
Visible-light-assisted selective catalytic reduction of NO with NH3 on porphyrin derivative-modified TiO2 photocatalysts†
Akira
Yamamoto
a,
Yuto
Mizuno
a,
Kentaro
Teramura
*abc,
Saburo
Hosokawa
ab,
Tetsuya
Shishido
 bd and
Tsunehiro
Tanaka
*ab
bd and
Tsunehiro
Tanaka
*ab
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto daigaku Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: teramura@moleng.kyoto-u.ac.jp; Fax: +81 75 383 2561; Tel: +81 75 383 2559
bElements Strategy Initiative for Catalysts & Batteries (ESICB), Kyoto University, 1-30 Goryo-Ohara, Nishikyo-ku, Kyoto 615-8245, Japan
cPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
dDepartment of Applied Chemistry, Graduate School of Urban Environmental Sciences, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo 192-0397, Japan
First published on 18th September 2014
Abstract
Porphyrin-derivative-modified TiO2 photocatalysts showed high photocatalytic activity for the selective catalytic reduction of NO with NH3 in the presence of O2 under visible-light irradiation. Tetra(p-carboxyphenyl)porphyrin (TCPP) was the most effective photosensitizer among the five porphyrin derivatives investigated. NO conversion and N2 selectivity of 79.0% and 100%, respectively, were achieved at a gas hourly space velocity of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. UV–Vis and photoluminescence spectroscopies revealed the presence of two species of TCPP on the TiO2 surface; one was a TCPP monomer and the other was an H-aggregate of the TCPP molecules. It was concluded that the TCPP monomer is an active species for the photo-assisted selective catalytic reduction (photo-SCR). Moreover, an increase in the fraction of H-aggregates with increasing TCPP loading amount resulted in a decrease in the photocatalytic activity of the photo-SCR.
000 h−1. UV–Vis and photoluminescence spectroscopies revealed the presence of two species of TCPP on the TiO2 surface; one was a TCPP monomer and the other was an H-aggregate of the TCPP molecules. It was concluded that the TCPP monomer is an active species for the photo-assisted selective catalytic reduction (photo-SCR). Moreover, an increase in the fraction of H-aggregates with increasing TCPP loading amount resulted in a decrease in the photocatalytic activity of the photo-SCR.
1. Introduction
NOx, which is present in the exhaust gas of stationary emission sources, is removed by selective catalytic reduction with NH3 (NH3-SCR) over vanadium oxide-based catalysts according to the following equation: (4NO + 4NH3 + O2 → 4N2 + 6H2O).1–3 The NH3-SCR process is performed at temperatures above 573 K. To save the energy used for heating the catalyst bed, novel catalysts are required for performing NH3-SCR at low temperatures.4–6 Photocatalysis is one of the promising candidates for NH3-SCR because photocatalytic reactions proceed at room temperature. We have reported the photo-assisted selective catalytic reduction (photo-SCR) of NO with NH3 in the presence of O2 over TiO2 photocatalysts under UV-light irradiation.7–10 In this system, the NO conversion and N2 selectivity of 90% and 99%, respectively, were achieved at a gas hourly space velocity (GHSV) of 8000 h−1, which is sufficient for the deNOx process in typical stationary sources such as power plants, blast furnaces, and incinerators. However, a very high GHSV was required in diesel engines owing to the limited installation space of the deNOx process and a high flow rate of the exhaust gas. The volume of the catalyst was required to be of the order of the volume of the diesel engine cylinder (typical GHSV in a three-way catalyst is approximately 100000 h−1).11 Unfortunately, the NO conversion decreased with increasing GHSV in the photo-SCR system and it decreased to 40% at a GHSV of 100000 h−1 (ref. 10). Therefore, the photocatalytic activity of the photo-SCR has to be improved at a high GHSV region in order to remove the NOx from the exhaust gas of diesel engines.
Expansion of the adsorption wavelength to the visible-light region is an effective way of improving the photocatalytic activity. TiO2 photocatalysts do not absorb visible light because of their wide bandgap (>3.2 eV). Surprisingly, the photo-SCR proceeds to some extent under visible-light irradiation over the TiO2 photocatalysts. This is due to the direct electron transfer from the electron donor level of the N 2p orbital of the adsorbed NH3 to the conduction band of the Ti 3d orbital of TiO2 (in situ doping).12,13 However, the photocatalytic activity under visible-light irradiation is not sufficient for the application of the photo-SCR technology to the system at a high GHSV region. Thus, the proposed study aims to increase the photocatalytic efficiency under visible-light irradiation.
Porphyrin derivatives have absorption bands in the visible region owing to the π–π* transitions. Porphyrin derivatives are widely used as sensitizers in dye-sensitized solar cells (DSSCs)14–17 and dye-sensitized photocatalysts18–20 under visible-light irradiation owing to the following properties: 1) they exhibit intense absorption bands in the visible-light region, and 2) their photochemical and electrochemical properties can be tuned by the modification of the substituents and selection of the central metal. In previous studies, the porphyrin-sensitized photocatalysts were used for performing liquid phase reactions such as hydrogen production from water20 and degradation of organic compounds.18,19,21 However, there are only a few reports on the reactions involving porphyrin-sensitized photocatalysts in the gas phase. Recently, Ismail et al. reported that the porphyrin-sensitized mesoporous TiO2 films exhibited an improved photocatalytic activity for the photodegradation of acetaldehyde in the gas phase under visible-light irradiation.22 The porphyrin derivative-modified photocatalyst works efficiently as a visible-light response photocatalyst in the gas phase. In this study, we used five types of porphyrins for the modification of the TiO2 photocatalyst and investigated their performance in the photo-SCR using a gas flow reactor at a high GHSV of 50000 h−1.
2. Results and discussion
2.1. Effects of the functional group on porphyrins
Fig. 1 shows the conversion of NO during the photo-SCR over the various porphyrin derivative-modified TiO2 photocatalysts at a GHSV of 50000 h−1 after 6 h of visible-light irradiation, because the steady-state reaction rate was obtained after 6 h. The conversion of NO over the unmodified TiO2 photocatalyst was 13.4% under visible-light irradiation. Modification of the TiO2 photocatalyst with the porphyrin derivatives greatly enhanced the photocatalytic conversion of NO under the visible-light irradiation as shown on the top of Fig. 1. Among the five porphyrin derivative-modified TiO2 photocatalysts, the TCPP-TiO2 photocatalyst showed the highest conversion of NO (71.4%). The conversion of NO decreased in the following order: TCPP-TiO2 > TPP-TiO2 > TSPP-TiO2 > TMPP-TiO2 > TAPP-TiO2 > TiO2. Hence, it can be seen that the porphyrin functional group used for the modification of TiO2 had a significant effect on the photo-SCR activity.
 | ||
| Fig. 1 Structures of porphyrin derivatives and conversion of NO in the photo-SCR over the various porphyrin derivative-modified TiO2 photocatalysts after 6 h of visible-light irradiation (loading of porphyrin derivative: 18 μmol g−1, catalyst amount: 110 mg, NO: 1000 ppm, NH3: 1000 ppm, O2: 2%, GHSV: 50000 h−1). | ||
Fig. 2 shows the UV–Vis DR spectra of the various porphyrin-modified TiO2 photocatalysts. The unmodified TiO2 photocatalyst did not absorb the visible light above 400 nm. The modification of the porphyrin derivatives increased the absorption in the visible-light region. The absorption at 420 nm (Soret band) decreased in the following order: TCPP-TiO2 > TPP-TiO2 ≒ TSPP-TiO2 > TMPP-TiO2 > TAPP-TiO2 > TiO2. The order of the absorption is consistent with that of the NO conversion. The photocatalytic activities of NO were strongly dependent on the absorbance in the visible-light region, which suggested that the porphyrin derivatives functioned as photosensitizers under visible-light irradiation.
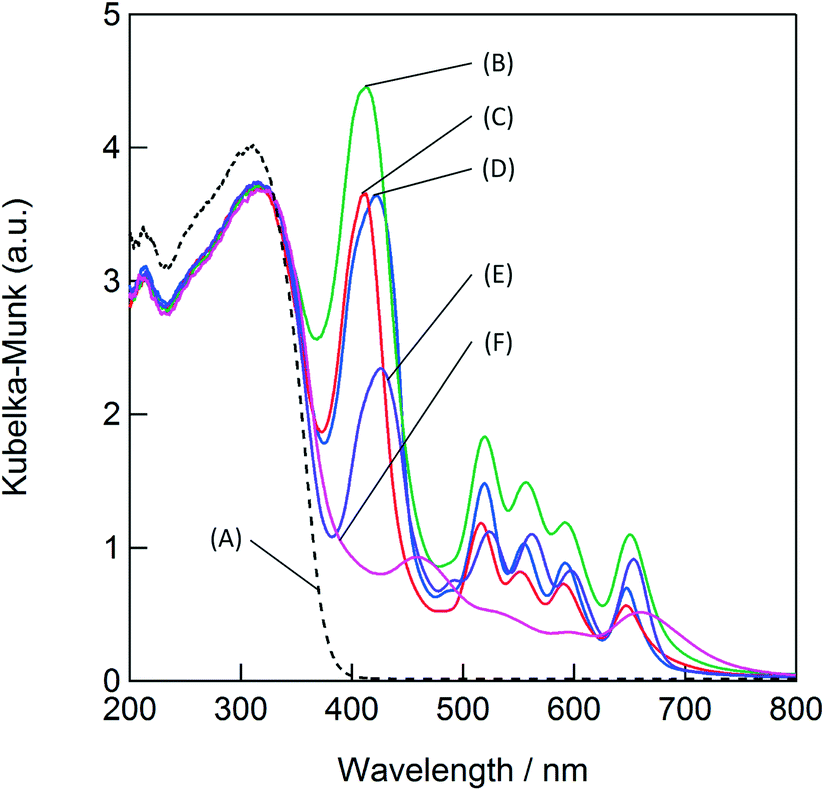 | ||
| Fig. 2 UV–Vis diffuse reflectance spectra of (A) TiO2, (B) TCPP-TiO2, (C) TSPP-TiO2, (D) TPP-TiO2, (E) TMPP-TiO2, and (F) TAPP-TiO2. | ||
2.2. Effect of TCPP loading on the activity of photo-SCR
Fig. 3 shows the UV/Vis DR spectra of the different loading amounts of the TCPP-TiO2 photocatalysts. One major peak and four minor peaks were observed in the visible region in each sample. The major peak is the Soret band (S2 ← S0 transition) and the four minor peaks are the Q bands (S1 ← S0 transition). The Q bands are attributed to the 0–0 and 0–1 components of the non-degenerated Qx and Qy bands (etio type),23 as expected for the D2h symmetry. The increase in the TCPP loading amount enhanced the capability of visible-light absorption. | ||
| Fig. 3 UV–Vis diffuse reflectance spectra of various loading amounts of TCPP-TiO2: (A) 0 μmol g−1, (B) 1.3 μmol g−1, (C) 6.3 μmol g−1, (D) 12.5 μmol g−1, (E) 37.5 μmol g−1, and (F) 62.5 μmol g−1. | ||
The peak positions of the Soret band and Q bands as a function of TCPP loading are shown in Table 1. The peak position of the Soret band of 1.3 μmol g−1 TCPP-TiO2-IMP (417 nm) corresponded to those of the TCPP molecules dissolved in CH3OH (418 nm). The TCPP molecules existed as a monomer in the CH3OH solution and the peak position of the TCPP monomer coincided with that reported previously.24 Thus, the TCPP molecules existed as monomers on the TiO2 surface at the low loading amount of 1.3 μmol g−1. The peak position of the Soret band was shifted from 417 to 407 nm (blue shift) when the loading amount of TCPP was increased from 1.3 to 62.5 μmol g−1. The origin of the blue shift can be explained on the basis of a “face to face” stacking pattern of TCPP (H-aggregate) according to Kasha's exciton theory.25 Thus, the blue-shifted peak observed at 407 nm can be attributed to the Soret exciton band in the H-aggregates of the TCPP. The gradual spectral shift can be explained as the result of the relative contribution of the monomers and H-aggregates. The H-aggregates were generated with an increase in the TCPP loading. The contribution of the H-aggregates became dominant for the Soret band of adsorption spectra of various loading amounts of TCPP-TiO2, which results in the gradual blue shift of the Soret band.
| Sample | Loading of TCPP/μmol g−1 | Soret/nm | Qy(0,1)/nm | Qy(0,0)/nm | Qx(0,1)/nm | Qx(0, 0)/nm |
|---|---|---|---|---|---|---|
| TCPP-TiO2-IMP | 1.3 | 417 | 519 | 555 | 592 | 649 |
| TCPP-TiO2-IMP | 6.3 | 416 | 520 | 557 | 592 | 651 |
| TCPP-TiO2-IMP | 12.5 | 411 | 521 | 559 | 593 | 652 |
| TCPP-TiO2-IMP | 37.5 | 411 | 522 | 562 | 595 | 654 |
| TCPP-TiO2-IMP | 62.5 | 407 | 522 | 560 | 594 | 653 |
| TCPP-TiO2-MIX | 12.5 | 419 | 529 | 565 | 600 | 658 |
| TCPP-SiO2-IMP | 12.5 | 422 | 521 | 556 | 593 | 649 |
| TCPP in MeOH | — | 418 | 515 | 549 | 590 | 646 |
On the other hand, the peak positions of the Q bands of 1.3 μmol g−1 TCPP-TiO2-IMP (519, 555, 592, and 649 nm) were slightly shifted to longer wavelengths compared to those of the TCPP monomer in CH3OH (515, 549, 590, and 646 nm). The slight red shift can be ascribed to the aggregation of TCPP24 and/or the interactions between the TCPP and a solid surface.26 The peak positions of the four Q bands of the TCPP-TiO2-IMP were shifted to longer wavelengths with an increase in the TCPP loading amount. The peak position of the Q bands of the H-aggregates of the TCPP is larger than that of the TCPP monomers.24 Thus, the red shift of the Q bands is due to the generation of the H-aggregates. Consequently, both the monomers and the H-aggregates of TCPP are generated on TiO2 and the fraction of the H-aggregates increased with increasing TCPP loading amount.
Fig. 4 shows the effect of various loading amounts of TCPP-TiO2-IMP on the photo-SCR. The conversion of NO increased with increasing TCPP loading amount up to 12.5 μmol g−1. The surface density of TCPP molecules on 12.5 μmol g−1 TCPP-TiO2-IMP is estimated to be 2.8 × 10−2 molecule nm−2. As shown in Fig. 3, increasing the loading amount of TCPP enhanced the capability of the visible-light absorption, which leads to the higher conversion of NO. On the other hand, the conversion of NO decreased when the TCPP loading was higher than 12.5 μmol g−1. UV–Vis spectroscopy revealed that the H-aggregates of TCPP were generated with an increase in the TCPP loading amount. The excited state lifetime of the H-aggregates was slightly shorter than those of the monomers.24,27 The generation of H-aggregates with a short excited state lifetime decreased the electron transfer efficiency from the excited state of TCPP to the conduction band of TiO2 due to non-radiative deactivation, which possibly resulted in the decrease of the photo-SCR activity.
 | ||
| Fig. 4 Dependence of conversion of NO on TCPP loading on TiO2 (catalyst amount: 110 mg, NO: 1000 ppm, NH3: 1000 ppm, O2: 2%, GHSV: 50000 h−1). | ||
2.3. Photo-SCR under various reaction conditions
Table 2 summarizes the concentrations of N2 in the outlet gas for the photo-SCR under various reaction conditions. The conversion of NO in the photo-SCR over TCPP-TiO2-IMP was 79.0% (entry 1), and the activity was stable for at least 6 h of visible-light irradiation (the time course is shown in Fig. S1 in the ESI†). N2 was the only product observed and N2O was not detected in any of the reactions. The utilization of visible light was advantageous for the high selectivity to N2, since N2O was generated as a by-product of the photo-SCR over the TiO2 photocatalyst under UV-light irradiation. Turnover number (TON) of TCPP was calculated to be 810 after 6 h of visible-light irradiation. Thus, the total N2 in the outlet gas originated from the nitrogen atoms of NO and the NH3 molecules in the gas phase and not from the TCPP molecules. The reaction hardly proceeded over the TCPP-TiO2 photocatalyst without a substrate such as NO, NH3, and O2 (entries 2, 3, and 4). The O2 concentrations did not affect the generation rate of N2 over 2% (entries 1, 5, and 6). TCPP-SiO2-IMP showed much lower activity under the same reaction conditions than TCPP-TiO2-IMP (entry 7), although the TCPP on SiO2 absorbed visible light as well as that on TiO2 as shown in Fig. 5. In addition, the activity of the photo-SCR over the TCPP-TiO2 photocatalyst prepared by a physical mixture method (TCPP-TiO2-MIX) was similar to that of the unmodified TiO2, although TCPP-TiO2-MIX absorbed in the visible region as shown in Fig. 5.| Entry | Catalyst | Inlet gas conc. (ppm) | N2 conc.a (ppm) | ||
|---|---|---|---|---|---|
| NO (ppm) | NH3 (ppm) | O2 (%) | |||
| a Concentration of N2 in the outlet gas. Catalyst amount: 110 mg (TiO2, TCPP-TiO2), 50 mg (TCPP-SiO2), b TCPP loading: 12.5 μmol g−1. | |||||
| 1 | TCPP-TiO2-IMPb | 1000 | 1000 | 2 | 790 |
| 2 | TCPP-TiO2-IMPb | 0 | 1000 | 2 | 57 |
| 3 | TCPP-TiO2-IMPb | 1000 | 0 | 2 | 24 |
| 4 | TCPP-TiO2-IMPb | 1000 | 1000 | 0 | 42 |
| 5 | TCPP-TiO2-IMPb | 1000 | 1000 | 5 | 780 |
| 6 | TCPP-TiO2-IMPb | 1000 | 1000 | 10 | 771 |
| 7 | TCPP-SiO2-IMP2b | 1000 | 1000 | 2 | 63 |
| 8 | TCPP-TiO2-MIXb | 1000 | 1000 | 2 | 140 |
 | ||
| Fig. 5 UV–Vis diffuse reflectance spectra of (A) TCPP-TiO2(IMP), (B) TCPP-SiO2, and (C) TCPP-TiO2(MIX) and adsorption spectra of 3.4 × 10−6 mol L−1 TCPP in methanol (D). | ||
Fig. 6 shows the photoluminescence spectra of TCPP-TiO2-IMP, TCPP-SiO2-IMP, TCPP-TiO2-MIX, and TCPP monomer in CH3OH. Two emission bands were observed in the TCPP monomer in CH3OH solution (650 and 714 nm), which coincided with the previously reported values.24 The emission bands at 650 and 714 nm can be attributed to the transition from the vibrational ground state of S1 to the vibrational ground state of S0 (Qx(0,0) transition) and to the vibrational excited state of S0 (Qx(0,1) transition) of the TCPP monomer, respectively.24,28 The peak positions of the emission bands in the TCPP-TiO2-IMP (654 and 715 nm) were almost similar to those of the TCPP monomer in CH3OH. It is reported that the emission bands of the H-aggregates of TCPP have lower intensity and are shifted to longer wavelengths than those of the monomers,29 which is totally different from the emission spectrum of the TCPP-TiO2-IMP. Accordingly, the emission bands of TCPP-TiO2-IMP were mainly composed of the TCPP monomer emissions.
 | ||
| Fig. 6 Photoluminescence spectra of (A) TCPP-TiO2-IMP, (B) TCPP in methanol, (C) TCPP-SiO2-IMP, (D) TCPP-TiO2-MIX, and (E) TCPP powder. The excitation wavelength was 410 nm. The spectra of (A), (C), (D), and (E) were measured at the voltage of the photomultiplier tube of 700 V and spectrum (B) was measured at 450 V. Loading of TCPP was 12.5 μmol g−1, and the concentration of TCPP in methanol was 1.0 × 10−6 mol L−1. | ||
Two emission bands were observed at 658 and 713 nm, and the peak positions were similar to those of the TCPP in CH3OH (650 and 714 nm) and TCPP-TiO2-IMP (654 and 715 nm) (Fig. 6). Thus, the TCPP species on SiO2 possesses a monomeric state, which is similar to that on TiO2. The low activity of TCPP-SiO2-IMP can be explained by an electron transfer mechanism, which is the key step in the DSSCs and dye-sensitized photocatalysts. In the first step of the photo-SCR over the TCPP-TiO2-IMP, the TCPP is excited by the visible-light irradiation. In the next step, the electron transfer occurs from the photo-excited TCPP to the conduction band of TiO2. However, the electron transfer cannot occur from the photo-excited TCPP to the conduction band of SiO2 because the energy level of the SiO2 conduction band is much higher than that of the lowest unoccupied molecular orbital (LUMO) of TCPP, which results in the low activity of the TCPP-SiO2-IMP. Hence, the photo-SCR over TCPP-TiO2-IMP under visible-light irradiation proceeds via the electron transfer from the photo-excited TCPP to the conduction band of TiO2.
No emission peak was observed for the TCPP-TiO2-MIX and TCPP powder (Fig. 6). The interaction between the TCPP molecules might lead to the non-radiative deactivation of the photo-excited states of TCPP, resulting in the luminescence quenching for the TCPP-TiO2-MIX and TCPP powder. These results explain the low activity of the TCPP-TiO2-MIX, i.e. although the TCPP molecules on the TCPP-TiO2-MIX absorb visible light, they do not function as a photosensitizer under visible-light irradiation because of the fast non-radiative quenching.
2.4. On–off response tests of visible-light irradiation
Fig. 7 shows the on–off response for the photo-SCR under visible-light irradiation over the TCPP-TiO2 photocatalyst at a GHSV of 50000 h−1. The conversion of NO was about 6% without visible-light irradiation. The conversion of NO significantly increased to 86.5% under visible-light irradiation, indicating the function of the TCPP-TiO2 photocatalyst as a visible-light-driven photocatalyst for the photo-SCR. The conversion of NO gradually decreased to 81.2% with increasing irradiation time, although the conversion was restored to the original level (85.5%) after the on–off action. The decrease in the conversion of NO with the irradiation time was not due to the decomposition of TCPP. If the TCPP on the TiO2 surface was decomposed under visible-light irradiation, the initial conversion of NO was expected to decrease gradually in the second, third, and fourth times. However, we did not observe a decrease in the initial conversion of NO. The recovery of the initial conversion of NO took place reversibly.
 | ||
| Fig. 7 Conversion of NO during several on/off cycles of visible-light irradiation over the TCPP/TiO2 photocatalyst: each cycle consisted of 2 hours of light on and off (TCPP loading: 12.5 μmol g−1, catalyst amount: 110 mg, NO: 1000 ppm, NH3: 1000 ppm, O2: 2%, GHSV: 50000 h−1). | ||
3. Experimental section
3.1. Catalyst preparation
TiO2 powder (ST-01, anatase, 273 m2 g−1) was purchased from Ishihara Sangyo Kaisha, Ltd. SiO2 powder (630 m2 g−1) was prepared by hydrolysis of tetraethyl orthosilicate (TEOS) in a water–ethanol mixture at boiling point, followed by calcination in dry air at 773 K for 5 h. Tetraphenylporphyrin (TPP), tetra(p-carboxyphenyl)porphyrin (TCPP), tetra(p-sulfonatephenyl)porphyrin (TSPP), tetra(p-methoxyphenyl)porphyrin (TMPP), and tetra(p-aminophenyl)porphyrin (TAPP) were purchased from Tokyo Chemical Industry Co., Ltd. and used without further purification (see Fig. 1). The porphyrin derivatives were impregnated over the TiO2 powder and the porphyrin-modified TiO2 photocatalysts were abbreviated to porphyrin-TiO2 (e.g. TCPP-TiO2). Various loading amounts of TCPP-TiO2 catalysts were prepared by an impregnation method using 1 M NH3 aqueous solution as a solvent (TCPP-TiO2-IMP). A TCPP-modified SiO2 catalyst was prepared by the impregnation method as shown above (TCPP-SiO2-IMP). A physical mixture catalyst of TCPP and the TiO2 powder was prepared as a reference (TCPP-TiO2-MIX).3.2. Photocatalytic reaction
The photo-SCR was carried out in a conventional fixed-bed flow system under atmospheric pressure. The catalyst was fixed with quartz wool and filled up in a quartz reactor with flat facets (12 mm (H) × 10 mm (W) × 1.0 mm (D)). The reaction gas composition was as follows: NO 1000 ppm, NH3 1000 ppm, O2 2–10%, He balance. A 300 W Xe lamp (PERKIN-ELMER PE300BF) equipped with an L-42 cut-off filter was used as a light source (λ > 400 nm), and the sample was irradiated from one side of the flat facets of the reactor. N2 and N2O were analyzed by SHIMADZU GC-8A TCD gas chromatographs equipped with MS-5A and Porapak Q.3.3. Characterization
UV–Vis transmission adsorption and diffuse reflectance spectra were obtained with a UV–Vis spectrometer (JASCO V-650). Transmission adsorption spectra were measured using a 1 cm quartz cell at room temperature in the scan range of 300–800 nm. Photoluminescence spectra were recorded on a Hitachi F-7000 fluorospectrometer in the scan range of 550–760 nm at an excitation wavelength of 410 nm. The concentrations of TCPP in methanol solution used in the adsorption and emission spectroscopy were 3.4 × 10−6 and 1.0 × 10−6 mol L−1, respectively.Conclusion
We found that porphyrin derivative-modified TiO2 functions as a visible-light-driven photocatalyst for the photo-SCR. The TCPP-modified TiO2 photocatalyst showed the highest activity of the photo-SCR among the TiO2 photocatalysts modified with the five porphyrin derivatives investigated. We elucidated that the state of TCPP on TiO2 affects the photocatalytic conversion of NO and the active species is a TCPP monomer adsorbed on TiO2 due to efficient electron transfer from the photo-excited TCPP monomer to the conduction band of TiO2.Acknowledgements
This study was partially supported by the Program for Element Strategy Initiative for Catalysts & Batteries (ESICB), commissioned by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and the Precursory Research for Embryonic Science and Technology (PRESTO), supported by the Japan Science and Technology Agency (JST). Akira Yamamoto would like to thank the JSPS Research Fellowships for Young Scientists.References
- H. Bosch and F. Janssen, Catal. Today, 1988, 2, 369–379 CrossRef CAS.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- P. Forzatti and L. Lietti, Heterog. Chem. Rev., 1996, 3, 33–51 CrossRef CAS.
- R. Q. Long, R. T. Yang and R. Chang, Chem. Commun., 2002, 452–453 RSC.
- J. Li, H. Chang, L. Ma, J. Hao and R. T. Yang, Catal. Today, 2011, 175, 147–156 CrossRef CAS PubMed.
- P. G. Smirniotis, D. A. Peña and B. S. Uphade, Angew. Chem., Int. Ed., 2001, 40, 2479–2482 CrossRef CAS.
- T. Tanaka, K. Teramura, K. Arakaki and T. Funabiki, Chem. Commun., 2002, 2742–2743 RSC.
- K. Teramura, T. Tanaka, S. Yamazoe, K. Arakaki and T. Funabiki, Appl. Catal., B, 2004, 53, 29–36 CrossRef CAS PubMed.
- S. Yamazoe, T. Okumura, K. Teramura and T. Tanaka, Catal. Today, 2006, 111, 266–270 CrossRef CAS PubMed.
- A. Yamamoto, Y. Mizuno, K. Teramura, T. Shishido and T. Tanaka, Catal. Sci. Technol., 2013, 3, 1771–1775 CAS.
- M. Koebel, M. Elsener and M. Kleemann, Catal. Today, 2000, 59, 335–345 CrossRef CAS.
- S. Yamazoe, K. Teramura, Y. Hitomi, T. Shishido and T. Tanaka, J. Phys. Chem. C, 2007, 111, 14189–14197 CAS.
- T. Shishido, K. Teramura and T. Tanaka, Catal. Sci. Technol., 2011, 1, 541–551 CAS.
- A. Kay and M. Graetzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363–1379 CrossRef CAS PubMed.
- H. Imahori, H. Norieda, H. Yamada, Y. Nishimura, I. Yamazaki, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2000, 123, 100–110 CrossRef PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- G. Mele, R. Del Sole, G. Vasapollo, E. García-López, L. Palmisano and M. Schiavello, J. Catal., 2003, 217, 334–342 CrossRef CAS.
- S. Murphy, C. Saurel, A. Morrissey, J. Tobin, M. Oelgemöller and K. Nolan, Appl. Catal., B, 2012, 119–120, 156–165 CrossRef CAS PubMed.
- H. Hagiwara, N. Ono, T. Inoue, H. Matsumoto and T. Ishihara, Angew. Chem., Int. Ed., 2006, 45, 1420–1422 CrossRef CAS PubMed.
- D. Li, W. Dong, S. Sun, Z. Shi and S. Feng, J. Phys. Chem. C, 2008, 112, 14878–14882 CAS.
- A. A. Ismail and D. W. Bahnemann, ChemSusChem, 2010, 3, 1057–1062 CrossRef CAS PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 Search PubMed.
- N. C. Maiti, S. Mazumdar and N. Periasamy, J. Phys. Chem. B, 1998, 102, 1528–1538 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- S. Cherian and C. C. Wamser, J. Phys. Chem. B, 2000, 104, 3624–3629 CrossRef CAS.
- N. C. Maiti, M. Ravikanth, S. Mazumdar and N. Periasamy, J. Phys. Chem., 1995, 99, 17192–17197 CrossRef CAS.
- M. Gouterman and G.-E. Khalil, J. Mol. Spectrosc., 1974, 53, 88–100 CrossRef CAS.
- S. Verma, A. Ghosh, A. Das and H. N. Ghosh, J. Phys. Chem. B, 2010, 114, 8327–8334 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00598h |
| This journal is © The Royal Society of Chemistry 2015 |