Preparation of Ru nanoparticles on TiO2 using selective deposition method and their application to selective CO methanation†
S.
Tada
ab and
R.
Kikuchi
*a
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: rkikuchi@chemsys.t.u-tokyo.ac.jp
bJapan Society for the Promotion of Science, 5-3-1 Kojimachi, Chiyoda-ku, Tokyo 102-0083, Japan
First published on 18th October 2013
Abstract
Selective CO methanation was investigated over Ru/TiO2 prepared using a selective deposition method with NaOH and NH3 aqueous solution as a pH adjuster. Control of pH by NH3 solution resulted in the small particle size of Ru (average 1.2 nm) and the formation of Na-free Ru/TiO2, leading to high CO methanation activity and a wide temperature window for selective CO methanation at low temperatures.
Polymer electrolyte fuel cells (PEFCs) have been developed as a promising power generator to convert chemical energy directly into electrochemical energy with high efficiency and low emission of pollution. In Japan, PEFC systems by the name of ENE-FARM have been commercially available since 2009, and 50
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units will have been installed by 2013.1 One of the problems is that the by-product CO in the reformates is required to be reduced preferably to less than 100 ppm, otherwise a drastic deactivation arises from CO poisoning of the anode.2 The water gas shift reaction (WGS reaction, CO + H2O → CO2 + H2)3 and the following preferential oxidation of CO (PROX, CO + 1/2O2 → CO2)4 get rid of a large amount of CO in the reformates from 10% to the CO-tolerant level of the PEFC anode. The PROX systems require addition of air to hydrogen-rich gas, leading to hydrogen consumption and efficient degradation of the PEFC systems.
000 units will have been installed by 2013.1 One of the problems is that the by-product CO in the reformates is required to be reduced preferably to less than 100 ppm, otherwise a drastic deactivation arises from CO poisoning of the anode.2 The water gas shift reaction (WGS reaction, CO + H2O → CO2 + H2)3 and the following preferential oxidation of CO (PROX, CO + 1/2O2 → CO2)4 get rid of a large amount of CO in the reformates from 10% to the CO-tolerant level of the PEFC anode. The PROX systems require addition of air to hydrogen-rich gas, leading to hydrogen consumption and efficient degradation of the PEFC systems.
Methanation of CO (CO + 3H2 → CH4 + H2O(g), ΔH0298 = −206 kJ) has become a promising candidate as an alternative technique to PROX. One of the merits of CO methanation is the reuse of produced CH4: this CH4 can be introduced into a burner for the reforming units. Another merit is its simple unit operation compared to PROX. On the other hand, as the demerit for CO methanation, a large amount of H2 is consumed via simultaneous CO2 methanation (CO2 + 4H2 → CH4 + 2H2O(g), ΔH0298 = −165 kJ) and the reverse water gas shift reaction (RWGS reaction, CO2 + H2 → CO + H2O(g), ΔH0298 = 41 kJ), leading to runaway of the methanation reactor. Suitable catalysts for selective CO methanation are, therefore, required to improve CO methanation at low temperatures and to suppress CO2 methanation and RWGS reaction at high temperatures.
A lot of studies have been conducted on selective CO methanation over Ru,5 Ni,6 and Ru–Ni7 catalysts. As for CO methanation, dissociation of CO on active metal promoted by adsorbed hydrogen and subsequent hydrogenation of surface carbonaceous species are the important steps,8 while for CO2 methanation CO2 was reduced to CO at the interfaces of active metals and supports and the CO was methanated.9 In our previous study, Ru/TiO2 catalysts prepared using a wet impregnation method exhibited high CO methanation activity and selectivity.9
A decrease in Ru particle size is considered to promote CO methanation due to an enlargement of active surface area for the reaction, resulting in avoidance of CO2 methanation at high temperatures and runaway of the reactor. Among various methods used to synthesize nanoparticles supported by metal oxides, the selective deposition method is one of the simplest procedures.10 In this process, the chemical composition and surface structure of the solid support are the dominant factors for the mean size, stability, and uniformity of the noble metal particles, and the following two points are important: (i) controlling pH of precursor solutions and stirring the solutions for 1 day to form a hydroxide complex (in this case Ru(OH)x) and (ii) aging the complex solutions to deposit nanoparticles on support materials. It has been reported that NaOH solution is used as a pH adjuster.10 As for the catalysts prepared using a selective deposition method, Na species are expected to reside on the catalysts, while after washing the catalyst powder with deionized water, Na species are anticipated to be eliminated. The contact of alkali metals with active metals such as Ru can enhance the electron density in the active metal, which means that CO adsorbed on the active metal is more easily dissociated by enhanced dπ–pπ* back bonding and consequently CO methanation is improved.11
In this study, Ru/TiO2 was prepared using a selective deposition method with NaOH aqueous solution as a pH adjuster, and then the relationship between Ru particle sizes on Ru/TiO2 catalysts and selective CO methanation activity was investigated. The influence of residual Na on selective CO methanation was also studied over unwashed and washed Ru/TiO2. In order to prepare Na-free Ru/TiO2 catalysts using the selective deposition method, NH3 aqueous solution was also used as a pH adjuster.
2 wt% Ru/TiO2 catalysts were prepared (see ESI†), and the abbreviations for the catalysts are listed in Table 1. Fig. 1(a) shows a plot of CO concentration against reaction temperature over Ru/TiO2. The order of the decreasing rate of CO concentration was SD-NH3 > IMP, SD-Na-wash > SD-Na. The CO concentration decreased rapidly with temperature over the catalysts: as for SD-NH3, the CO concentration fell to 110 ppm at 150 °C, whereas over IMP and SD-Na-wash, the concentration of CO dropped to ca. 600 ppm, and for SD-Na to 1700 ppm at 150 °C. Over all catalysts except for IMP, CO concentration started to increase slightly at temperatures above 225 °C, which means that CO promotion via the RWGS reaction became faster than CO consumption via CO methanation. It is noteworthy that SD-NH3 showed higher CO conversion and/or CO conversion rate than the reported Ru catalysts (see Table S1, ESI†).5f–h
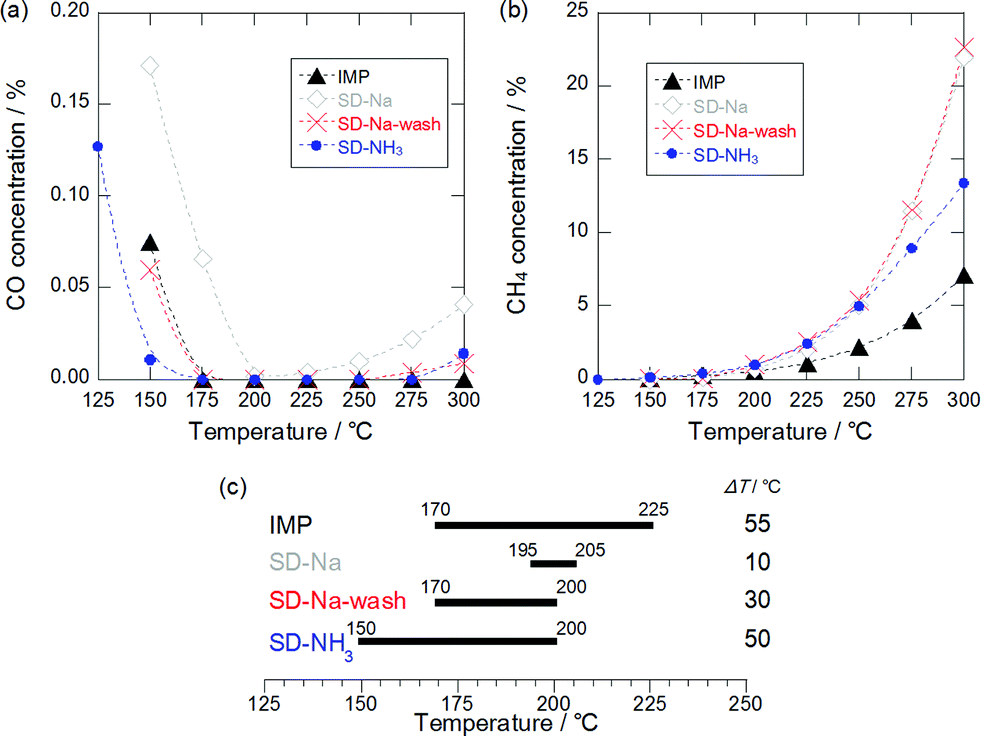 | ||
| Fig. 1 (a) CO and (b) CH4 concentration over Ru/TiO2 prepared using a wet impregnation method and a selective deposition method. (c) Temperature window for selective CO methanation (ΔT) over Ru catalysts. Definition of the window: CO concentration <100 ppm and CH4 concentration <1%. | ||
Fig. 1(b) exhibits the CH4 concentration curves over IMP, SD-Na, SD-Na-wash, and SD-NH3 as a function of reaction temperature. For all Ru/TiO2 catalysts, the CH4 concentration increased rapidly at temperatures above 175 °C. This increase in CH4 production at high temperatures is derived from CO2 methanation. Consequently, the order of CO2 methanation activity was SD-Na, SD-Na-wash and SD-NH3 > IMP below 250 °C.
Fig. 1(c) illustrates the temperature windows of the Ru/TiO2 catalysts for selective CO methanation in which the CO and CH4 concentrations are less than 100 ppm and 1%, respectively. Under these conditions, H2 conversion is estimated to be less than 9%. Catalysts for selective CO methanation are required to have a wide temperature window at a low temperature range for application to CO removal units in commercial PEFC systems. The window of SD-NH3 was as wide as that of IMP (ca. 50 °C) and was located at low temperatures compared to the other catalysts, which means that the SD-NH3 catalyst is the most suitable for selective CO methanation
To investigate the crystalline phases of Ru/TiO2 catalysts, the powder X-ray diffraction (XRD) measurements of IMP, SD-Na, SD-Na-wash, and SD-NH3 were carried out (see Fig S1, ESI†). All peaks were identified using ICDD files. All catalysts had multiple peaks assignable to anatase- and rutile-type TiO2. Thus, there is no apparent difference in the crystalline characteristics of TiO2 among the four catalysts based on the crystalline data about TiO2. As for IMP, a weak peak due to Ru species was detected at 28° in the XRD patterns, while for SD-Na, SD-Na-wash, and SD-NH3, no peaks were detected. The latter results indicate that Ru species on the catalysts prepared using a selective deposition method are in an amorphous state or have particle sizes smaller than the XRD detection limit (ca. 4 nm).
Fig. 2(a) presents the Na 1s XP spectra for SD-Na and SD-Na-wash reduced by H2. The spectra from SD-Na have a single strong peak at 1072 eV, while the spectra from SD-Na-wash have almost no distinct peaks. In the Ru 3d region shown in Fig. 2(b), for both catalysts, the peak at 280 eV assignable to metallic Ru appeared.12 The Ti 3d spectra were also measured (see Fig S2, ESI†), and there is no difference between SD-Na and SD-Na-wash. According to these XP spectra, by washing SD-Na with deionized water, Na species were removed without changing the electronic states of Ru and Ti species.
 | ||
| Fig. 2 XP spectra of (a) Na 1s and (b) Ru 3d of SD-Na and SD-Na-wash. | ||
The CO uptake of metallic Ru on the catalysts was measured using the CO chemisorption method, and the amounts of CO chemisorption of IMP, SD-Na-wash, and SD-NH3 were 222, 301, and 288 μl gcat−1, respectively. While it is difficult to determine CO surface coverage on metallic Ru due to the complex adsorption states of CO on Ru/TiO2,13 this result implies that Ru species of the catalysts prepared using a selective deposition method were highly dispersed compared to the IMP catalysts. As summarized in Fig. 3 and S3 (see ESI†), transmission electron microscopy (TEM) was used to examine the IMP, SD-Na-wash, and SD-NH3. In Fig. S3 (see ESI†), ca. 14 nm Ru particles were uniformly distributed on the surface of the IMP catalysts. In Fig. 3(a), a lot of darker spots, assignable to Ru particles, with a size of ca. 4 nm can be observed, and the secondary particles of Ru were formed, indicated by two dotted arrows. In Fig. 3(b), Ru particles (average size: 1.2 nm, see Fig. S4, ESI†) were well dispersed on the TiO2 surface, resulting in the high CO methanation activity presented in Fig. 1(a).
 | ||
| Fig. 3 TEM images of Ru/TiO2 prepared using a selective deposition method with (a) NaOH (SD-Na-wash) and (b) NH3 solution (SD-NH3). | ||
It is noteworthy that washing SD-Na with deionized water promoted CO methanation. In addition, SD-Na-wash showed almost the same activity of CO methanation as IMP despite the high Ru surface area. The amount of Na residual must be recalled here, and the order of the amount is SD-Na > SD-Na-wash > SD-NH3 and IMP. These do not indicate that Na residual on Ru/TiO2 suppressed CO methanation. Numerous attempts have been made by researchers to show the effects of alkali and alkali earth metal addition to metal catalysts on COx methanation and RWGS reaction.14 The addition of K to Ru catalysts promoted CO methanation due to the increase in the back donation of electrons from Ru d-orbitals to the π* antibonding molecular orbitals of CO. In addition, K and MgO can function as CO2 supply sources, leading to improvement of CO production via RWGS reaction and CO2 methanation. If the Na residual plays a role similar to those of MgO and K, Ru/TiO2 with a small amount of Na can also boost RWGS reaction and CO2 methanation. Actually, SD-Na and SD-Na-wash exhibited a rapid increase in CH4 concentration (Fig. 1(b)) and a narrow temperature window for selective CO methanation (Fig. 1(c)). The production rate of CO by RWGS reaction is higher than the CO consumption rate by CO methanation, giving rise to a slower decrease in CO concentration as illustrated in Fig. 1(a).
In conclusion, Ru/TiO2 catalysts were prepared using a selective deposition method and a wet impregnation method, and the influence of preparation method on selective CO methanation was studied. The catalysts prepared using a selective deposition method with NH3 aqueous solution as a pH adjuster exhibited the high activity and selectivity of CO methanation because of the following two reasons:
(i) Ru particle size was small (average 1.2 nm), leading to increase in CO methanation active sites.
(ii) Na-free Ru/TiO2 was prepared, resulting in low activity of CO2 methanation. Na residual on Ru/TiO2 is considered to enhance the CO production via reverse water gas shift reaction and the successive CO methanation.
Acknowledgements
This study was supported by the New Energy and Industrial Technology Development Organization (NEDO). TEM observation was conducted at the Center for Nano Lithography & Analysis, The University of Tokyo, which was supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- I. Staffell and R. Green, Int. J. Hydrogen Energy, 2013, 38, 1088 CrossRef CAS PubMed.
- E. Antolini, J. Appl. Electrochem., 2004, 34, 563 CrossRef CAS.
- S. Nishimura, T. Shishido, T. Ohyama, K. Teramura, A. Takagaki, T. Tanaka and K. Ebitani, Catal. Sci. Technol., 2012, 2, 1685 CAS.
- Z. Zhao, X. Lin, R. Jin, Y. Dai and G. Wang, Catal. Sci. Technol., 2012, 2, 554 CAS.
- (a) S. Eckel, Y. Denkwitz and R. J. Behm, J. Catal., 2010, 269, 255 CrossRef PubMed; (b) R. A. Dagle, Y. Wang, G.-G. Xia, J. J. Strohm, J. Holladay and D. R. Palo, Appl. Catal., A, 2007, 326, 213 CrossRef CAS PubMed; (c) S. Takenaka, T. Shimizu and K. Otsuka, Int. J. Hydrogen Energy, 2004, 29, 1065 CrossRef CAS PubMed; (d) P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, J. Phys. Chem. C, 2011, 115, 1220 CrossRef CAS; (e) P. Djinović, C. Galletti, S. Specchia and V. Specchia, Catal. Today, 2011, 164, 282 CrossRef PubMed; (f) K. Urasaki, K.-I. Endo, T. Takahiro, R. Kikuchi, T. Kojima and S. Satokawa, Top. Catal., 2010, 53, 707 CrossRef CAS; (g) S. Eckle, H.-G. Anfang and R. J. Behm, Appl. Catal., A, 2011, 391, 325 CrossRef CAS PubMed; (h) C. Galletti, S. Specchia, G. Saracco and V. Specchia, Chem. Eng. Sci., 2010, 65, 590 CrossRef CAS PubMed.
- (a) K. Urasaki, Y. Tanpo, T. Takahiro, J. Christopher, R. Kikuchi, T. Kojima and S. Satokawa, Chem. Lett., 2010, 39, 972 CrossRef CAS; (b) K. Urasaki, Y. Tanpo, Y. Nagashima, R. Kikuchi and S. Satokawa, Appl. Catal., A, 2013, 452, 174 CrossRef CAS PubMed.
- (a) S. Tada, R. Kikuchi, A. Takagaki, T. Sugawara, S. T. Oyama, K. Urasaki and S. Satokawa, Appl. Catal., B, 2013, 140–141, 258 CrossRef CAS PubMed; (b) A. Chen, T. Miyao, K. Higashiyama, H. Yamashita and M. Watanabe, Angew. Chem., Int. Ed., 2010, 49, 9895 CrossRef CAS PubMed.
- F. Solymosi, I. Tombácz and M. Kocsis, J. Catal., 1982, 75, 78 CrossRef CAS.
- S. Tada, R. Kikuchi, K. Urasaki and S. Satokawa, Appl. Catal., A, 2011, 404, 149 CrossRef CAS PubMed.
- (a) Y. Sunagawa, K. Yamamoto, H. Takahashi and A. Muramatsu, Catal. Today, 2008, 132, 81 CrossRef CAS PubMed; (b) T. Sugimoto, Monodispersed Particles, Elsevier, Amsterdam, 2001, p. 322 Search PubMed.
- I. Rossetti, N. Pernicone and L. Forni, Appl. Catal., A, 2001, 208, 278 CrossRef.
- T. Mitsui, K. Tsutsui, T. Matsui, R. Kikuchi and K. Eguchi, Appl. Catal., B, 2008, 81, 56 CrossRef CAS PubMed.
- C. Elmasides, D. I. Kondarides, W. Grünert and X. E. Verykios, J. Phys. Chem. B, 1999, 103, 5227 CrossRef CAS.
- (a) J.-N. Park and E. W. McFarland, J. Catal., 2009, 266, 92 CrossRef CAS PubMed; (b) R. D. Gonzalez and H. Miura, J. Catal., 1982, 77, 338 CrossRef CAS; (c) C.-S. Chen, W.-H. Cheng and S.-S. Liu, Appl. Catal., A, 2003, 238, 55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy00723e |
| This journal is © The Royal Society of Chemistry 2014 |