Efficient H2 generation from formic acid using azole complexes in water†
Yuichi
Manaka
a,
Wan-Hui
Wang
*ab,
Yuki
Suna
a,
Hide
Kambayashi
a,
James T.
Muckerman
c,
Etsuko
Fujita
c and
Yuichiro
Himeda
*ab
aNational Institute of Advanced Industrial Science and Technology, Tsukuba Central 5-2, 1-1-1 Higashi, Ibaraki, Tsukuba, 305-8565 Japan. E-mail: himeda.y@aist.go.jp; ou-wanhui@aist.go.jp
bJST, ACT-C, 4-1-8 Honcho, Saitama, Kawaguchi, 332-0012 Japan
cChemistry Department, Brookhaven National Laboratory, Upton, NY 11973-5000, USA
First published on 23rd October 2013
Abstract
Iridium azole-containing complexes are demonstrated to catalyze the dehydrogenation of formic acid into H2–CO2 (1/1) mixtures in aqueous solution in the absence of organic additives, and with a maximum turnover frequency (TOF) of 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 at 80 °C.
000 h−1 at 80 °C.
Formic acid is a potentially valuable hydrogen carrier in the context of renewable fuels for the solution of today's energy problems. It is a non-toxic liquid at room temperature and contains H2 in 4.3 wt%, thereby offering an easy-handling H2 storage material compared with direct H2 storage as a compressed gas. Moreover, the free energy for conversion of formic acid into H2 and CO2 in aqueous solution is quite low (Scheme 1).1
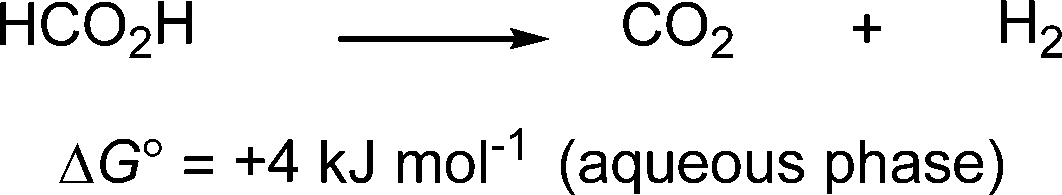 | ||
| Scheme 1 Dehydrogenation of formic acid. | ||
The generation of formic acid by hydrogenation of CO2 has been widely investigated during the last four decades.2 In combination with the hydrogenation of CO2 for H2 storage, the dehydrogenation of formic acid for H2 supply may provide an energy-efficient H2 storage/supply system. Therefore, H2 generation through dehydrogenation of formic acid has been studied intensively in the past few years (Scheme 1).3
Recently, we have investigated the catalytic dehydrogenation of formic acid using iridium catalysts with 2,2′-bipyridine (bpy) derivative ligands in water without additional organic base or organic solvents.4,5 Formic acid was selectively decomposed into H2 and CO2 without CO generation. CO causes a major problem for hydrogen fuel cells because it acts as a Pt anode catalyst poison at concentrations on the order of ppm. The strategy of increasing the catalytic activity of the iridium catalyst with bpy derivative ligands is based on controlling the electron donating ability of the ligand through introduction of electron donating substituents. For example, complex 2 [Cp*Ir(4DHBP)(OH2)]2+ (4DHBP: 4,4′-dihydroxy-2,2′-bipyridine), Fig. 1, bearing two electron donating hydroxyl groups, produced a remarkable initial TOF of 14000 h−1 at 90 °C,4 and we have achieved the best TOF value (TOF: 228000 h−1, 90 °C) on the dehydrogenation of formic acid by introducing four hydroxyl groups to the dinuclear Ir complex.2f
With a view toward developing a more efficient catalyst, we explored a new design for increasing the electron donating ability of the ligand through the use of five-membered aromatic N-heterocycles (azoles) as a ligand in an iridium or rhodium complex. Pyridine, which was chosen in our previous studies,4,5 is well known as an electron deficient aromatic heterocyclic ligand. In contrast, azoles, such as imidazole and pyrazole, have an electron rich character because of having six π-electrons delocalized on each five-membered ring. The electron donating ability of imidazole to a coordinated metal atom is stronger than that of pyridine, as has been demonstrated on the basis of measured Mossbauer spectra,6 absorption spectra,7,8 and cyclic voltammetry.7 This prompted us to investigate the azole complexes as potentially good catalysts in the dehydrogenation of formic acid. In addition, some groups have already attempted to use azole-containing complexes in catalytic reactions, e.g., hydroamination,9 hydrogenation of CO2,10 and dehydrogenation of formic acid.11 In particular, the dehydrogenation of formic acid using a phenylazole complex yielded a TOF of 536 h−1 in formic acid–triethylamine (5/2) for the first 3 min at 25 °C.11 Herein, we report the highly efficient dehydrogenation of formic acid in water with simple azole-containing complexes.
Fig. 1 shows the iridium and rhodium complexes employed in this work. Imidazole and pyrazole were selected as basic azole structures. These complexes are divided into three categories according to the structure of the chelated ligand. The first one is composed of bi-six membered nitrogen heterocycles containing complexes 1 and 2. Complex 2 has two hydroxyl groups for enhancing the electron donating ability of the bpy ligand. The second series consists of ligands with six and five membered nitrogen heterocycles (pyridinylazole) containing complexes 3, 4 and 5. The last series contains biazole complexes 6, 7 and 8. Complexes 7 and 8 have four electron donating methyl groups on the biimidazole ligand. These complexes were prepared according to literature procedures (see the ESI†).12
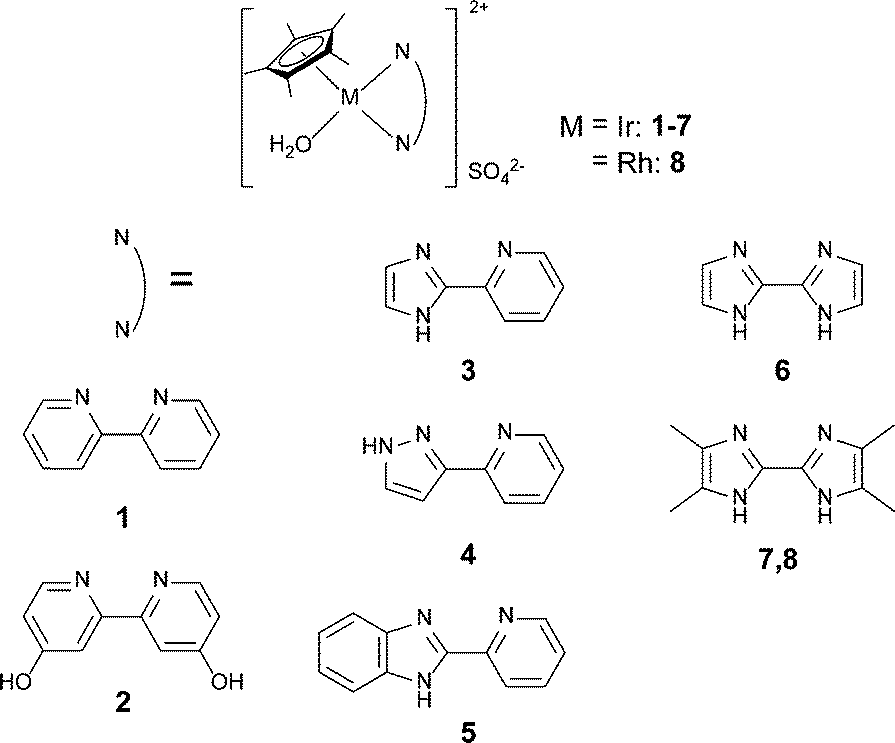 | ||
| Fig. 1 Complexes for dehydrogenation of formic acid and hydrogenation of carbon dioxide. | ||
All the azole complexes were investigated for dehydrogenation of formic acid in 1 M aqueous formic acid solution at 60 °C, and CO contamination was not detected by GC (Table 1, entries 1–8). Initial TOF values of pyridinylazole complexes 3, 4 and 5 were 810, 400, and 570 h−1, respectively (entries 3, 4, and 5). In comparison with the TOF value of non-substituted complex 1 (entry 1), these TOF values are higher by more than a factor of 13. The TOF value of 3 was twice as high as that of 4. This difference in activity is attributed to the nitrogen position on the azole. The two adjacent nitrogen atoms in the pyrazole withdraw electrons to the aromatic ring, causing the electron donating ability of the coordinating nitrogen atom of pyrazole to be less than that of imidazole. When using pyridinyl benzimidazole complex 5 (entry 5), the TOF value was slightly lower than that of 3 owing to the electron withdrawing effect of the benzene ring. The TOF value of biazole complex 6 was 3980 h−1 (entry 6), higher than that of pyridinylazole complexes and 130 times higher than that of 1. As expected, the exchange of pyridine with azole enhanced the catalytic activity remarkably. It is noteworthy that the TOF value was even higher than that of complex 2, in which electron donating ability was enhanced by hydroxyl substituents on the bpy ligand (entry 2). The iridium tetramethyl biimidazole complex 7 (entry 7) shows a high catalytic activity (TOF = 7020 h−1). This high value is attributed to the electron donating ability of the ligand in 7 that is enhanced by the four methyl substituents. Thus, we choose 7 as a catalyst in the following experiments. We tried using rhodium catalyst 8 (entry 8), but its activity was much lower than that of iridium complex 7. The same tendency was observed when using bpy derivatives as a ligand.5
| Entry | Catalyst | Amount (mM) | TOFa (h−1) | Time (h) | Minimum TON | Conversion (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: the reaction was carried out at 60 °C in 10 mL of 1 M aqueous formic acid (pH 1.75). a The average value during the first 10 min.b The average value during the first 2 h.c Used 2 M aqueous formic acid.d Carried out at 80 °C.e Used 1 M aqueous sodium formate–formic acid (1/4) pH 2.8. | ||||||
| 1b | 1 | 2.0 | 30 | — | — | — |
| 2c | 2 | 0.2 | 2400 | 5 | 5000 | 100 |
| 3 | 3 | 0.5 | 810 | 4 | 1970 | 98 |
| 4 | 4 | 0.5 | 400 | 7 | 2000 | >99 |
| 5 | 5 | 0.5 | 570 | 6 | 2000 | >99 |
| 6 | 6 | 0.1 | 3980 | 5 | 10000 |
100 |
| 7 | 7 | 0.1 | 7020 | 2 | 10000 |
100 |
| 8c | 8 | 0.5 | 250 | 8 | 1020 | 26 |
| 9d | 7 | 0.1 | 34000 |
0.5 | 10000 |
100 |
| 10e | 7 | 0.1 | 9600 | 2.5 | 8000 | 80 |
The temperature dependence of the TOF values was also investigated. Fig. 2a shows the initial part of the time course of gas evolution using 7 in aqueous formic acid at 40–80 °C. The initial TOF value of 7 was observed to increase with increasing reaction temperature. Fig. 2b shows the Arrhenius plots of the TOF values of 7. The apparent activation energy of 7 is 78 kJ mol−1. This value is almost the same as that of complex 2 (76 kJ mol−1).4 The highest TOF value for 7 (34000 h−1) was obtained at 80 °C (Table 1, entry 9).
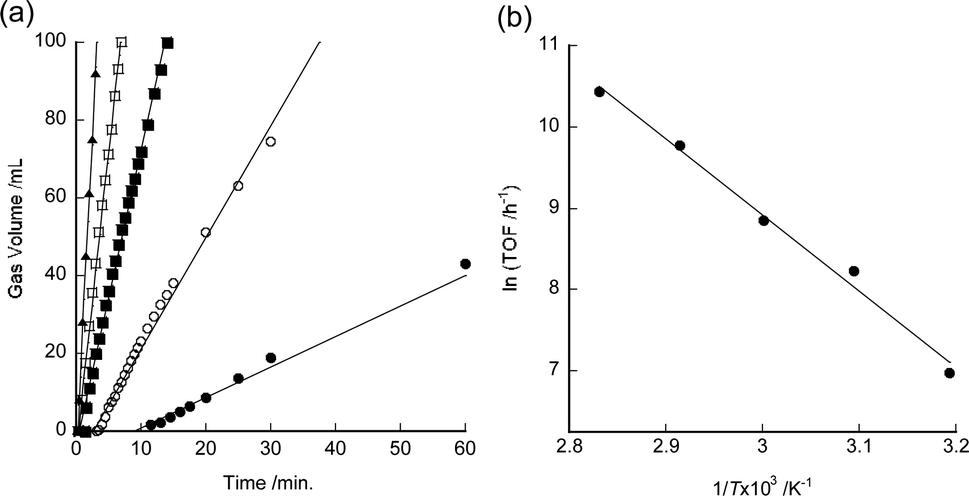 | ||
| Fig. 2 (a) Time course of gas evolution using 0.1 mM of 7 in 10 mL of 1 M aqueous formic acid under 0.1 MPa (closed circles: 40 °C, open circles: 50 °C, closed squares: 60 °C, open squares: 70 °C, closed triangles: 80 °C). Experimental details are available in the ESI.† The induction periods observed at low temperatures are due to the detection limit of the gas meter used. (b) Arrhenius plot of initial TOF values for dehydrogenation of formic acid using 7. | ||
Dehydrogenation of formic acid using 7 under various catalyst concentrations and formic acid concentrations was investigated (Table S2†). Catalytic dehydrogenation of formic acid also proceeded smoothly in a closed system under pressurized conditions (Fig. 3a). The pressure increased up to 1.8 MPa after 1.5 h in this system. Formic acid was dehydrogenated by 7 almost completely (>98%). These results show that the pressure in the system did not inhibit the reaction. It indicates that high pressure H2 gas can be supplied using 7. Moreover, no CO contaminant was observed despite the high-pressure reaction conditions (Fig. 3b).
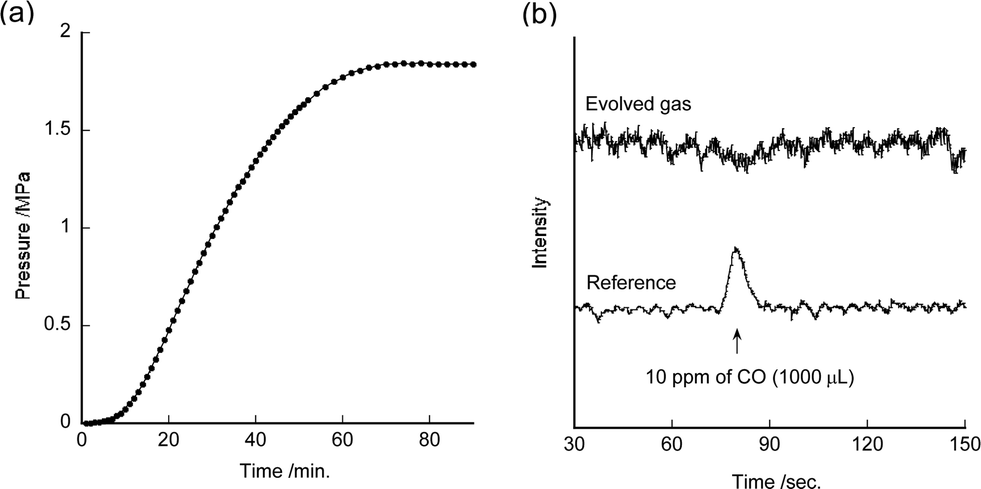 | ||
| Fig. 3 (a) Time course of pressure value during dehydrogenation processes using 7 (0.1 mM) at 80 °C in a 1 M formic acid aqueous solution (7 mL) in a glass autoclave. (b) Gas chromatogram of the evolved gas and CO containing gas using a FID with methaniser. 1000 μL of 10 ppm of CO gas and 500 μL of evolved gas were injected. | ||
The pH dependence of TOF values using catalyst 7 was also investigated. The TOF values from a 1 M aqueous sodium formate–formic acid solution were measured at various pH values (Fig. 4). The highest TOF value of 9600 h−1 was obtained at pH 2.8 at 60 °C (Table 1, entry 10). When the pH was higher than 7, no gas generation was observed. The UV/Vis spectral changes of 7 were measured at pH 1.5 to 12.5 (see Fig. S1, ESI†). The average pKa1 of 7 for single deprotonation of the imidazole and pKa2 for deprotonation of the aqua ligand were respectively 8.8 and 11.4 as determined from these measurements. These values are similar to the reported pKa values of 9.0 and 11.3 of its ruthenium analogue.8 Since the protonation status of 7 did not change below pH 7, the pH dependence of dehydrogenation by 7 was not due to the deprotonation status of the imidazole ligand. It is inferred that the pKa of formic acid (3.75) affected the pH dependence as previously reported.2f,13 The TOF value of other azole complexes at pH 3.5 (close to the pKa of formic acid, 1 M aqueous sodium formate–formic acid (1/1) solution) was also measured (Table S1†). At pH 3.5, TOF values for the dehydrogenation of formic acid were lower than that at pH 1.75 except for rhodium complex 8.
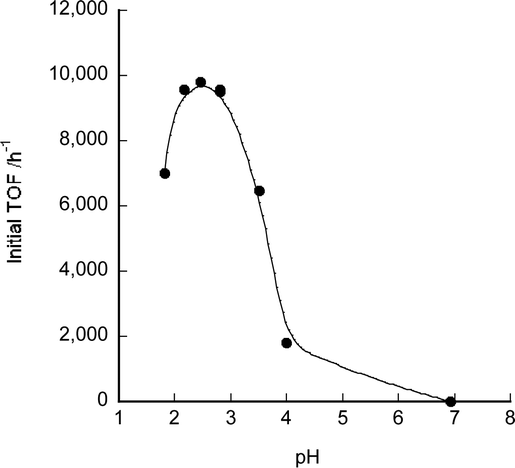 | ||
| Fig. 4 pH dependence of initial TOF values at 60 °C in 1 M HCO2H/HCO2Na using 7. | ||
The hydrogenation reaction of CO2 to formate using azole complexes was also carried out under 1 MPa of H2–CO2 (1/1) in aqueous solution (Table S3†). The results of hydrogenation using azole complexes were higher compared to 1. Therefore, these azole complexes exhibit the possibility of catalysing an interconversion cycle between HCO2H and CO2 using only one catalyst.
In summary, the newly designed azole-containing complexes exhibit higher catalytic activities than the unsubstituted bpy analogues in the dehydrogenation of formic acid in aqueous solutions. Especially, the iridium tetramethyl biimidazole complex 7 achieved a TOF value as high as 34000 h−1 (80 °C) which is higher than the TOF values of any other mononuclear complexes yet reported. The extraordinary activity of complex 7 is only inferior to the most active dinuclear complex that we previously reported. Nevertheless, complex 7 is more robust in concentrated aqueous solutions of formic acid. The remarkable activity and stability are strongly affected by the imidazole structure and electron donating methyl groups. A study of the detailed activation mechanism with azole ligands is under way.
Acknowledgements
Y. H. and W.-H. W. acknowledge JST, ACT-C for financial support. Work at Brookhaven National Laboratory was carried out under contract DE-AC02-98CH10886 with the U.S. Department of Energy and supported by its Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences.References
- P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS PubMed.
- (a) W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC; (b) C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254–6257 CrossRef CAS PubMed; (c) P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS; (d) R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed; (e) Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702–712 CrossRef CAS; (f) J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed; (g) W.-H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923–7926 RSC; (h) W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, ACS Catal., 2013, 3, 856–860 CrossRef CAS.
- (a) M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC; (b) M. Czaun, A. Goeppert, R. May, R. Haiges, G. K. S. Prakash and G. A. Olah, ChemSusChem, 2011, 4, 1241–1248 CrossRef CAS PubMed; (c) S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC; (d) B. Loges, A. Boddien, F. Gartner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS; (e) A. Boddien, B. Loges, H. Junge and M. Beller, ChemSusChem, 2008, 1, 751–758 CrossRef CAS PubMed; (f) C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS PubMed; (g) S. Fukuzumi, T. Kobayashi and T. Suenobu, ChemSusChem, 2008, 1, 827–834 CrossRef CAS PubMed; (h) D. J. Morris, G. J. Clarkson and M. Wills, Organometallics, 2009, 28, 4133–4140 CrossRef CAS; (i) S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496–1497 CrossRef CAS PubMed; (j) A. Majewski, D. J. Morris, K. Kendall and M. Wills, ChemSusChem, 2010, 3, 431–434 CrossRef CAS PubMed; (k) A. Boddien, D. Mellmann, F. Gartner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed; (l) N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed; (m) R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742–6750 CrossRef CAS; (n) G. Papp, J. Csorba, G. Laurenczy and F. Joó, Angew. Chem., Int. Ed., 2011, 50, 10433–10435 CrossRef CAS PubMed; (o) M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 RSC; (p) P. Sponholz, D. Mellmann, H. Junge and M. Beller, ChemSusChem, 2013, 6, 1172–1176 CrossRef CAS PubMed; (q) Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7367 RSC; (r) E. Fujita, J. T. Muckerman and Y. Himeda, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1031–1038 CrossRef CAS PubMed.
- Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- Y. Himeda, S. Miyazawa and T. Hirose, ChemSusChem, 2011, 4, 487–493 CrossRef CAS PubMed.
- L. M. Epstein, D. K. Straub and C. Maricond, Inorg. Chem., 1967, 6, 1720–1724 CrossRef CAS.
- M. Haga, Inorg. Chim. Acta, 1983, 75, 29–35 CrossRef CAS.
- M. Okamura, M. Yoshida, R. Kuga, K. Sakai, M. Kondo and S. Masaoka, Dalton Trans., 2012, 41, 13081–13089 RSC.
- Y. Kashiwame, M. Watanabe, K. Araki, S. Kuwata and T. Ikariya, Bull. Chem. Soc. Jpn., 2011, 84, 251–258 CrossRef CAS.
- K. Muller, Y. Sun, A. Heimermann, F. Menges, G. Niedner-Schatteburg, C. van Wullen and W. R. Thiel, Chem.–Eur. J., 2013, 19, 7825–7834 CrossRef CAS PubMed.
- J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Chem. Sci., 2013, 4, 1234–1244 RSC.
- W.-H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita, T. Hirose and Y. Himeda, Chem.–Eur. J., 2012, 18, 9397–9404 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496–1497 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy00830d |
| This journal is © The Royal Society of Chemistry 2014 |