A dinuclear iron complex with a single oxo bridge as an efficient water-oxidizing catalyst in the presence of cerium(IV) ammonium nitrate: new findings and current controversies†
Mohammad Mahdi
Najafpour
*ab,
Atefeh Nemati
Moghaddam
a,
Davood Jafarian
Sedigh
a and
Małgorzata
Hołyńska
c
aDepartment of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, 45137-66731, Iran. E-mail: mmnajafpour@iasbs.ac.ir; Tel: +98 241 415 3201
bCenter of Climate Change and Global Warming, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, 45137-66731, Iran
cFachbereich Chemie und Wissenschaftliches Zentrum für Materialwissenschaften (WZMW), Philipps-Universität Marburg, Hans-Meerwein-Straße, D-35032 Marburg, Germany
First published on 4th October 2013
Abstract
In this paper a dinuclear Fe complex, [tpa(H2O)FeOFe(H2O)tpa](ClO4)4 (tpa: tris(2-pyridylmethyl)amine) is reported as an efficient catalyst for water oxidation in the presence of cerium(IV) ammonium nitrate. The turnover frequency of this dimer is 6 times greater than the turnover frequency reported for the [Fe(OTf or Cl)2(tpa)] complex (OTf: trifluoromethanesulfonate). The role of FeO42− ions in the water oxidation reaction is also discussed.
Hydrogen is a very important energy carrier, as it is reliable and important for future centralized energy systems.1,2 Water splitting is a promising approach for the production of hydrogen.3,4 However, the development of an efficient water-oxidizing catalyst is fundamental to achieving a breakthrough in the technology of water splitting.3,4 Mn,5–11 Fe,12–16 Co17–19 and Cu20,21 compounds are used as water-oxidizing catalysts because of the wide availability and low toxicity of these metals. In 2010, Stefan Bernhard, Terrence J. Collins and co-workers presented an Fe-centered tetraamido macrocyclic ligand (Fe-TAML) that can efficiently catalyze water oxidation in the presence of ceric ammonium nitrate (Ce(IV)) with a turnover frequency (TOF) that exceeds 1.3 s−1.14 However, the catalyst’s activity vanishes after a few seconds (turnover number (TON) = 16).14
In 2011, Fillol, Costas and co-workers reported that Fe complexes with neutral tetradentate nitrogen-donor ligands could catalyse water oxidation with high efficiency for a period of hours. TONs >350 and >1000 were obtained using Ce(IV) at pH = 1 and sodium periodate at pH = 2, respectively (Scheme S1†).13
Complexes of Ni, Co, and Mn with these ligands do not catalyze water oxidation.13 Gas chromatography (GC) and mass spectrometry (MS) studies have shown that the reaction catalyzed by the Fe complexes is a real water oxidation process with regard to the statistical distribution of the oxygen produced. It was also reported that the Fe(III) oxohydroxo 2μ-(O,OH) diferric dimer shows a lower reaction rate and a different kinetic behavior.13 This result indicates that the oxohydroxo dimeric compounds do not act as active intermediates for the studied Fe(II) complexes. On the other hand, the complexes with one or two available trans coordination sites are inactive. In other words, the presence of the cis labile sites is the key structural feature for application in water oxidation catalysis. Based on their experiments, Fillol, Costas and co-workers proposed a mechanism for the corresponding reaction (Scheme S2†).13 The complexes do not cause any oxygen evolution in the presence of the oxidant [Ru(bpy)3](ClO4)3 (bpy: bipyridine).16 The same group proposed a similar mechanism with an Fe(IV)(μ-OH)–Ce(IV) intermediate.12
Tai-Chu Lau and co-workers investigated the catalytic activity of a small number of Fe complexes, [Fe(bpy)2Cl2]Cl, [Fe(tpy)2]Cl2, cis-[Fe(cyclen)Cl2]Cl and trans-Fe(tmc)Br2 (where tpy: 2,2′:6′,2′′-terpyridine, cyclen: 1,4,7,10-tetraazacyclodecane and tmc: 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) towards water oxidation at pH = 7–9. The study was carried out by chemical methods, using [Ru(bpy)3](ClO4)3 (bpy = bipyridine) as an oxidant.16a It was found that the simple Fe(III) salts, such as Fe(ClO4)3, are even more active than the other Fe complexes under these conditions. The group showed that in the presence of [Ru(bpy)3](ClO4)3, Fe complexes are decomposed, under oxidizing conditions, to metal oxides, which in turn form the actual catalysts.16a The particles formed after photocatalytic water oxidation were isolated and identified as Fe2O3 using various techniques. The results reported for these two systems in the presence of Ce(IV) and [Ru(bpy)3](ClO4)3 suggest that the active Fe catalysts are different for water oxidation at low pH values compared to high pH values.16a At low pH values, water oxidation by Fe complexes bearing multi-dentate N-donor ligands appears to go via a molecular oxo-Fe active intermediate, with no evidence for the formation of Fe oxide nanoparticles. As discussed by Fillol and Costas, the reason for this is most probably that under acidic conditions the Fe(III) ions do not oxidize water and also do not convert to Fe oxide. Another intermediate species present is FeO42−. FeO42− ions are strong oxidants (E0 = FeO42−/Fe(III) = 2.2 V) that can oxidize water in acidic conditions.22,23 However, Tai-Chu Lau and co-workers suggested that Ce(IV) is not capable of oxidizing Fe(III) to FeO42−.16a On the other hand, at a higher pH, Fe2O3 is the actual catalyst for water oxidation.16 Herein, we answer two important questions regarding the Fe complexes reported by Fillol, Costas and their co-workers:
Fillol and Costas showed13 that Fe(III) 2μ-(O,OH) diferric dimer complexes do not catalyze water oxidation. A good question is therefore whether an Fe(III) complex with only one bridge (oxo or hydroxo) can act as a water-oxidizing catalyst?
The second important question is: could the FeO42− ions be the actual catalyst for water oxidation in these reactions?
To answer these questions, we selected the tris(2-pyridylmethyl)amine (tpa) ligand, which is commercially available. We synthesized an Fe(III) oxo diferric dimer with only one μ-O bridge using this ligand, [tpa(H2O)FeOFe(H2O)tpa](ClO4)4 (1), using a modification of the previously reported method.24
1 is isomorphous with the reported crystal structure of (μ-oxo)bis{aquatris(2-pyridylmethyl)amine}iron(III) perchlorate24 (Fig. 1) (see the ESI† for further crystallographic details; Table S1). The only difference in composition is in the solvent molecules that are present: contrary to the previous report, compound 1 does not contain ethanol but does contain an increased number of solvating water molecules. Also, in the case of 1, it was necessary to model the disorder of the perchlorate anions. Moreover, the structure determination reported here was carried out at 100 K. Our re-determination essentially helped to reproduce all the previously reported structural features of the [{Fe(tpa)(H2O)}2O]4+ cation, including the unsymmetric FeOFe bridge (Table S2 †). It was also possible to determine, in part, the water H atom positions and thus the hydrogen bonding scheme (Table S3†). It is interesting to note that when we investigated water oxidation with the complex in the presence of Ce(IV), the maximum TOF was 6 times higher than that for the monomer reported by Fillol, Costas and their co-workers (Fig. 2). If the monomer is the actual catalyst for water oxidation, we expect that the dimer should show lower water oxidizing activity than the monomer.
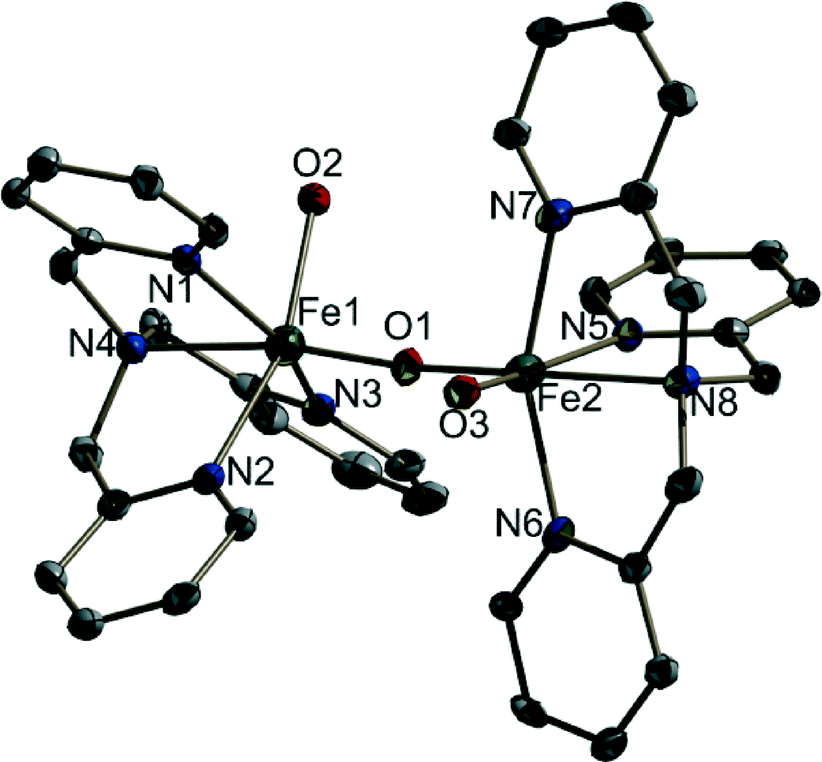 | ||
| Fig. 1 The structure of one of the symmetry-independent binuclear cations in 1. The thermal ellipsoids are plotted at a 30% probability level. H atoms are omitted for clarity. | ||
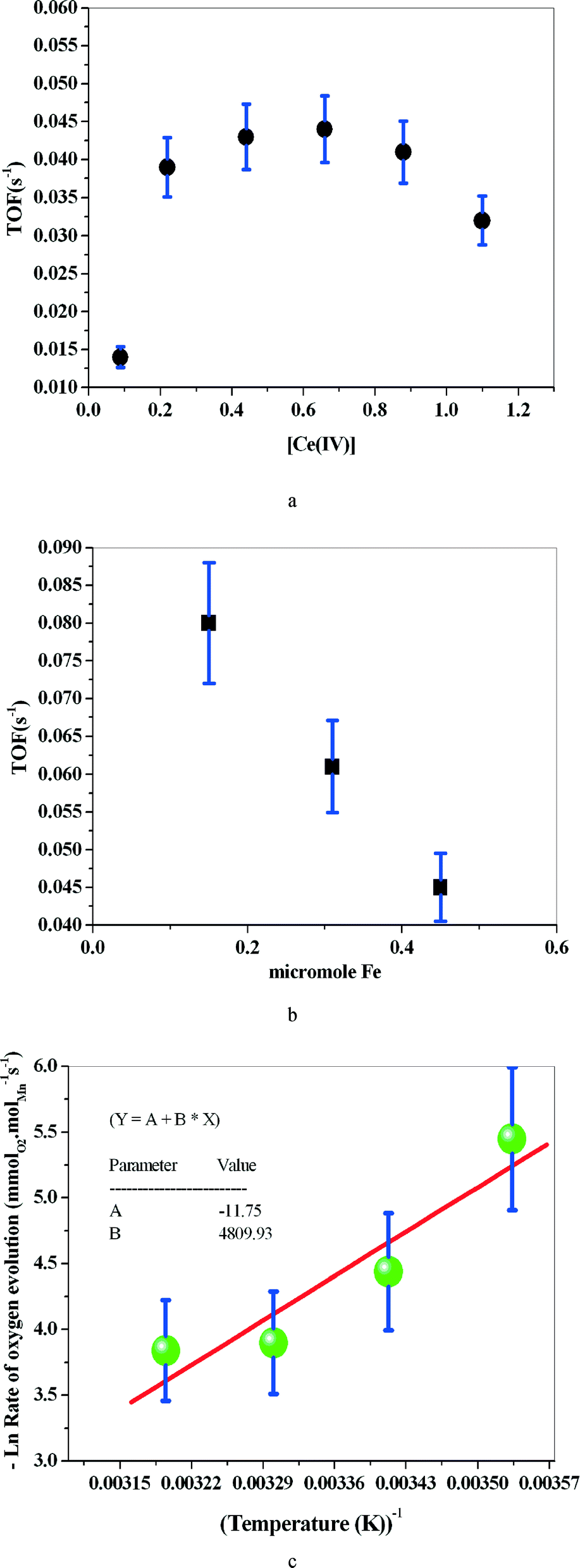 | ||
| Fig. 2 TOF for water oxidation by 1 at different concentrations of Ce(IV) ([1] = 8.23 μM) (a). TOF for water oxidation with different Fe quantities of the catalyzing complex, in the presence of Ce(IV) (0.33 M) (b). Water oxidation by 1 ([1] = 8.23 μM) in the presence of Ce(IV) (67 mM) at different temperatures (c). | ||
The activation energy (Ea) computed based on the Arrhenius equation is 40.0 kJ mol−1. However, the complex supported on montmorillonite shows no water oxidizing activity (ESI†).
The mechanism of water oxidation by complex 1 is not known, but it was reported that a very similar dimer shows intramolecular attack by a hydroxide ion coordinated to one Fe center onto the carbon atom of the acetonitrile coordinated to the adjacent Fe atom (Scheme 1a).25 A similar mechanism is proposed for water oxidation catalyzed by this dimer (Scheme 1b).
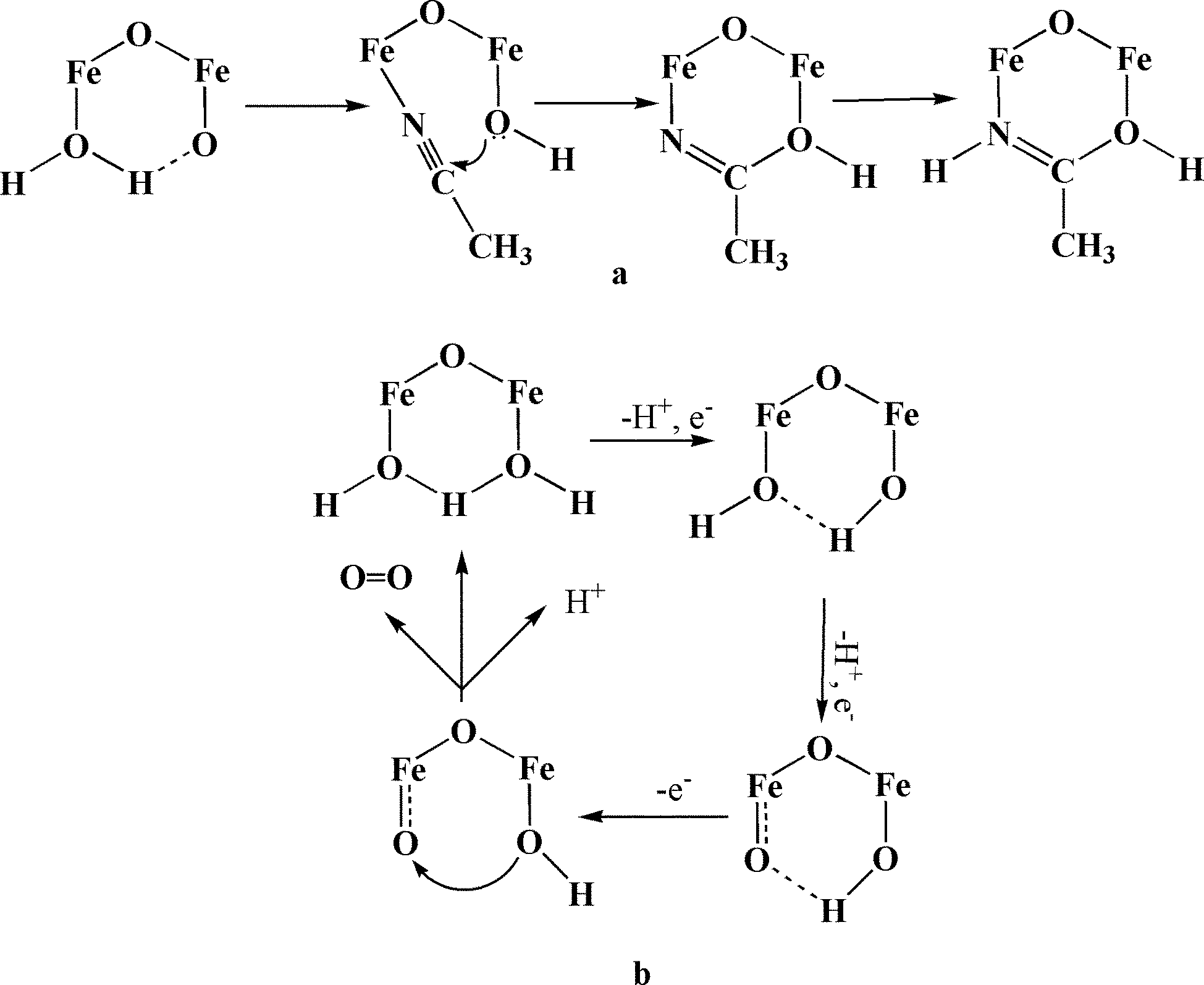 | ||
| Scheme 1 The proposed mechanisms for oxidation of the coordinated acetonitrile (a) and for water oxidation (b), catalyzed by 1. | ||
The rate of oxygen evolution is of first order for both Ce(IV) and 1 under our experimental conditions.
It is important to note that turnover numbers for the iron complexes are not high due to the ligand oxidation that occurs under the harsh conditions of water oxidation. On the other hand, the FeO42− ions were reported to be efficient water oxidizing catalysts under acidic conditions.23 To assess the role of the FeO42− ions in these water oxidation reactions, we added a solution of these ions to the solution containing Ce(IV). A very fast rate of oxygen evolution was observed, but the turnover number (TON) was less than one (mol O2/mol Fe) (Fig. 3). Studies of the UV–Vis spectra also showed immediate loss of these ions under these conditions. The results show that:
• A high-valent Fe compound can evolve oxygen very quickly.
• If Fe complexes decompose in the presence of Ce(IV) and the Fe ions convert to the FeO42− ions, the FeO42− ions cannot oxidize water catalytically but act non-catalytically with a low TON (less than 1).
It is important to point out that although the Ce(IV) compound has no effect on the water oxidation reaction induced by the FeO42− ions, under acidic conditions (pH ~ 1) its presence is needed. This is confirmed by the observation that a solution containing FeO42− ions shows similar oxygen evolution in the presence of HNO3 (pH ~ 1). On the other hand, less rapid oxygen evolution is observed in water (pH ~ 7).
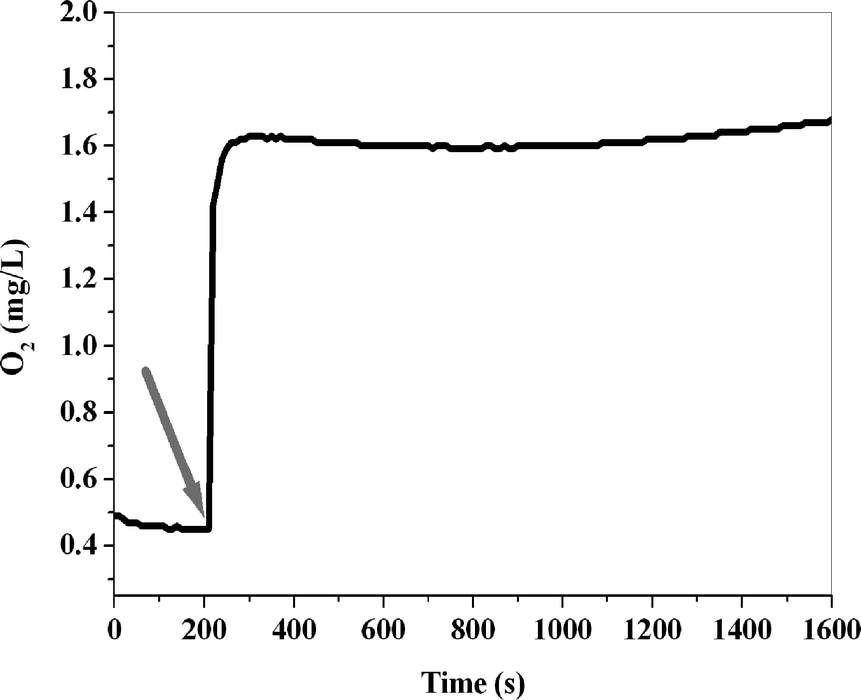 | ||
| Fig. 3 Oxygen evolution in the reaction of K2FeO4 (1 mL, 2.0 mM) with Ce(IV) (40 mL, 0.11 M) at 25 °C. The arrow indicates the time at which K2FeO4 was added to the Ce(IV) solution. | ||
In conclusion, we found that 1, with only one oxo or hydroxo bridge is a more efficient water oxidizing catalyst than the corresponding monomeric Fe(III) complex in the presence of Ce(IV). In other words, the monomer is a precursor to the active catalyst which might be a di- or even a multi-nuclear iron compound. On the other hand, we showed that the FeO42− ion does not play the role of an intermediate in this reaction.
Acknowledgements
The authors are grateful to the Institute for Advanced Studies in Basic Sciences and the National Elite Foundation for financial support. M. H. is grateful to Prof. Dr. Stefanie Dehnen for her generous support and helpful discussions.Notes and references
- J. Barber, Chem. Soc. Rev., 2009, 38, 185 RSC.
- N. S. Lewis, Science, 2007, 315, 798 CrossRef CAS PubMed.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141 CrossRef CAS.
- J. K. Hurst, Science, 2010, 328, 315 CrossRef CAS PubMed.
- M. M. Najafpour, F. Rahimi, E. Aro, C. Lee and S. I. Allakhverdiev, J. R. Soc. Interface, 2012, 9, 2383 CrossRef CAS PubMed; M. Hirahara, H. Yamazaki, S. Yamada, K. Matsubara, K. Saito, T. Yuia and M. Yagi, Catal. Sci. Technol., 2013, 3, 1776 Search PubMed.
- M. M. Najafpour and S. I. Allakhverdiev, Int. J. Hydrogen Energy, 2012, 37, 8753 CrossRef CAS PubMed.
- M. M. Najafpour, B. Pashaei and S. Nayeri, Dalton Trans., 2012, 41, 7134 RSC; M. M. Najafpour and A. Nemati Moghaddam, Dalton Trans., 2012, 41, 10292 RSC; M. M. Najafpour, M. Amouzadeh Tabrizi, B. Haghighi and Govindjee, Dalton Trans., 2012, 41, 3906 RSC; M. M. Najafpour, B. Pashaei and S. Nayeri, Dalton Trans., 2012, 41, 4799 RSC; M. M. Najafpour, Geomicrobiol. J., 2011, 28, 714 CrossRef CAS.
- W. Ruttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1 CrossRef PubMed.
- M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21 CrossRef CAS PubMed.
- H. J. M. Hou, Materials, 2011, 4, 1693 CrossRef CAS.
- M. M. Najafpour, B. Haghighi, M. Zarei Ghobadi and D. Jafarian Sedigh, Chem. Commun., 2013, 49, 8824 RSC.
- Z. Codolà, I. Garcia-Bosch, F. Acuña-Parés, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem.–Eur. J., 2013, 19, 8042 CrossRef PubMed.
- J. Lloret-Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807 CrossRef PubMed.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990 CrossRef CAS PubMed.
- D. Hong, S. Mandal, Y. Yamada, Y. Lee, W. Nam, A. Llobet and S. Fukuzumi, Inorg. Chem., 2013, 52, 9522 CrossRef CAS PubMed.
- (a) G. Chen, L. Chen, S. M. Ng, W. L. Man and T. C. Lau, Angew. Chem., Int. Ed., 2013, 52, 1789 CrossRef CAS PubMed; (b) M. T. Mayer, C. Du and D. Wang, J. Am. Chem. Soc., 2012, 134, 12406 CrossRef CAS PubMed; (c) Y. Lin, S. Zhou, S. W. Sheehan and D. Wang, J. Am. Chem. Soc., 2011, 133, 2398 CrossRef CAS PubMed; (d) K. M. H. Young, B. M. Klahr, O. Zandia and T. W. Hamann, Catal. Sci. Technol., 2013, 3, 1660 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- D. Shevchenko, M. F. Anderlund, A. Thapper and S. Styring, Energy Environ. Sci., 2011, 4, 1284 CAS.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841 CrossRef CAS PubMed.
- Z. Chen and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 700 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498 CrossRef CAS PubMed.
- H. Goff and R. K. Murmann, J. Am. Chem. Soc., 1971, 93, 6058 CrossRef CAS.
- R. Sarma, A. M. Angeles-Boza, D. W. Brinkley and J. P. Roth, J. Am. Chem. Soc., 2012, 134, 15371 CrossRef CAS PubMed.
- B. R. Whittlesey, Z. Pang and R. A. Holwerda, Inorg. Chim. Acta, 1999, 284, 124 CrossRef CAS.
- A. Hazell, K. B. Jensen, C. J. McKenzie and H. Toftlund, Inorg. Chem., 1994, 33, 3127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 955723. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cy00644a |
| This journal is © The Royal Society of Chemistry 2014 |