the other P-atom.
10 was not observed. The NMR spectra of the yellow solid, with a marked upfield 31P coordination chemical shift in C6D6 (Δδ31 = −84.8 ppm), indicated complex formation, but attempts to grow single crystals for a detailed structural characterization failed. A crystalline 1,3-benzazaphosphole–HgCl2 complex was finally obtained from the recently reported σ3P,σ2P-hybrid benzazaphosphole 411 and HgCl2 in THF (Scheme 2). XRD analysis of the yellow single crystals of 5 (Fig. 2), grown from concentrated THF solution, showed a P,P′-chelate structure as expected. However, whereas in the recently reported Mo(0)(CO)4(P,P′)-chelate complex with 4 the zero-valent metal is only slightly (by 14.4°) tilted out of the ring plane with shorter ring-P–Mo than Ph2P–Mo(0) distances and marginal changes within the ligand,11 the HgCl2 complex 5 coordinates mercury almost perpendicular to the benzazaphosphole ring plane with much longer Hg–P3 (2.6978(7) Å) than Hg–P1 bonds (2.4219(5) Å) and altered P–C and C–N bond lengths within the ring. The angle of the Hg–P vector to the ring plane of 80.3° (deviation from plane 2.62 Å) and the angles C2–P–Hg (86.40(8)) and C3a–P–Hg (98.92(8)), average 92.7°, hint at a π-donor bond from the ring-P atom to Hg(II). In combination with the elongated C2–P and shortened N–C2 bond (1.770(3) and 1.344(3) Å compared to 1.7173(17) and 1.369(2) Å in the Mo(0) complex11) this implies a high weight for a zwitterionic P-coordinated phosphidomethylenimmonium “resonance structure” (cf. Scheme 2). The increased NH acidity favours N–H⋯Cl hydrogen bonds (H01⋯Cl(2)#1 2.42(3), N1⋯Cl(2)#1 3.238(2) Å, N–H⋯Cl 169(3)°) in the crystal packing and in solution, the latter indicated by a strongly downfield shifted NH proton signal (Δδ = 3.0 ppm). The coordination of HgCl2 at the two P-atoms is indicated in solution by a downfield shift for the phosphanyl group Δδ = 31.0 ppm, similar to that of (Ph3P)2HgCl2 (Δδ = 39 ppm12), and upfield shift for the ring-P atom, which is however much smaller than for the μ-bridging coordination in 3 and even smaller than for the two terminal benzazaphosphole ligands in 2. The coordination at the ring-P atom is labile, displaying a rather sharp doublet by two-bond P–P-coupling but no 1J(119Hg31P) satellites, whereas the coordinated phosphanyl group exhibits a broad signal associated with the dynamic behaviour at the other P-atom.
 | ||
| Scheme 2 Synthesis of the chelate complex 5. | ||
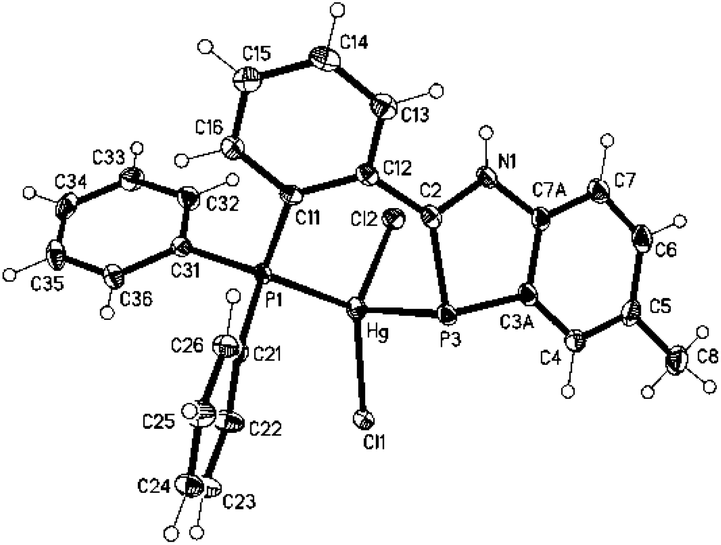 | ||
| Fig. 2 Structure of 5 in the crystal (ellipsoids with 50% probability). | ||
The coordination of two benzazaphosphole ligands at each Ag(I) atom, the tilting of the P–Ag bond out of the ring plane in 2, the μ-coordination mode in 3 and the unprecedented side-on coordination in the chelate complex 5 distinguish the complexes of the π-excess-type PC–N ligands 1 and 4 from phosphabenzene complexes, which usually coordinate non-zero-valent transition metals within the ring plane.1 To shed light on the background of these differences and to understand the unusual bonding in more detail, density functional calculations were carried out at the ωB97D/6-31G* level.13 First a second order perturbation analysis of the donor–acceptor interaction between NBOs at the crystal structures of 2 and 5 was performed. While in the case of 2 both P lone pairs at each atom exhibit about 60.5 kcal mol−1 stabilization donating to Ag, an additional 6.4 kcal mol−1 stabilization interaction was obtained for the (PC bond)–Ag interaction in the case of the 32.4° tilted azaphosphole ring, showing some donor contribution of the π electron pair to the dative bonding. In the case of 5 the interaction between the σ3P lone pair and mercury amounts to 129.0 kcal mol−1, while the interaction between the σ2P and mercury is only 12.1 kcal mol−1. The small interaction with the mainly s-type14 σ2P lone pair is in agreement with the near perpendicular position of the Hg atom with respect to the benzazaphosphole ring. The apparently lost (P lone pair)–Hg interaction, however, is amply compensated by the 40.0 kcal mol−1 stabilization arising from the interaction between the PC π-orbital and mercury. This interaction is also clearly seen on the HOMO of 5 with the involvement of both Hg and the mainly PC bonded delocalised π orbital (the HOMO of the benzazaphosphole ligand) as is shown in Fig. 3.
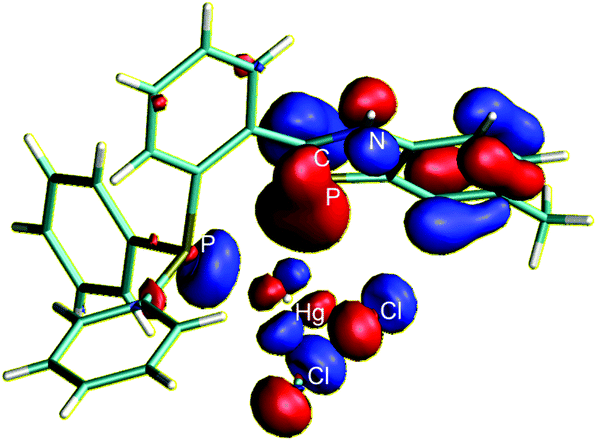 | ||
| Fig. 3 The HOMO of 5 showing the involvement of one Hg d-orbital and the delocalised PC π-orbital in the bonding. | ||
The involvement of the (largely) PC bonded orbital in the dative bonding is in agreement with the lengthening of the PC distance (see above). It is noteworthy that no back-bonding from mercury to π* type orbitals was detected. Optimization of the structure of 5 at the ωB97D/6-31G* level resulted in a shift of the Hg atom from phosphorus towards the ring nitrogen, together with the formation of an intramolecular Cl⋯H bond (see Fig. S3.3 in the ESI†). This behaviour is in agreement with the conclusions drawn from the solution NMR data discussed above. To understand why mercury prefers to interact with the PC bond rather than the P lone pair, further model calculations were carried out. In order to exclude the effect of any steric constraint caused by the interlocking of the two phosphorus atoms of 4, the structure of various L(PMe3)HgCl2 complexes was investigated, where L contained σ2P in different bonding environments.
During the geometry optimization of the benzazaphosphole the HgCl2 unit occupies an out-of-plane position similar to that in 5 (albeit with larger P–Hg distances), even if the geometry optimization is started from a structure where Hg lies in the plane of the benzazaphosphole15 ring. The complexes with the other ligands exhibit similar structural characteristics, with the exception of the in-plane complexed phosphabenzene ligand. The optimized structures can be seen in Fig. S3.4 of the ESI.† It is noteworthy that the σ2P–Hg distance (between 2.57 and 3.49 Å) is related to the interaction energy between the π-system and mercury. Strongest interactions are obtained with ligands (L) having high energy (and localised) HOMO (Table 1) as can easily be explained by simple perturbation theory arguments.
C ligands with HgCl2

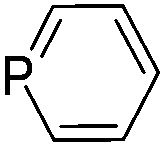
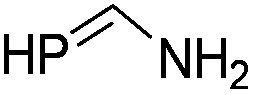
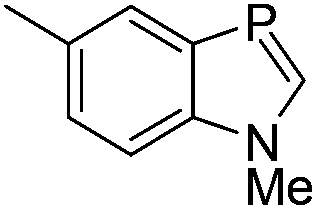
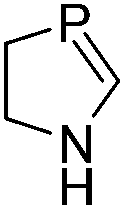
Accordingly, while the phosphabenzene ligand with stabilized PC π-orbitals (in agreement with the aromaticity) has a limited π-interaction and thus prefers the “classical” nP → Hg bonding, the electron-rich PC–N units (high energy HOMO) prefer the π interaction. The aromatic stabilization on the PC π-orbital16 of the 1,3-azaphosphole (compared to the 1,3-azaphospholine), together with the delocalization, weakens the interaction with mercury. It should be noted that a phenyl mercury compound with intermolecular phenyl Hg π-interactions is known;17 however, its linear geometry is not distorted by the weak interaction.
In conclusion, we have presented the first complexes of π-rich σ2P-aromatic 1,3-azaphosphole ligands with non-zerovalent late transition metals and revealed unprecedented significant π-donor contributions in the coordinative bond. Our findings suggest to search for related P(π)-donor complexes which open up new tuning possibilities in the highly important field of phosphorus-based late-transition metal catalysis.
This work was supported by the Deutsche Forschungs-gemeinschaft. We thank Dr M. K. Kindermann, G. Thede and M. Steinich for NMR, mass spectra and elemental analyses. Support from OTKA K 105417 and the Jedlik fellowship (L. K.) is also gratefully acknowledged.
Notes and references
- (a) C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 187 CrossRef; (b) C. Müller and D. Vogt, Dalton Trans., 2007, 5505 RSC; (c) P. Le Floch, Coord. Chem. Rev., 2006, 250, 627 CrossRef CAS.
- (a) M. Freytag, S. Ito and M. Yoshifuji, Chem.–Asian J., 2006, 1, 693 CrossRef CAS PubMed; (b) L. Weber, Angew. Chem., Int. Ed., 2002, 41, 563 CrossRef CAS.
- (a) A. Schmidpeter, in Phosphorus-Carbon Heterocyclic Chemistry: The Rise of a New Domain, ed. F. Mathey, Pergamon, Amsterdam, 2001, p. 363 Search PubMed; (b) R. K. Bansal and J. Heinicke, Chem. Rev., 2001, 101, 3549 CrossRef CAS PubMed.
- M. Ghalib, B. Niaz, P. G. Jones and J. W. Heinicke, Tetrahedron Lett., 2012, 53, 5012 CrossRef CAS.
- (a) K. C. Dash, H. Schmidbaur and A. Schmidpeter, Inorg. Chim. Acta, 1980, 41, 167 CrossRef; (b) J. G. Kraaijkamp, D. M. Grove, G. van Koten and A. Schmidpeter, Inorg. Chem., 1988, 27, 2612 CrossRef CAS.
- K. Angermund, A. Eckerle, J. Monkiewicz, C. Krüger and G. Wilke, Inorg. Chim. Acta, 1998, 270, 273 CrossRef CAS.
- D. Gudat, M. Schrott, V. Bajorat, M. Nieger, S. Kotila, R. Fleischer and D. Stalke, Chem. Ber., 1996, 129, 337 CrossRef CAS.
- (a) E. Deschamps, B. Deschamps, J. L. Dormieux, L. Ricard, N. Mezailles and P. Le Floch, Dalton Trans., 2006, 594 RSC; (b) A. Hayashi, M. Okazaki, F. Ozawa and R. Tanaka, Organometallics, 2007, 26, 5246 CrossRef CAS.
- B. R. Aluri, M. K. Kindermann, P. G. Jones, I. Dix and J. W. Heinicke, Inorg. Chem., 2008, 47, 6900 CrossRef CAS PubMed.
- H. Kanter and K. Dimroth, Tetrahedron Lett., 1975, 541 CrossRef CAS.
- B. Niaz, M. Ghalib, P. G. Jones and J. W. Heinicke, Dalton Trans., 2013, 42, 9523 RSC.
- P. S. Pregosin and R. W. Kunz, 31P and 13C NMR of Transition Metal Complexes, Springer, Berlin, 1979, p. 133 Search PubMed.
- Density functional calculations (see details in the ESI†) were carried out at the ωB97D/6-31G* level (for Hg and Ag the pseudopotential defined for the def2-ccPVTZ basis was applied) using the Gaussian 09 suite of programs. M. J. Frisch et al. , Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- L. Nyulászi, Chem. Rev., 2001, 101, 1229 CrossRef.
- Since the Cl⋯HN interaction modifies the geometry, we used the N-methylated benzazaphosphole as L.
- It has been shown that the stabilization of the lone pair orbital energies – as judged from the corresponding measured ionization energies compared to the saturated counterpart – in five membered heterocycles is related to the aromaticity. L. Nyulászi, Gy. Keglevich and L. D. Quin, J. Org. Chem., 1996, 61, 7808 CrossRef . This is because of the favourable HOMO–LUMO interaction between the fragments in the case of the cyclic conjugated compounds.
- Y. Sarazin, J. A. Wright and M. Bochmann, J. Organomet. Chem., 2006, 691, 5680 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses and NMR spectra of 2, 3, and 5, crystallographic details for 2 and 5 and quantum chemical calculations. CCDC 926714 and 926715. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52909f |
| This journal is © The Royal Society of Chemistry 2014 |