Heterolytic cleavage of Si–H bonds: reduction of imines using silane/high-valent oxo-molybdenum MoO2Cl2 as a catalyst†
Yiou
Wang
,
Piao
Gu
,
Wenmin
Wang
and
Haiyan
Wei
*
Jiangsu Key Laboratory of Biofunctional Materials, School of Chemistry and Materials Science, Jiangsu Provincial Key Laboratory for NSLSCS, Nanjing Normal University, Nanjing 210097, China. E-mail: weihaiyan@njnu.edu.cn
First published on 31st October 2013
Abstract
This work describes a DFT investigation into reducing imines using a high-valent MoO2Cl2/silane system, proposing that a recently introduced mechanistic model, the heterolytic Si–H bond cleavage upon imine substrates attacking the η1-silane molybdenum adduct, accounts for this catalytic reactivity, rather than the [2 + 2] addition mechanism.
The role of high-valent oxo-molybdenum and oxo-rhenium complexes, traditionally employed as excellent catalysts for oxidation reactions, has been totally reversed in recent studies.1 A series of reductions of a variety of functional groups (aldehyde/ketones, imines, esters, sulfoxides and pyridine-N-oxides, nitriles, amides, alkynes, etc.) promoted by these oxo-complexes in high oxidation states (MoO2Cl2, CpMoO2Cl, ReO2I(PPh3)2, Re(O)Cl3(PPh3)2, ReCH3O3, Re2O7, HReO4) has been reported, which opens an entirely new area of catalysis for these complexes.2,3
For the di-oxo molybdenum(VI)/rhenium(V) catalyst, MoO2Cl2 and Re(O)2(I)(PPh3)2 mediate the hydrosilylation of aldehyde and ketones; the most commonly accepted mechanism describing this process assumes that the reaction starts from the Si–H bond adding across one of the two Mo/Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds to afford a siloxy molybdenum/rhenium hydride, which is then followed by coordination of carbonyl compounds to the molybdenum center. This process allows migratory insertion into the Mo/Re–H bond to generate the alkoxide intermediate. This unconventional [2 + 2] addition mechanism has been verified experimentally,2,3 as well as theoretically using DFT calculations.4,5 In this [2 + 2] addition mechanism, as shown in Fig. 1, the optimized structure of TS[2 + 2] (for a MoO2Cl2 catalyst (dioxomolybdenum dichloride)) has a four-membered cyclic (O⋯Si⋯H⋯Mo) ring, representing the silane hydrogen abstracted by the molybdenum center and silyl to form a bond with multiple oxo ligands.
O bonds to afford a siloxy molybdenum/rhenium hydride, which is then followed by coordination of carbonyl compounds to the molybdenum center. This process allows migratory insertion into the Mo/Re–H bond to generate the alkoxide intermediate. This unconventional [2 + 2] addition mechanism has been verified experimentally,2,3 as well as theoretically using DFT calculations.4,5 In this [2 + 2] addition mechanism, as shown in Fig. 1, the optimized structure of TS[2 + 2] (for a MoO2Cl2 catalyst (dioxomolybdenum dichloride)) has a four-membered cyclic (O⋯Si⋯H⋯Mo) ring, representing the silane hydrogen abstracted by the molybdenum center and silyl to form a bond with multiple oxo ligands.
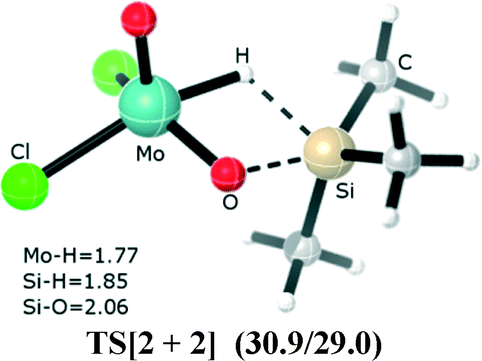 | ||
| Fig. 1 The optimized geometry of the [2 + 2] addition transition state. The calculated free energy barrier of TS[2 + 2] is 30.9 kcal mol−1 (29.0 kcal mol−1 with the effect of the solvent included) higher than the reactants (1 (MoO2Cl2) + trimethylsilane). Bond lengths, Å. | ||
The di-oxo molybdenum(VI) complex MoO2Cl2 is likewise confirmed to be an effective catalyst for hydrosilylation of imines (shown in Scheme 1). This has provided an attractive alternative approach to hydrogenation of imines due to its experimental simplicity.6 However, our DFT computational study indicates that the [2 + 2] addition mechanism is not the favorable pathway for MoO2Cl2 mediating the hydrosilylation of imines to give an N-silylamine. Instead, we propose that the active promoting species is the heterolytic Si–H bond cleavage based on the organic substrate–imine nucleophilically attacking the η1-silane molybdenum complex. As a result, the multiple oxo ligands do not participate in activating the Si–H bond process. This heterolytic cleavage of the Si–H bond is analogous to the ionic mechanism proposed for the hydrosilylation of carbonyl compounds by tris(pentafluorophenyl)borane systems7 and a cationic iridium catalyst.8
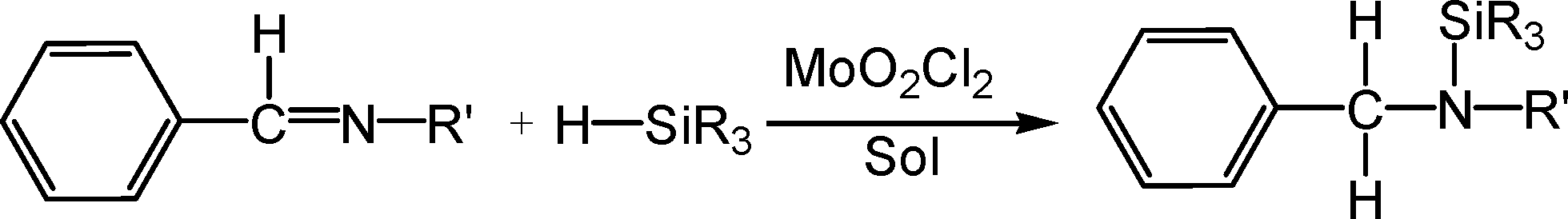 | ||
| Scheme 1 | ||
All DFT calculations were carried out using the Gaussian 09 program package.9 The geometry optimizations of all the minima and transition states were performed at the M06 level of theory10 with the 6-311++G(d,p) basis set used for the C, H, N, Cl, O, and Si atoms.11 The effective core potentials (ECPs) of Hay and Wadt with double-ζ valence basis sets (LanL2DZ) with one set of augmented f-polarization functions were used for modeling the molybdenum.12,13 The vibrational frequencies were computed to determine whether each optimized structure was an energy minimum or a transition state, and IRC calculations were carried out to confirm that each transition state was connected to its corresponding reactant and product.14 Solvent effects were computed with the SMD model using gas phase optimized structures.15 The values for ΔGsol and ΔGgas reported in this paper are relative Gibbs free energies calculated at 298 K in solution and in the gas phase, respectively, by carrying out a single-point calculation using the larger basis set of the quintuple zeta set with extra polarization basis sets (cc-QVZP) for the molybdenum16 and 6-311++G(2d,p) for the C, H, N, Cl, O, and Si atoms.
The first step of the ionic hydrosilylation pathway involves the coordination of silane (here trimethylsilane) to the molybdenum center in the MoO2Cl2 catalyst. The optimized structure of complex 3 displays a significantly long Mo⋯H distance (2.45 Å) and a normal Si–H bond length (1.50 Å), indicating that silane is weakly η1 coordinated to the molybdenum center. Complex 3 is 4.0 kcal mol−1 above the separation of MoO2Cl2 and a free silane.
The next step in the process is an imine molecule (here CH3NCCH3Ph, N-methyl-1-phenylethanimine, referred to as im) carrying out a nucleophilic attack on the silicon center in 3 in a “backside” fashion, which represents the imine anti attacking the silane at the face opposite to the metal center. Initially, a loosely bound adduct 3+im forms, in which the geometries of the η1-silane molybdenum adduct and imine are barely perturbed from their isolated structures. The formation of this adduct is endergonic by 14.3 kcal mol−1 compared to the reactants (1 + im + trimethylsilane).
From adduct 3+im, taking either H⋯Si or Si⋯N (im) separation as the reaction coordinate, we can identify an approximate consecutive process representing the heterolytic cleavage of the Si–H bond (the optimized structures are shown in Fig. 2). The first transition state TS4anti corresponds to the approach of the nitrogen atom (im) to the silicon center, which is accompanied by an elongation of the Si–H bond. TS4anti exhibits an early geometry state in which the Si–H separation lengthens by 0.13 Å, the forming H–Mo distance is 1.93 Å, and the Si⋯N separation is 2.54 Å. The calculated vibrational motion of the imaginary frequency associated with TS4anti corresponds to the attack of the nucleophilic imine group on the silicon center.
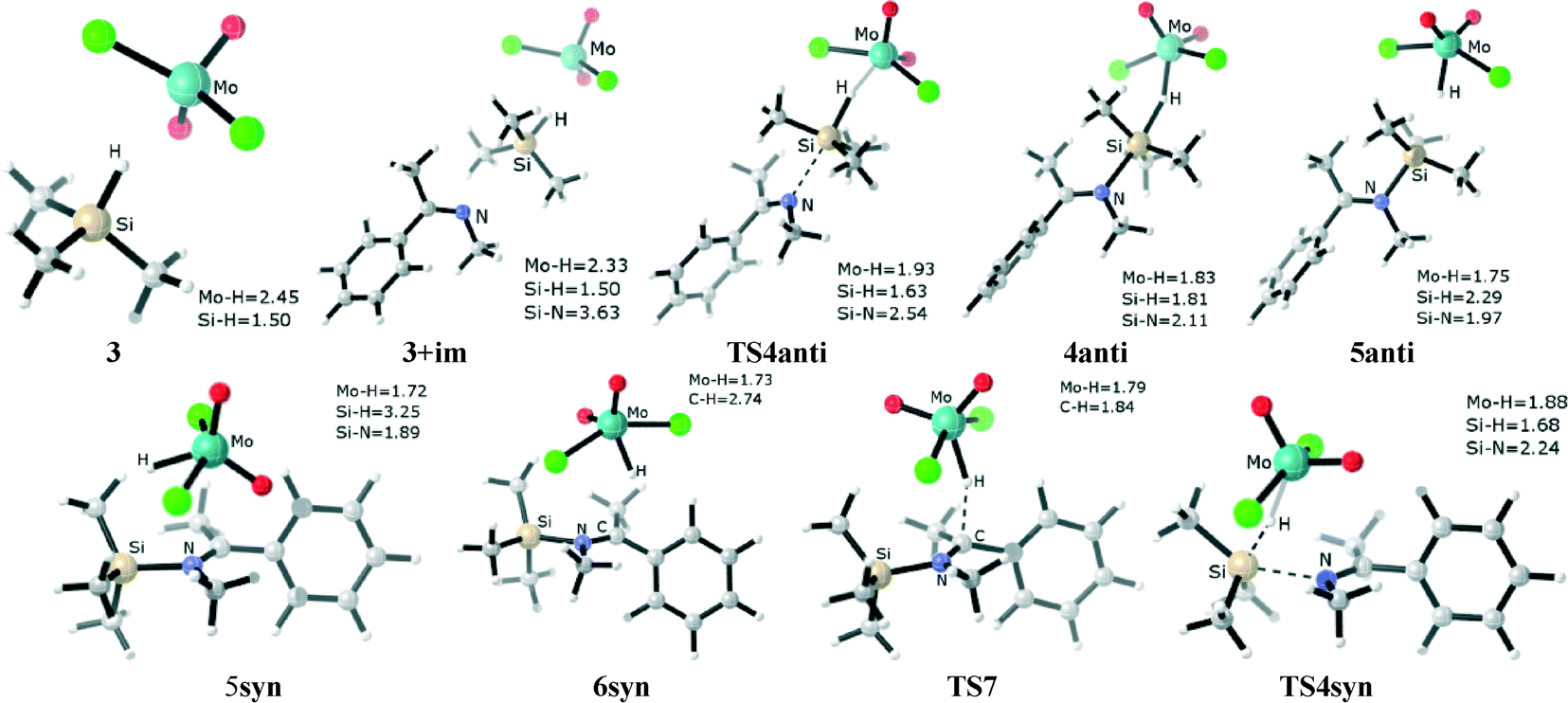 | ||
| Fig. 2 The optimized geometry of the η1-silane molybdenum complex 3, adduct 3+im, and the transition states (TS4anti, TS7, TS4syn) and intermediates (4anti, 5anti, 5syn, 6syn) along the ionic hydrosilylation pathway. Bond lengths, Å. | ||
The transition state TS4anti leads to a shallow intermediate, 4anti, calculated at 0.1 kcal mol−1 (gas-phase) below TS4anti. The optimized structure of 4anti exhibits a SN2-Si reaction mode. The coordination geometry around the silicon center is a penta-coordinated trigonal bipyramid, with the silyl group (SiMe3) acting as the plane and the departing hydrogen atom and the incoming N(im) atom occupying the apical positions (d(Mo–H) = 1.83 Å, d(Si–H) = 1.81 Å and d(Si–N) = 2.11 Å, respectively). The four atoms of Mo⋯H⋯Si⋯N are roughly in a straight line, and the Mo–H–Si and H–Si–N angles are 158.8° and 169.8°, respectively.
Proceed from 4anti, our calculations found that the cleavage of the Si–H bond easily provides the intermediate 5anti (the corresponding transition state fails to be located due to the flat potential surface). In the formed intermediate 5anti, the Si–H bond distance elongates to 2.29 Å, which indicates that the silane hydrogen is transferred to the molybdenum center while the silyl ion (Me3Si+) couples to the imine. Thus, the two moieties of [Mo(O)2(Cl)2H]−[Me3SiNCH3CCH3Ph]+ are largely separated. Our analysis shows that the intermediate 5anti is 3.3 kcal mol−1 higher (gas-phase) than 4anti.
The N(im) atom approaches the silicon center along the 3+im → TS4anti → 4anti → 5anti process, with d(N⋯Si) decreasing by 3.63 Å → 2.54 Å → 2.11 Å → 1.97 Å, which prompts the stretching of the Si–H bond by 1.50 Å → 1.63 Å → 1.81 Å → 2.29 Å, respectively. At the same time, the silane hydrogen progressively binds to the molybdenum center as the Mo⋯H distance decreases by 2.33 Å → 1.93 Å → 1.83 Å → 1.75 Å. Correspondingly, the electron density from the silane hydrogen progressively drains toward the molybdenum center. The calculated natural bond orbital (NBO) negative charge on the silane hydrogen decreases by 0.28e → 0.24e → 0.21e → 0.13e with the transitions. However, the NBO charge on the molybdenum center becomes less positively charged, i.e. 0.98e → 0.93e → 0.89e → 0.82e with the transitions, while the NBO charge on the silicon center increases along 1.55e → 1.72e → 1.75e → 1.88e during the path. Accordingly, the CN bond of imine becomes more polar, with the NBO negative charge on the nitrogen atom of CN increasing along 0.50e → 0.52e → 0.57e → 0.62e, and the NBO charge on the carbon atom of CN increasing along 0.32e → 0.36e → 0.42e → 0.45e.
To complete the catalytic cycle, another form of the ion pair 5syn is generated by repositioning the two moieties of the molybdenum hydride anion [Mo(O)2(Cl)2H]− and the silylated imine ion [Me3SiNCH3CCH3Ph]+. In the structure of ion pair 5syn, the imine ion resides at the same face as the molybdenum center, representing the imine “frontside” attacking at the silane center. From 5syn, the silylated imine ion abstracts the hydride attached at the molybdenum center to produce N-silylamine and regenerate the MoO2Cl2 catalyst.
The calculated free-energy profiles of the above ionic hydrosilylation pathway for reducing imines using a high-valent MoO2Cl2/silane system are shown in Scheme 2 (black line). In summary, the pathway can be divided into three steps: (1) the addition of silane to the metal center, 1 → 3; (2) the heterolytic cleavage of the Si–H bond through the nucleophilic attack of imine, TS4anti → 4anti → 5anti; (3) the hydride transfer, 5syn → 6syn → TS7 → 1. Scheme 2 shows that the Si–H cleavage step is endergonic, with an activation free energy barrier of 25.4 (21.4 in sol-phase) kcal mol−1, and thus is not favored. However, note that the solvent greatly stabilizes the ion pair.17 Once the intermediate 5anti ion pair is generated, the pair can readily undergo isomerization (5syn with the hydride pointing to the Si atom and 6syn with the hydride pointing to the carbon atom (CN) are 4.5 and 4.2 kcal mol−1 more stable than 5anti, respectively), as well as undergo hydride transfer reaction from the molybdenum center to the silylated imine viaTS7 (a small barrier of 5.0 kcal mol−1, TS7 above 5syn). Furthermore, the driving force for the hydride transferring reaction is large, ΔG(5syn → 1+N-silylamine) = 10.2 kcal mol−1 (sol-phase), which likely originates from the strong electrostatic attraction between the hydride on the molybdenum center and the silylcarbonium carbon atom.
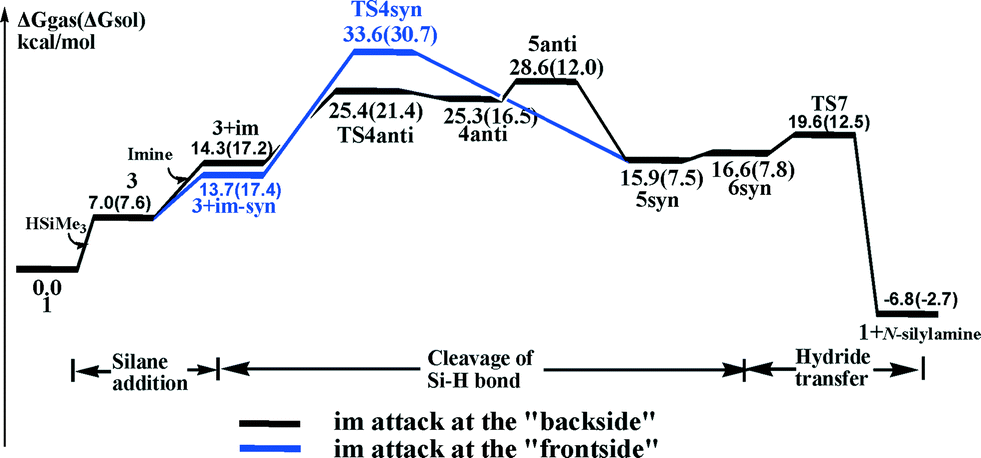 | ||
| Scheme 2 The energy profile for MoO2Cl2 catalyzing the hydrosilylation process through the ionic pathway. The solvation energies are included in parentheses, and the gas-phase energies are the values without parentheses. | ||
Alternatively, it is worth noting that a “frontside” attack of the imine molecule at the silicon center in 3 can directly generate the 5syn ion pair. A concerted transition state (TS4syn, Fig. 2, and Scheme 2, blue line) for such an arrangement is located. The calculated vibrational motion of the imaginary frequency associated with TS4syn represents an attack of the nucleophilic imine group on the silicon center, with the simultaneous cleavage of the Si–H bond and the formation of the Mo–H bond, indicated by bond distances of 2.24 Å (d(Si–N)), 1.68 Å (d(Si–H)), and 1.88 Å (d(Mo–H)), respectively. However, the calculated free energy barrier is high at 33.6 kcal mol−1 (30.7 kcal mol−1 in sol-phase), which makes an imine syn attacking unfavorable by ca. 8.0 kcal mol−1 compared to the imine anti attacking the silyl group.
In summary, our theoretical analysis shows that the imine substrate carrying out a nucleophilic attack on the η1-silane molybdenum adduct, which prompts the heterolytic cleavage of the Si–H bond, is the most favorable pathway for the MoO2Cl2/silane system mediating the hydrosilylation of imines. The activation free energy of the ionic process is at 21.4 kcal mol−1, which is favored by 7.6 kcal mol−1 (TS4anti above TS[2 + 2]) compared to the [2 + 2] addition mechanism.18
There are two clear differences between the proposed ionic mechanism and the [2 + 2] addition mechanism, which center on whether multiple oxo ligands participate in the reaction and whether the reduced species is initially coordinated to the metal. For the reaction proceeding via the ionic pathway, the coordination of the organic substrate to the metal center and subsequent insertion into the TM–H bond are not needed. This new understanding of the process involved will help chemists to design new catalysts and reactions.
Acknowledgements
We acknowledge the support of the National Natural Science Foundation of China (no. 21103093), the Jiangsu Province Science and Technology Natural Science Project (no. BK2011780), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials. We also thank the Shanghai Supercomputer Center and the Nanjing University HPCC for their technical support.References
- R. H. Holm, Chem. Rev., 1987, 87, 1401 CrossRef CAS.
- (a) W. R. Thiel, Angew. Chem., Int. Ed., 2003, 42, 5390 CrossRef CAS PubMed; (b) B. Royo and C. C. Romão, J. Mol. Catal. A: Chem., 2005, 236, 107 CrossRef CAS PubMed; (c) A. C. Fernandes, R. Fernandes, C. C. Romão and B. Royo, Chem. Commun., 2005, 213 RSC; (d) A. C. Fernandes and C. C. Romão, J. Mol. Catal. A: Chem., 2006, 253, 96 CrossRef CAS PubMed; (e) P. M. Reis, C. C. Romão and B. Royo, Dalton Trans., 2006, 1842 RSC.
- (a) B. D. Sherry, A. T. Radosevich and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 6076 CrossRef CAS PubMed; (b) J. J. Kennedy-Smith, K. A. Nolin, H. P. Gunterman and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 4056 CrossRef CAS PubMed; (c) K. A. Nolin, R. W. Ahn and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 12462 CrossRef CAS PubMed; (d) K. A. Nolin, R. W. Ahn and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 17168 CrossRef PubMed; (e) A. T. Radosevich, C. Musich and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 1090 CrossRef CAS PubMed; (f) K. A. Nolin, J. R. Krumper, M. D. Pluth, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 14684 CrossRef CAS PubMed.
- (a) P. J. Costa, C. C. Romão, A. C. Fernandes, B. Roya, P. M. Reis and M. J. Calhorda, Chem.–Eur. J., 2007, 13, 3934 CrossRef CAS PubMed; (b) D. Markus and T. Strassner, Inorg. Chem., 2007, 46, 10850 CrossRef PubMed.
- L. W. Chung, H. G. Lee and Y. W. Wu, J. Org. Chem., 2006, 71, 6000 CrossRef CAS PubMed.
- (a) A. C. Fernandes and C. C. Romão, Tetrahedron Lett., 2005, 46, 8881 CrossRef CAS PubMed; (b) S. C. A. Sousa, I. Cabrita and A. C. Fernandes, Chem. Soc. Rev., 2012, 41, 5641 RSC.
- (a) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440 CrossRef CAS; (b) D. J. Parks, W. E. Piers, M. Parvez, R. Atencio and M. J. Zaworotko, Organometallics, 1998, 17, 1369 CrossRef CAS; (c) D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090 CrossRef CAS PubMed; (d) F. Bertini, V. Lyaskovskyy, B. J. J. Timmer, M. Lutz, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, J. Am. Chem. Soc., 2012, 134, 201 CrossRef CAS PubMed.
- (a) V. K. Dioumaev and R. M. Bullock, Nature, 2003, 424, 530 CrossRef CAS PubMed; (b) D. V. Gutsulyak, S. F. Vyboishchikov and G. I. Nikonov, J. Am. Chem. Soc., 2010, 132, 5950 CrossRef CAS PubMed; (c) W. H. Bernskoetter, S. K. Hanson and M. Brookhart, J. Am. Chem. Soc., 2009, 131, 8603 CrossRef CAS PubMed; (d) S. Park and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 640 CrossRef CAS PubMed; (e) J. Yang and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 12656 CrossRef CAS PubMed; (f) S. Park, D. Bezier and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11404 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) Z. Yan and D. G. Truhlar, J. Phys. Chem. A, 2008, 112, 6794 CrossRef PubMed; (b) J. Denis, A. Eric, A. Carlo, V. Rosendo, Z. Yan and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2071 CrossRef; (c) O. N. Faza, R. Á. Rodríguez and C. S. López, Theor. Chem. Acc., 2011, 128, 647 CrossRef CAS PubMed.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS; (b) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS; (c) A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Hoöllwarth, V. Jonas, K. F. Koöhler, R. Stegmenn and G. Frenking, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS; (d) A. Hoöllwarth, M. Boöhme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Koöhler, R. Stegmenn, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237 CrossRef.
- For Mo, the triple-ζ SDD basis set with Stuttgart–Dresden ECP was also employed, the results show that the dependence on the basis set is insignificant, shown in the ESI.†.
- (a) K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS; (c) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- (a) SDM computes that solvation, even the weakly polar CH2Cl2, provides tremendous stabilization for the ion pair of 4anti, 5anti, 5syn and 6syn. Therefore, the transformation (shown in the ESI†) between those ion pairs occurs easily in the sol-phase; (b) The ion pair 5anti is reoptimized in the solvent environment, and the results give a similar Gibbs free energy of 8.3 kcal mol−1.
- (a) The transition state TS4anti is favored by 18.8 (16.2 sol-phase) kcal mol−1 than the [2 + 2] addition mechanism calculated at the MP2 level of theory, shown in the ESI.†; (b) The entropic effects in the gas phase tend to overestimate and underestimate the energetic costs of multimolecular associative and dissociative processes, respectively. Therefore, we have compared the energies for the activation barrier (ΔE‡, only consider the electric energies) for the ionic transition state of TS4anti and the [2 + 2] addition transition state TS[2 + 2]; our results have shown that the activation barrier (ΔE‡) for TS[2 + 2] is 18.2 kcal mol−1, while it is significantly higher than the ionic transition state of TS4anti (−3.0 kcal mol−1) and 5anti (0.2 kcal mol−1).
Footnote |
| † Electronic supplementary information (ESI) available: Full citation for ref. 9 and Cartesian coordinates of all optimized structures discussed in the paper. See DOI: 10.1039/c3cy00727h |
| This journal is © The Royal Society of Chemistry 2014 |