NiAl and CoAl materials derived from takovite-like LDHs and related structures as efficient chemoselective hydrogenation catalysts
Constantin
Rudolf
a,
Brindusa
Dragoi
*a,
Adrian
Ungureanu
a,
Alexandru
Chirieac
a,
Sébastien
Royer
b,
Alfonso
Nastro
c and
Emil
Dumitriu
a
aTechnical University of Iasi, Faculty of Chemical Engineering and Environmental Protection, 71A D. Mangeron Bvd, 700050, Iasi, Romania. E-mail: bdragoi@tuiasi.ro; Fax: +40 232 271 311; Tel: +40 232 278683
bUniversité de Poitiers, UMR 7285 CNRS, IC2MP, 4 Rue Michel Brunet, 86022 Poitiers, France
cDipartimento di Chimica, Università della Calabria, via P. Bucci, Arcavacata di Rende, 87036, Cosenza, Italy
First published on 10th October 2013
Abstract
The catalytic performance of metallic catalysts derived from layered double hydroxide (LDH) precursors with nickel or cobalt incorporated in the brucite-like layers besides aluminium (i.e., NiAl takovite and related CoAl) was investigated for the first time in the chemoselective hydrogenation of cinnamaldehyde. The precursors in the as-synthesized and calcined forms were thoroughly analysed by ICP, XRD, nitrogen physisorption, DR UV-vis spectroscopy, TPR and in situ XRD after temperature-programmed reduction. According to the XRD results, both as-synthesized samples contained purely LDH phases with degrees of crystallization depending on the nature of incorporated metal cations (i.e., the CoAl sample presents larger crystallites than the NiAl takovite-like sample). For the calcined NiAl and CoAl samples, well-crystallized oxide phases of NiO and Co3O4, respectively, besides amorphous alumina and corresponding spinels were evidenced by XRD. The TPR and in situ XRD results for the calcined samples indicated strong metal–alumina interactions, which resulted in depressed sintering of evolved metal nanoparticles in the high-temperature range of 600–800 °C, in well agreement with TEM analysis. Materials derived from the studied LDH systems are found to be efficient catalysts for the hydrogenation of cinnamaldehyde, showing enhanced control over the activity or selectivity, depending on the nature of active metals and the thermal regime of the catalyst activation.
Introduction
The hydrogenation of cinnamaldehyde (CNA) is one of the most studied catalytic reductive processes. Though CNA molecule has high degree of unsaturation (i.e., aromatic ring, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC double bonds), its partial hydrogenation preferentially occurs in the side aliphatic chain forming hydrocinnamaldehyde (HCNA) and cinnamyl alcohol (CNOL), respectively.1–3 Both hydrogenated products are of industrial interest and therefore it is challenging to develop high performance catalysts which are selective either to HCNA or CNOL. HCNA is used as inhibitor of the potato tubers sprouting4 or for the preparation of pharmaceutical products such as HIV protease inhibitors,5 while CNOL is largely used in perfumery and flavour industries.3 For instance, it is frequently used in blossom compositions such as lilac, jasmine and lily of the valley, hyacinth and gardenia, to impart balsamic and oriental notes to the fragrances. It could be noted that hydrocinnamyl alcohol (HCNOL), the product of total hydrogenation, has practical applications as well. Therefore, it is a component in perfumes,6 while in adequate chemicals mixture has bactericidal properties.7
O and CC double bonds), its partial hydrogenation preferentially occurs in the side aliphatic chain forming hydrocinnamaldehyde (HCNA) and cinnamyl alcohol (CNOL), respectively.1–3 Both hydrogenated products are of industrial interest and therefore it is challenging to develop high performance catalysts which are selective either to HCNA or CNOL. HCNA is used as inhibitor of the potato tubers sprouting4 or for the preparation of pharmaceutical products such as HIV protease inhibitors,5 while CNOL is largely used in perfumery and flavour industries.3 For instance, it is frequently used in blossom compositions such as lilac, jasmine and lily of the valley, hyacinth and gardenia, to impart balsamic and oriental notes to the fragrances. It could be noted that hydrocinnamyl alcohol (HCNOL), the product of total hydrogenation, has practical applications as well. Therefore, it is a component in perfumes,6 while in adequate chemicals mixture has bactericidal properties.7
The majority of the catalysts used for the hydrogenation of CNA to HCNA and CNOL are based on noble metals such as Rh,8,9 Pd,10–12 Pt,13,14 Au,15–17 Ru,18etc. In the presence of such catalysts high selectivities towards HCNA or CNOL were obtained (e.g., 100% unsaturated aldehyde at 79% conversion of CNA on Rh(III)-hexamolybdate/γ-Al2O3;9 ~88% HCNA at total conversion of CNA on Pd/polyketone;8 50% unsaturated alcohol at 50% conversion of CNA on Pt supported on carbon aerogel;11 48% unsaturated alcohol at 60% conversion of CNA on Ru/carbon nanofiber)18 but the catalytic phases based on noble metals are very expensive and in limited amounts as offer.
The use of mono- and bimetallic catalysts based on non-noble metals such as Cu, Ni and Co for selective hydrogenation reactions, including the hydrogenation of α,β-unsaturated aldehydes such as cinnamaldehyde, was also reported in several papers, either in liquid or gas phase.19–25 Such materials presented interesting catalytic performances which depend on a wide variety of factors such as the nature of active sites, interaction with the support, reaction and catalyst activation conditions, nature of the second metal and so on. For instance, a CNOL selectivity of ~45% was reported on Cu–Co/SiO2 bimetallic catalyst.20 It was shown that the rate of CNOL formation is essentially increased on spinel Cu–Ni(Co)–Zn–Al catalysts compared to Cu/SiO2 or binary Cu–Al samples due to the formation of surface Cu0–M2+ dual sites (M2+ = Co, Ni, Zn). Ni0/TiO2 also manifested high activity in the hydrogenation of CNA with very high selectivity to HCNA (98% at CNA conversion of 66.2% in iso-propanol and under 2 MPa hydrogen pressure22 while Ni0/MCM-41 converted 94.6% CNA with a selectivity to HCNA of 44.2%).25
In this contribution we report an original process to obtain highly active and chemoselective catalysts based on non-noble metals whose preparation is easy to perform and not expensive. Usually, the mixed oxides of reducible cations obtained via layered double hydroxides (LDHs) are effective catalytic precursors because the LDHs are obtained as well-organized and compositional versatile crystalline materials. Indeed, interesting catalytic performances were previously reported in the hydrogenation of CNA over materials derived from as-synthesized hydrotalcite-like materials with Ni, Cu or Co incorporated in MgAl or ZnAl matrixes (e.g., 60% HCNA at total conversion of CNA on MgCuAl or 90% CNOL at 9% conversion of CNA on ZnNiCoAl (Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co = 8:2)).26 However, it was demonstrated that the acid–basic sites associated with the magnesium or zinc cations promote the secondary reactions of aldol condensation of aldehydes, which negatively influences the yield to the hydrogenation products.27 To circumvent these drawbacks, in the present investigation we report for the first time the use of Mg(Zn)-free Ni- and Co-based catalysts derived from binary takovite (NiAl) and related CoAl-layered double hydroxides (M:Al ratio of 2:1) in the chemoselective hydrogenation of cinnamaldehyde. As it is known, takovite-based catalysts are able to activate various reactions such as methanation of CO, Fischer–Tropsch reactions and steam reforming of methane.28 Interestingly, the first patent about the application of layered double hydroxides for the catalytic hydrogenations mainly claimed the use of reduced oxide derived from takovite and related hydrotalcite-like structures.29 In addition, these bicomponent catalysts allow the preparation of a large amount of metals highly dispersed on alumina, which is difficult to achieve for metal supported catalysts obtained by conventional impregnation. Materials derived from takovite and related materials are found to be efficient hydrogenation catalysts, showing enhanced control over the activity or selectivity, depending on the nature of active metals and the thermal regime of the catalyst activation.
:Co = 8:2)).26 However, it was demonstrated that the acid–basic sites associated with the magnesium or zinc cations promote the secondary reactions of aldol condensation of aldehydes, which negatively influences the yield to the hydrogenation products.27 To circumvent these drawbacks, in the present investigation we report for the first time the use of Mg(Zn)-free Ni- and Co-based catalysts derived from binary takovite (NiAl) and related CoAl-layered double hydroxides (M:Al ratio of 2:1) in the chemoselective hydrogenation of cinnamaldehyde. As it is known, takovite-based catalysts are able to activate various reactions such as methanation of CO, Fischer–Tropsch reactions and steam reforming of methane.28 Interestingly, the first patent about the application of layered double hydroxides for the catalytic hydrogenations mainly claimed the use of reduced oxide derived from takovite and related hydrotalcite-like structures.29 In addition, these bicomponent catalysts allow the preparation of a large amount of metals highly dispersed on alumina, which is difficult to achieve for metal supported catalysts obtained by conventional impregnation. Materials derived from takovite and related materials are found to be efficient hydrogenation catalysts, showing enhanced control over the activity or selectivity, depending on the nature of active metals and the thermal regime of the catalyst activation.
Experimental
Catalyst preparation
All chemicals were used as received: aluminium nitrate (Al(NO3)3·9H2O, 98%, Sigma-Aldrich), nickel nitrate (Ni(NO3)2·6H2O, 98%, Sigma-Aldrich) and cobalt nitrate (Co(NO3)2·6H2O, 98%, Sigma-Aldrich) as metal precursors, sodium hydroxide (NaOH) and sodium carbonate (Na2CO3 99.8%, Merck) as precipitating agents. For catalytic runs, the chemicals were also used as purchased: trans-cinnamaldehyde (C6H5CHCHCHO, 98%, Merck) as reagent and propylene carbonate (C4H6O3, 99%, Sigma-Aldrich) as solvent.
Two samples of layered double hydroxides, CoAl and NiAl with a molar ratio 2:1, were prepared by co-precipitation at low supra-saturation method. An aqueous solution containing the corresponding metal sources and a solution containing Na2CO3 (0.8 M solution) and NaOH (1.6 M solution) were simultaneously added at a flow rate of 0.7 mL min−1 under vigorous stirring and constant temperature (30 °C) and pH (9.0).
The precipitates were recovered by filtration and then washed with deionized water to remove Na+ ions. After washing, the resulting solid was dried at 40 °C up to constant weight. Before use, the samples were calcined at 450 °C for 5 h under static air (heating ramp of 1.5 °C min−1) and then reduced for 10 h at 500 °C under H2 flow (1 L h−1), with a heating ramp of 1.5 °C min−1. CoAl sample was also reduced at 700 °C.
Catalyst characterization
Chemical composition (Co, Ni and Al) of the samples was performed on a Perkin sequential scanning inductively coupled plasma optical emission spectrometer (Optima 2000 DV). Before analysis, a known amount of sample was digested in a diluted HCl solution and then heated under microwave until complete dissolution. XRD measurements were performed on a Bruker AXS D8 apparatus with monochromatic CuKα radiation (λ = 0.15406 nm wavelength) at room temperature. The patterns were recorded from 5 to 70° 2θ, with a step of 0.02°. The crystallite sizes were calculated using the Scherrer equation: dhkl = λ/βcosθ, where λ = incident ray wavelength (0.15406 nm); β = peak width at half height (rad) after Warren's correction for instrumental broadening; and θ = Bragg angle. Nitrogen physisorption was carried out at −196 °C on an Autosorb MP-1 instrument from Quantachrome. Surface areas and pore volumes were calculated from the corresponding isotherms using conventional algorithms. Diffuse reflectance UV-visible (DR UV-vis) spectra were recorded on a Shimadzu UV-2450 spectrometer equipped with an integrating sphere unit (ISR-2200). The spectra were collected in the range between 200–800 nm by using BaSO4 as the reflectance standard. In situ powder XRD patterns were recorded on a Bruker D8 ADVANCE X-ray diffractometer equipped with a VANTEC-1 detector, using a CuKα radiation (λ = 1.54184 Å) as X-ray source. The powder samples were placed on a kanthal filament (FeCrAl) cavity. The data were collected in the 2θ range from 10 to 80° with a step of 0.05° (step time of 2 s). Phase identification was made by comparison with ICDD database. The diffractograms were recorded after in situ reduction from 30 to 800 °C under a flow of 3 vol.% H2 in He (30 mL min−1). The data were collected 3 times at each temperature. TEM images were obtained using a on a JEOL 2100 instrument (operated at 200 kV with a LaB6 source and equipped with a Gatan Ultra scan camera). Temperature programmed reduction experiments were performed on an Autochem chemisorption analyser from Micromeritics, equipped with a TCD and MS (Omnistar, Pfeiffer) detectors. About 100 mg of as-synthesized LDH were inserted in a U-shape microreactor. Before each TPR run, the catalyst was activated at 120 °C for 4 h under a flow of simulated air (30 mL min−1). After cooling to 50 °C, the H2 containing flow was stabilized (30 mL min−1, 3 vol.% H2 in Ar) and the TPR was performed from 50 to 800 °C with a temperature ramp of 2 °C min−1.Hydrogenation of trans-cinnamaldehyde
The catalytic runs were carried out in a round-bottom three-neck glass reactor equipped with a bubbler, magnetic stirrer and a reflux condenser. The reaction was performed at atmospheric pressure under the following conditions: 0.265 g of catalyst, 1.0 mL of trans-cinnamaldehyde, 25.0 mL of propylene carbonate as solvent, hydrogen flow of 1.0 L h−1, reaction temperature of 150 °C, and agitation speed of 900 rpm. Samples of reaction mixture were periodically withdrawn and analyzed with a gas-chromatograph (HP 5890 equipped with a DB-5 capillary column (25 m × 0.20 mm × 0.33 μm) and FID detector). The identification of the products from the reaction mixture was accomplished from the retention time of the pure compounds and verified by GC-MS (Agilent 6890N system equipped with an Agilent 5973 MSD detector and a 30-m DB-5-ms column). The quantitative analyses were performed by taking into consideration the FID response factor for each compound. The total conversion of cinnamaldehyde and the selectivity towards different hydrogenation products were calculated as previously reported.30 For the recyclability tests, the catalysts were recovered by centrifugation from the end reaction mixture, washed with fresh propylene carbonate and ethanol, and then dried under vacuum before the reactivation.Results and discussion
Catalysts characterization
The chemical compositions of LDH precursors were evaluated by ICP and are presented in Table 1.| Sample | M/Ala (molar ratio) | Ionic radius for M2+b (pm) | d 003 (Å) | a (Å) | c (Å) | D (003) (nm) | D (110) (nm) | |
|---|---|---|---|---|---|---|---|---|
| Synthesis | ICP | |||||||
| a M = Co, Ni. b 33 c d 003 = λ/2sinϑ. d a = 2d110. e c = 3d003. f D (003) = crystallite size in the c direction. g D (110) = crystallite size in the a direction. | ||||||||
| CoAl | 2 | 2.18 | 74.5 | 7.82 | 3.06 | 23.46 | 17.41 | 32.64 |
| NiAl | 2 | 2.05 | 69.0 | 7.75 | 2.96 | 23.25 | 4.33 | 9.73 |
As noted, the Co/Al and Ni/Al ratios in the final solids are slightly different from those of the synthesis mixture (2.18 and 2.05 for Co/Al and Ni/Al, respectively vs. 2.0). These small variations of the final M2+/Al ratios are usual and are due to the preferential precipitation of one cation to another.31 According to these values, the complete incorporation of aluminium at pH = 9 did not occur during the synthesis, especially for the Co/Al sample.
The crystalline structure of LDH precursors was investigated by powder X-ray diffraction. As shown in Fig. 1, the diffraction patterns for the as-synthesized LDH samples display the characteristic diffraction peaks of crystalline hydrotalcite-like LDHs, which are indexed in a hexagonal lattice with R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m rhombohedral symmetry.1 It should be noted that for CoAl sample the diffraction peaks are sharper and narrower suggesting improved crystallinity (larger particle size) than for NiAl sample. On the basis of the diffractograms, the structural parameters (i.e., basal distances −d003, a and c parameters and the crystallite sizes) were calculated and are provided in Table 1. As it can be observed, d003 displays values of 7.82 and 7.75 Å for CoAl and NiAl, respectively, which are higher than that of the MgAl hydrotalcite (7.65 Å).32 However, it should be taken into account that the thickness of the brucite layer of hydrotalcite is 4.8 Å,32 the amount of water (data are not shown here) is almost the same for both samples while the counteranion is carbonate in both cases. Hence, the increase of the d003 value for NiAl and CoAl can be explained by the increase in the thickness of brucite-like sheets due to the substitution of Al3+ by Ni2+ and Co2+.31 These structural results confirm the incorporation of the metallic cations into the brucite-like layers of the samples.
m rhombohedral symmetry.1 It should be noted that for CoAl sample the diffraction peaks are sharper and narrower suggesting improved crystallinity (larger particle size) than for NiAl sample. On the basis of the diffractograms, the structural parameters (i.e., basal distances −d003, a and c parameters and the crystallite sizes) were calculated and are provided in Table 1. As it can be observed, d003 displays values of 7.82 and 7.75 Å for CoAl and NiAl, respectively, which are higher than that of the MgAl hydrotalcite (7.65 Å).32 However, it should be taken into account that the thickness of the brucite layer of hydrotalcite is 4.8 Å,32 the amount of water (data are not shown here) is almost the same for both samples while the counteranion is carbonate in both cases. Hence, the increase of the d003 value for NiAl and CoAl can be explained by the increase in the thickness of brucite-like sheets due to the substitution of Al3+ by Ni2+ and Co2+.31 These structural results confirm the incorporation of the metallic cations into the brucite-like layers of the samples.
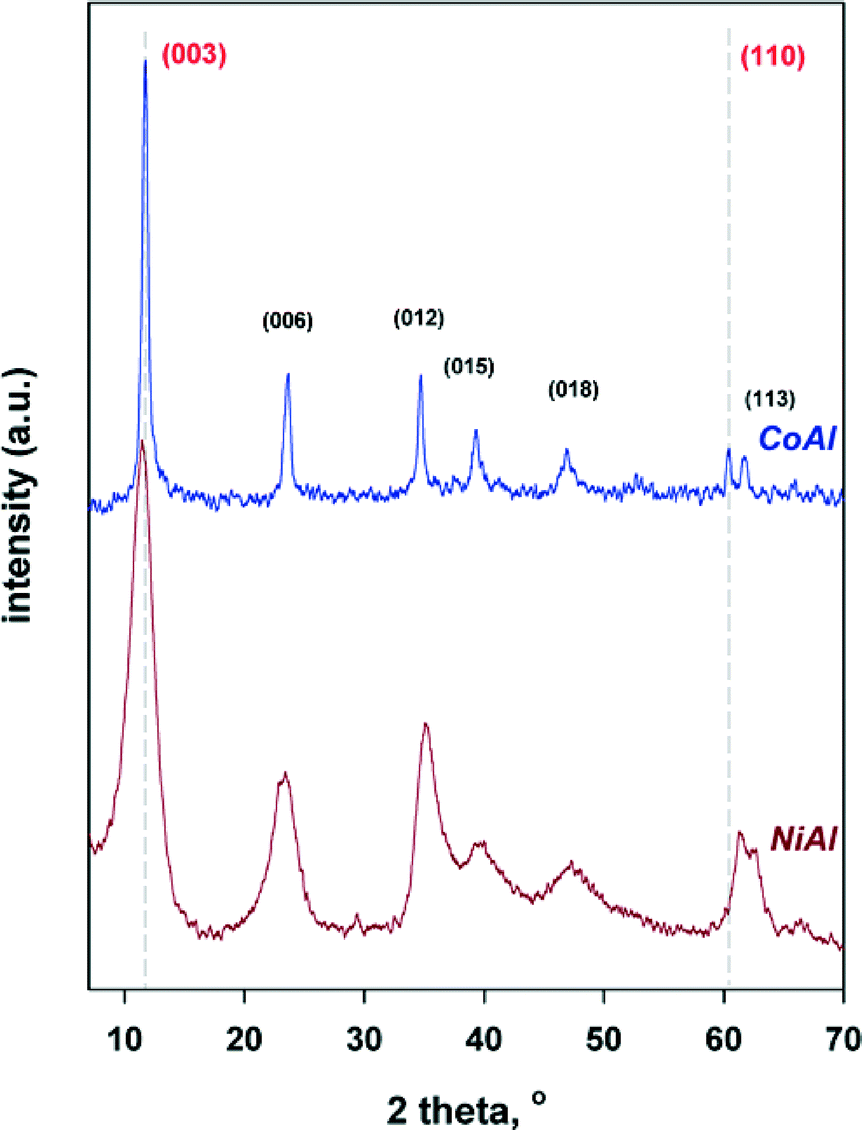 | ||
| Fig. 1 Powder X-ray diffraction for the as-synthesized LDHs. | ||
Information on the oxidation state as well as the coordination of the cations into the brucite-like sheets was provided by DR UV-vis spectrometry. The recorded spectra are displayed in Fig. 2. Because Al3+ has d0 configuration, it does not show absorption bands in the UV-vis spectra. Therefore, the spectra show the bands mainly associated to the d–d transitions of Ni2+ (d8) and Co2+ (d7) cations. As illustrated in Fig. 2, a characteristic band around 205 and 250 nm for NiAl and CoAl, respectively due to the O2− → Mn+ charge transfer transitions was identified in each corresponding spectrum.34–37
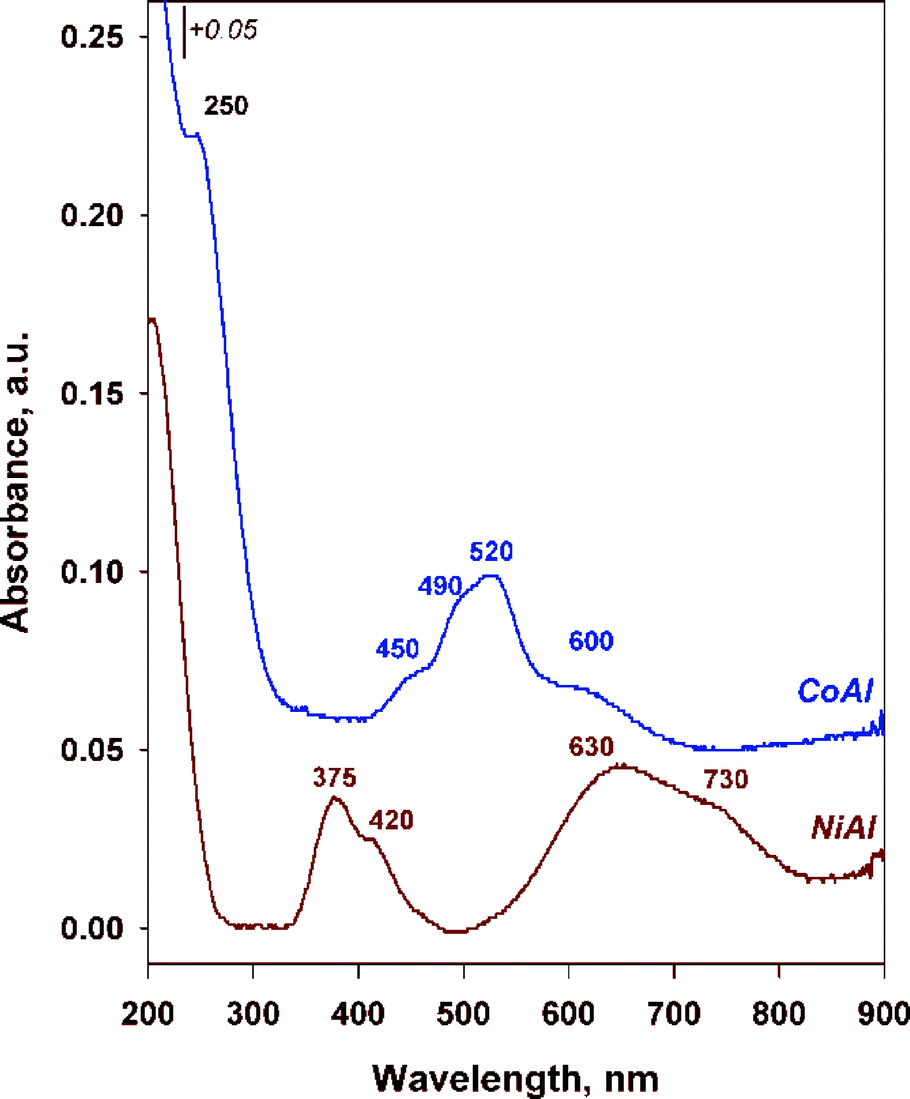 | ||
| Fig. 2 DR UV-vis spectra for the as-synthesized LDHs. | ||
The incorporation of cobalt in CoAl sample resulted in the formation of a three component absorption band positioned at ~450, 490 and 520 nm which was ascribed to Co2+ in octahedral coordination.38 Additionally, an absorption band centered at ~600 nm is observed and it may be attributed to d–d transitions in low spin of octahedrally coordinated Co3+ cations.39 Nevertheless, this band is much less intense in comparison to the band at ~520 nm, indicating that a minor amount of cobalt exists as Co3+.
For NiAl sample, two double important bands are observed. The first one is located at ~375 and 420 nm and the second one at ~630 and 730 nm. Two spin-allowed transitions 3A2g(F) → 3T1g(F) and 3A2g(F) → 3T1g(P) were assigned to the bands at 375 and 730 nm, respectively, while the bands at 420 and 630 nm were assigned to spin forbidden transitions 3A2g(F) → 1Eg(D) and 3A2g(F) → 1T2g(D).28 Both bands correspond to Ni2+ in octahedral coordination.
The textural properties of the samples were evaluated from the nitrogen adsorption–desorption isotherms (Fig. 3A). The isotherms are of type IIb45,46 and originate from the adsorption of nitrogen in the void spaces between the plate-like particles. The shape of the hysteresis loops (type H3) indicates a large distribution of the slit-shaped mesopores for both samples. The textural properties, i.e. the specific surface areas and pore volumes are summarized in Table 2. The obtained values are in agreement with those already reported for these kinds of materials.31,32 As expected, the calcination of these solids at 450 °C gave rise to changes in their texture. Indeed, for the calcined CoAl (denoted as CoAl-MO) and NiAl (denoted as CoAl-MO) samples the isotherms illustrated in Fig. 3B are of type IV with larger hysteresis loops, especially for NiAl sample. This indicates improved mesoporosity, which is a positive feature for catalysis as the diffusional limitations are restricted. As a result, the surface areas of MO-materials are almost three times the surface areas calculated for the as-synthesized materials. These increases might also indicate that the formation of highly stable spinels was avoided, which typically occurs for LDHs containing transition metals cations in the layer and carbonate as counteranion.46
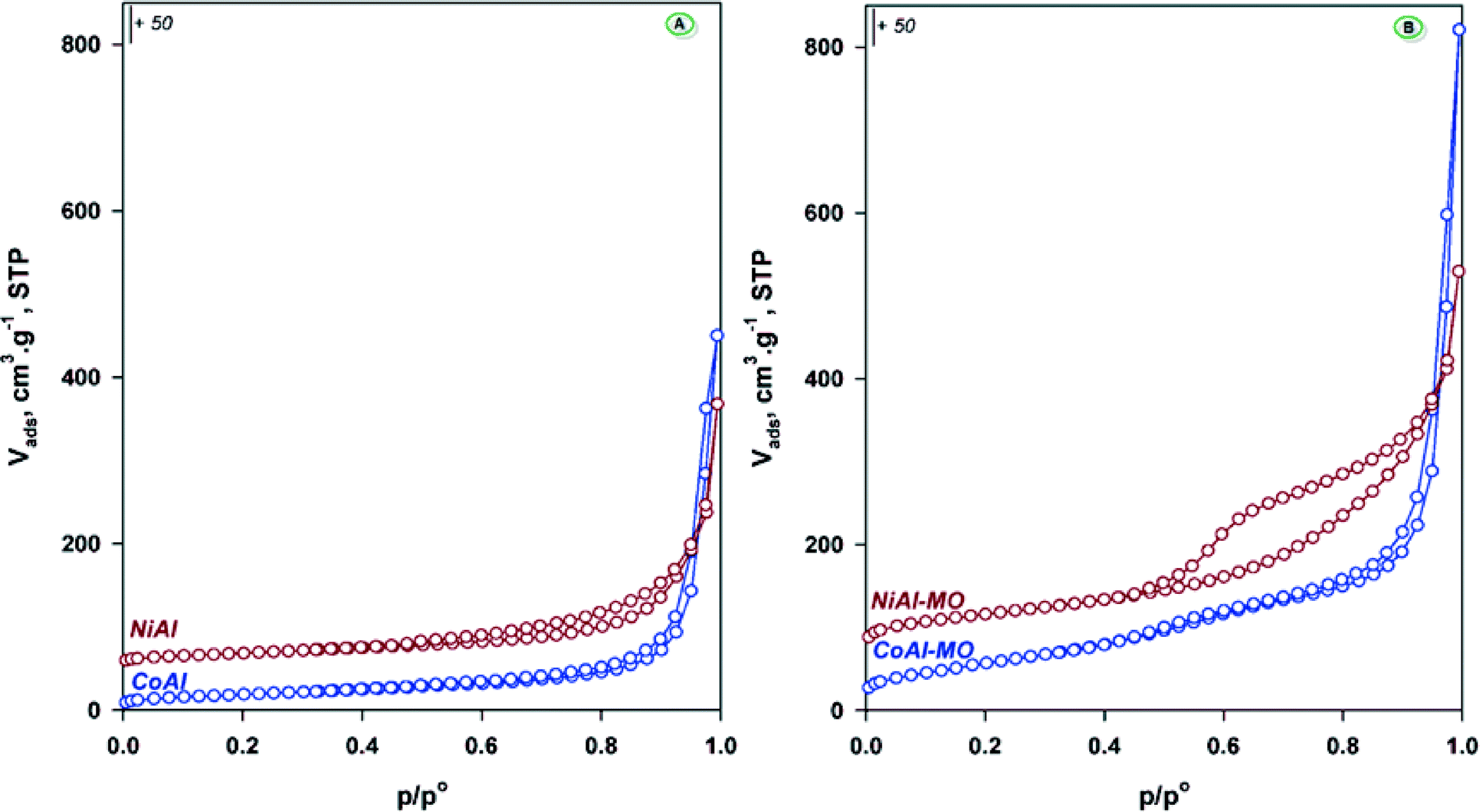 | ||
| Fig. 3 N2 adsorption–desorption isotherms for the as-synthesized (A) and calcined-mixed oxides (B) samples. | ||
The reducibility and extent of metal–alumina interactions for the calcined LDHs precursors were investigated by TPR (Fig. 4). The TPR profile for CoAl-MO sample displays two main maxima of reduction. Based on in situ XRD results (see below), the first peak, centered at ~350 °C, was assigned to the reduction of Co3O4 spinel to CoO while the second one, centered at ~700 °C, was attributed to the reduction of CoO to Co0. Nevertheless, the reduction of Co2+ in amorphous CoAl2O4 cannot be totally ruled out at 700 °C,40,41 although the characteristic peaks of the spinel were not detected by in situ XRD (see below). The TPR profile of NiAl-MO sample shows one minor and broad peak between 200 and 400 °C and a major peak at 400–800 °C with maximum at ~630 °C and a shoulder at ~560 °C. The first peak is attributed to the reduction of Ni2+ in a minor NiO phase with no interaction with the support.42 The reduction temperatures at ~560 °C and ~630 °C are assigned to the reduction of Ni2+ in NiO in strong interaction with alumina and/or NiAl2O4,41,43,44 also noticing that the characteristic peaks of the spinel were not detected by in situ XRD (see below). Because the TCD signal baseline is recovered after the reduction of nickel and cobalt cations, it can be affirmed that (i) there is good accessibility of H2 to completely reduce nickel and cobalt cations and (ii) metallic Ni0 and Co0 are in some extent supported on alumina and accessible to gas phase.
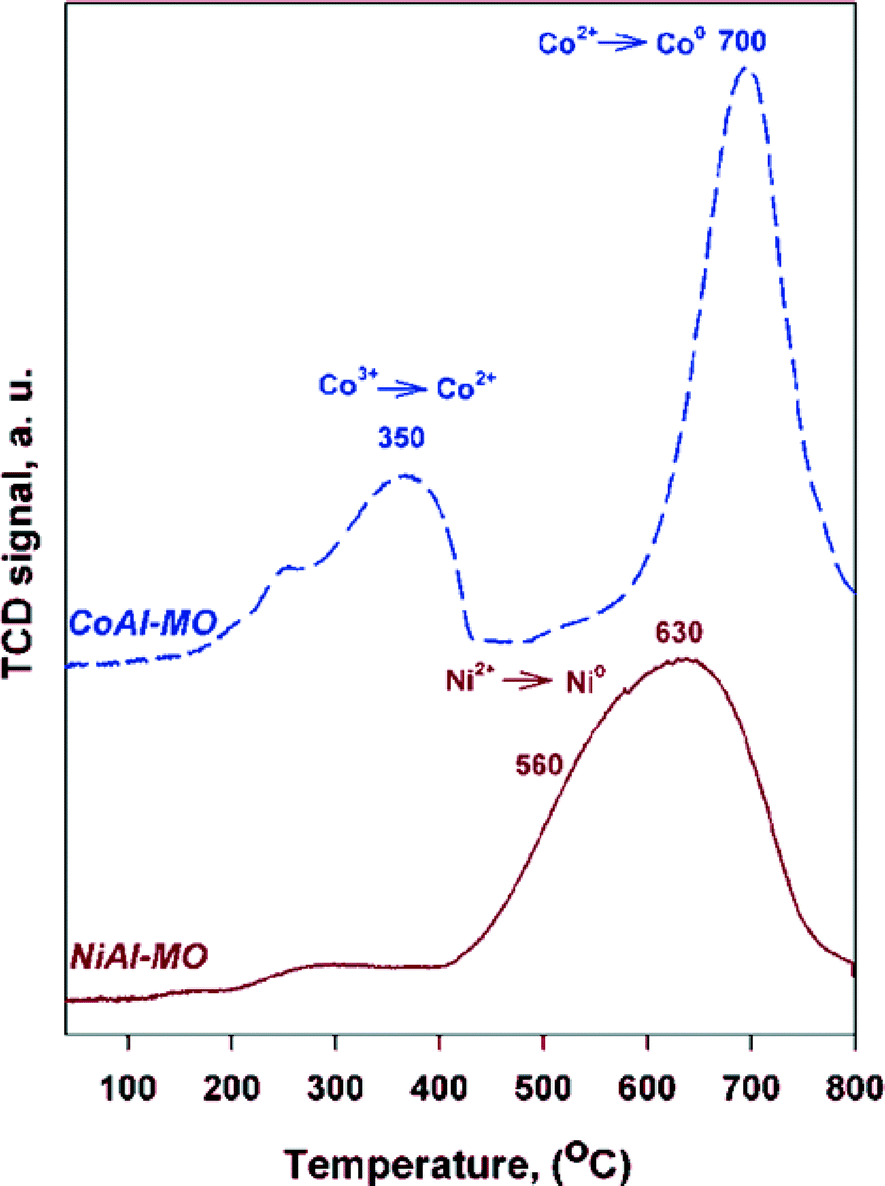 | ||
| Fig. 4 TPR profiles of the calcined LDHs. | ||
In order to elucidate the nature of intermediate crystalline phases formed during the reduction process, the calcined CoAl-MO and NiAl-MO samples were reduced under hydrogen flow at different temperatures in the range of 30–800 °C and analyzed in situ by XRD. The recorded diffractograms are displayed in Fig. 5A (for CoAl-MO) and Fig. 5B (for NiAl-MO). The XRD patterns registered at 30 °C for NiAl-MO and CoAl-MO (enlarged in Fig. 5C) show diffraction lines that fit the characteristic patterns for NiO (ICDD 047-1049)47 and Co3O4 (ICDD 42-1467)43,48,49 phases, respectively. The Co3O4 spinel is detected in the XRD patterns of CoAl-MO up to 300 °C. After reduction at 400 °C, the typical diffraction lines of the Co-spinel disappeared while new diffraction peaks appeared at 2θ = 36.47, 42.37 and 61.47°. These peaks are unambiguously assigned to the diffraction from the (111), (200) and (220) planes of CoO crystal lattice (ICDD 48-1719)50 (Fig. 5D). As observed from Fig. 5A, the intermediate CoO phases are not reducible up to a reduction temperature of 700 °C, providing clear evidence that the peak at 700 °C in the TPR profile for CoAl-MO (Fig. 4) is due to the reduction of CoO to metallic cobalt. Therefore, it is obvious that the calcination of CoAl generated mainly Co3O4 whose reduction under H2 takes place in two steps. However, this high reduction temperature (i.e., 700 °C) suggests a strong interaction between cobalt and alumina. Additionally, XRD showed that the alumina generated by the calcination of the CoAl sample (and NiAl sample) is amorphous.51 As noted, the crystalline CoAl2O4 spinel is still not observed in the diffractograms recorded at 700 °C reinforcing the idea of the amorphous state of alumina in the sample. The strong interaction between cobalt and alumina can also explain the absence of the typical diffraction peaks of the metallic Co0 in the diffractograms after further reduction at 800 °C, which indicates that the generated nanoparticles are stable against sintering. For NiAl-MO sample, traces of NiO can be still observed in the XRD patterns after reduction at 600 °C (Fig. 5D), which is in accord with TPR showing that the complete reduction of NiO takes place at ~750 °C. After reduction at 500 °C, new small diffraction lines attributed to metallic Ni0 phase (ICDD 04-0850)52 appeared in the diffractograms. Thus, the most intense peak (2θ = 44.5°) is attributed to (111) plane of Ni0 overlapping with the diffraction peak of the kanthal sample holder. The other two broad peaks observed at ~52 and ~76° and associated to the (200) and (220) planes,52 respectively, are well defined but they are too small to accurately calculate the average crystallite size of Ni0 nanoparticles. The reduction at 600 °C generated new Ni0 phases and the diffraction peaks are more intense. The calculated average crystallite size of the resulting Ni0 nanoparticles is 3.6 nm according to Scherrer's equation. Further increase in the reduction temperature to 700 and 800 °C results in the formation of slightly larger Ni0 nanoparticles with average crystallite sizes are of 5.5 and 5.8 nm, respectively. In similarity with the CoAl sample, the results obtained for NiAl suggest a limited sintering of nickel nanoparticles in the high-temperature range of 600–800 °C because of the strong interaction between nickel and alumina.52
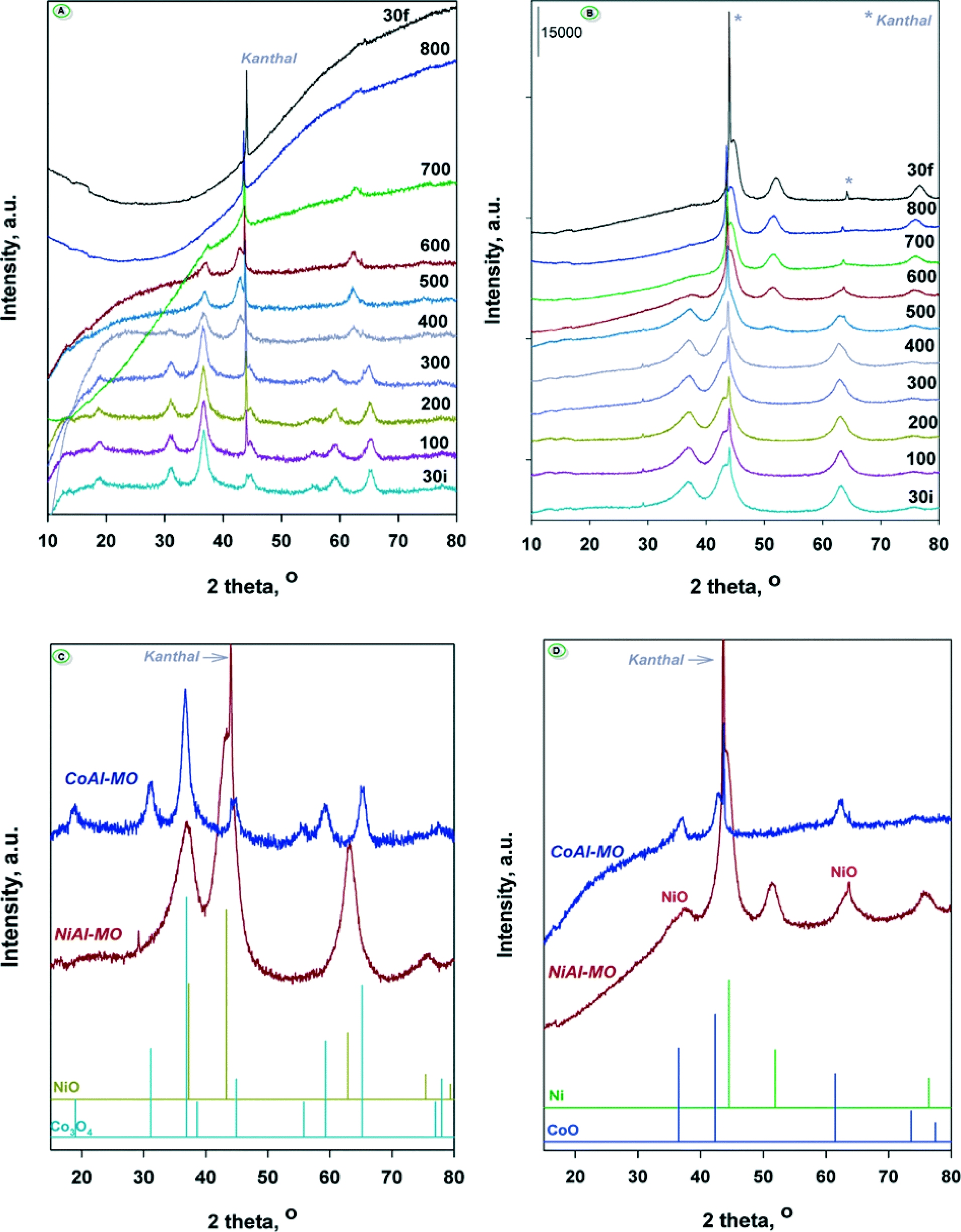 | ||
| Fig. 5 In situ XRD after reduction at different temperatures for the calcined samples: A) CoAl-MO; B) NiAl-MO; C) XRD patterns recorded for both samples after thermal treatment at 30 °C; D) XRD patterns recorded for both samples after thermal treatment at 600 °C. | ||
The morphostructural properties of the NiAl and CoAl catalysts after reduction under hydrogen were investigated by TEM analysis. Representative images are displayed in Fig. 6.
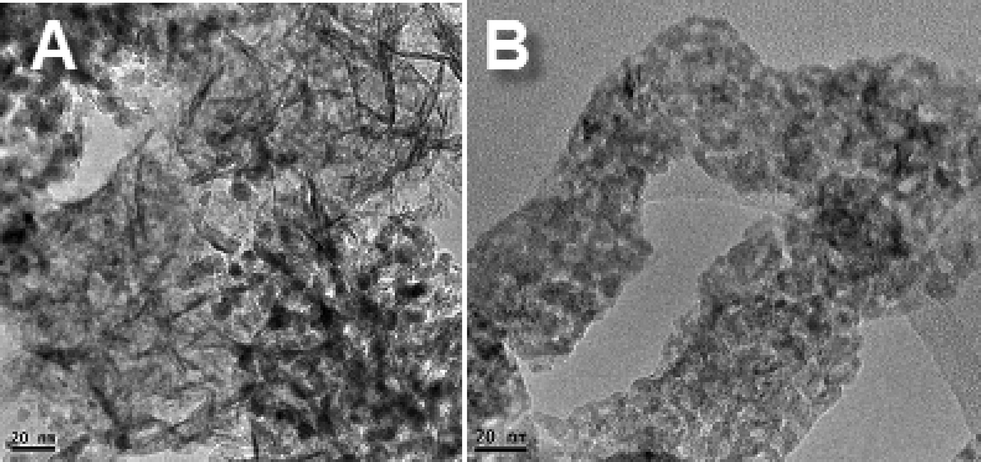 | ||
| Fig. 6 TEM images for NiAl (A) and CoAl (B) after reduction at 800 °C. | ||
As a first remark, traces of typical morphology with hexagonal plate-shaped particles are observed for NiAl sample, while for CoAl the brucite-like sheets are no longer maintained. This difference suggests higher stability of the brucite-like sheets when they contain nickel besides aluminium. Likewise, a monodisperse distribution of the metal particles with a size of 5–6 nm is observed for NiAl sample (Fig. 6A). For CoAl sample, very small metal particles are observed in Fig. 6B, those sizes are difficult to evaluate. These results are in good agreement with XRD data which indicated nickel crystallite size of ~6 nm for NiAl sample and highly dispersed cobalt metal particles in the case of CoAl sample.
Hydrogenation of the cinnamaldehyde
The catalytic properties of calcined NiAl and CoAl samples after reduction at 500 °C were evaluated for the liquid phase hydrogenation of cinnamaldehyde (CNA).It is worth remembering that (i) these two catalysts contain alumina that bring some acidity in the reaction medium and (ii) depending on the nature of the active sites (redox and/or acid–base sites), the reaction scheme of the conversion of cinnamaldehyde can be very complex.53 Despite the presence of alumina, the reaction products identified by GC-MS are mainly the result of the hydrogenation reaction. More precisely, in the case of NiAl-MO, condensation products below 2 mol% were identified whereas no traces of condensation or any other additional products were observed in the case of CoAl-MO sample. Consequently, the results of the hydrogenation reaction are exclusively discussed herein. The evolution of CNA conversion with the reaction time as well as the selectivities to the hydrogenation products are comparatively shown for both catalysts in Fig. 7.
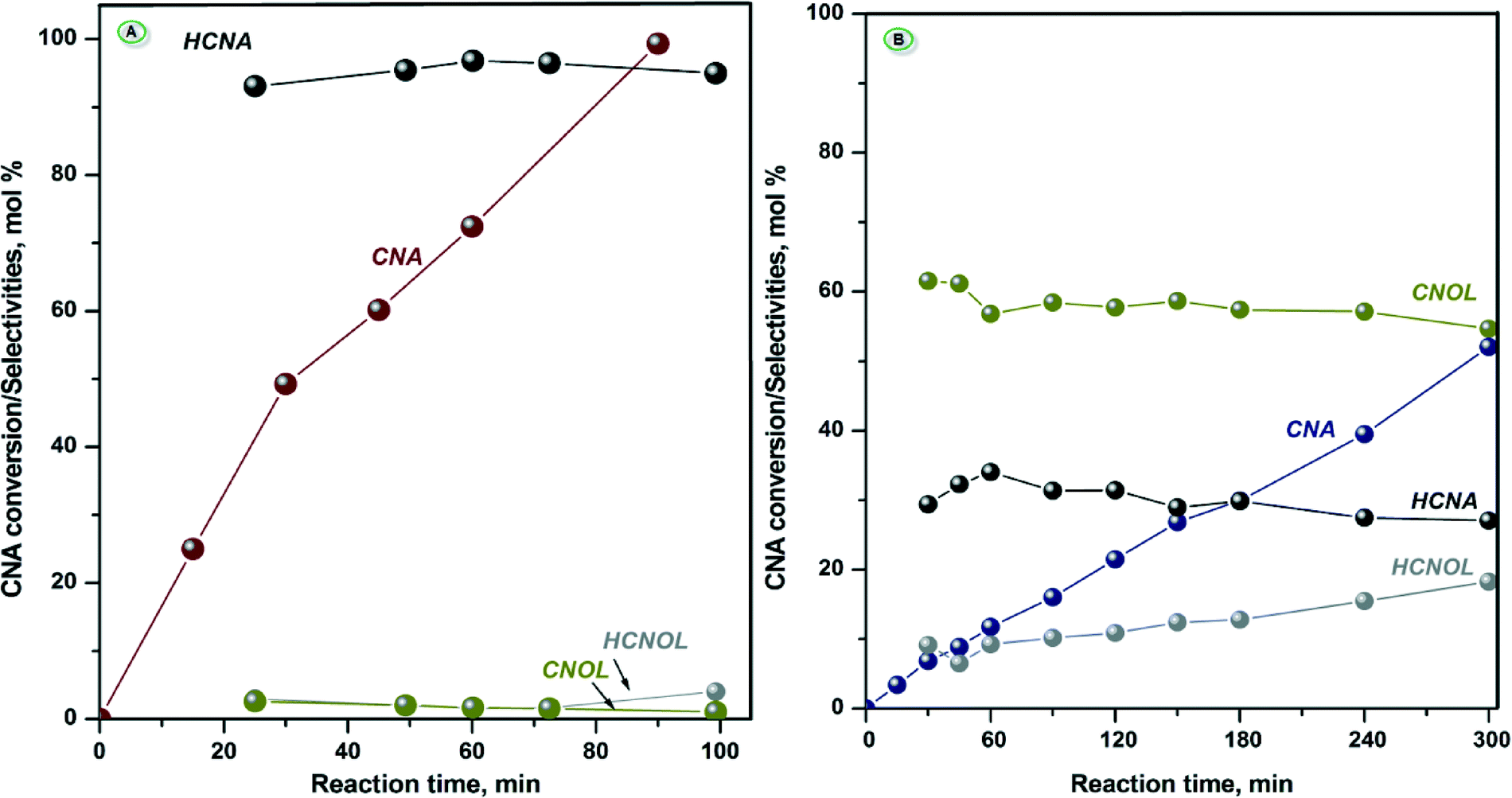 | ||
| Fig. 7 CNA conversion vs. time and selectivity to reactions products for NiAl-MO (A) and CoAl-MO (B) samples (Test conditions: Treduction = 500 °C; Treaction = 150 °C, 0.265 g catalyst; 1 mL of CNA, 25 mL propylene carbonate as solvent, H2 flow = 1 L h−1, stirring rate = 900 rpm). | ||
As mentioned in the introduction, CNA molecule has two double bonds in the side chain that makes the hydrogenation reaction to follow two possible ways: (i) the hydrogenation of CC bond with the formation of hydrocinnamaldehyde (HCNA) and (ii) the hydrogenation of CO bond when cinnamyl alcohol (CNOL) is formed (Scheme 1). The chemoselective hydrogenation towards the unsaturated alcohol is difficult to achieve since the thermodynamics favors the hydrogenation of the CC over the CO by ~35 kJ mol−1 and for kinetic reasons the reactivity of the CC group is higher than CO group in the hydrogenation reactions.54
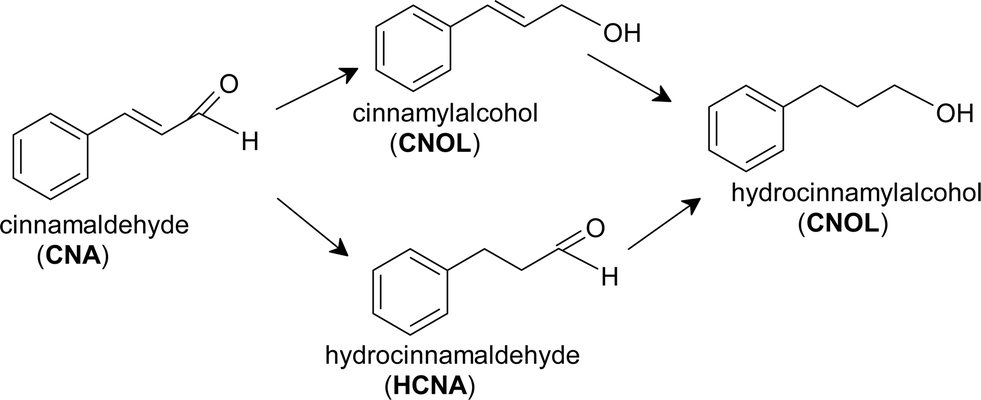 | ||
| Scheme 1 Reaction pathways for hydrogenation of cinnamaldehyde. | ||
Depending on the catalyst composition, different catalytic behaviors can be noted (Fig. 7). Therefore, NiAl-MO sample exhibited very high catalytic activity while CoAl-MO sample was moderately active. These results clearly show that a reduction temperature of 500 °C was sufficient to generate very active metallic sites in the case of NiAl-MO catalyst (complete conversion of CNA after 90 min of reaction), albeit the TPR results suggested that the nickel cations are not fully reduced to metallic nickel under these conditions. However, since the catalyst used in the catalytic runs were kept much longer under hydrogen at this reduction temperature, the degree of reduction is likely to be more elevated. For CoAl-MO sample, the generated metallic sites converted ~50% of CNA after 300 min of reaction. Moreover, it can be observed that the main product is HCNA on NiAl sample, while CNOL is the main reaction product on CoAl sample (Fig. 7). Also, the catalytic curves displayed in Fig. 7 show that in the case of NiAl-MO sample, a selectivity of ~95% HCNA can be achieved. The distribution of products is more equilibrated on CoAl-MO sample, with the highest selectivity obtained for CNOL at ~60% regardless of the CNA conversion.
It is well documented that the catalytic performance of a solid in the hydrogenation of CNA is influenced by several factors, between them the particle sizes and the electronic structure of the metallic active sites,1 both being considered to explain the catalytic results reported herein. As it was shown by in situ XRD, larger Ni0 nanoparticles are supposed to form in comparison with Co0 nanoparticles at similar reduction temperature. Although the calculation of the particle sizes after reduction at 500 °C was not feasible, the difference in size was suggested by the presence of the weak diffraction peaks of Ni0 and absence of those for Co0. The electronic structure of the metal is taken into consideration because it defines the interaction between the metal surface and the reactant molecule. Previous studies stated that nickel has similar behavior as Pd and Rh meaning that it favors the hydrogenation of CC bond while cobalt is included in the same group with Pt and Ru having a moderate selectivity to CNOL.55
From theoretical considerations56 and studies of the reaction mechanism,53 the formation of CNOL should be favored by enhancing the electron density of the metal that will determine the repulsive four electron interaction of the metal with the CC bond to decrease. The electron density can be correlated with the metal d-band width. Indeed, cobalt with larger d-band width than nickel (4.0 vs. 3.0 eV)57 is more selective toward the unsaturated alcohol. However, this factor cannot be independently discussed because the electron density depends also on the particle size. Thus, the electron density of the d-orbitals declines with the decrease of the particle size with direct consequence on the selectivity to CNOL. Nitta et al.58 found for supported Co nanoparticles prepared from cobalt chrysotile and cobalt precipitated on silica, that the selectivity to CNOL increases with the particle size, up to ~95 mol% for cobalt nanoparticle of ~24 nm in size. Similar size-dependent effects were observed in the case of platinum supported on carbon and graphite.55
For the catalytic results presented herein, the size of the cobalt nanoparticles lower than the detection limit of the XRD (<3 nm) should be taken into consideration to explain the moderate selectivity to CNOL (~60%) and the low selectivity to HCNA (~30%). It can be assumed that the adsorption of both double bonds is allowed on the small particles of cobalt but the adsorption of the CO bond is predominant. Indeed, Fig. 7 right shows that hydrogenation of CC and CO bonds takes place via parallel reactions, even when the total hydrogenation to HCNOL becomes obvious (i.e., at iso-conversion of ~30%).
On the other side, the adsorption of CNA on NiAl-MO sample predominantly occurs via CC bond, as also observed for highly dispersed nickel nanoparticles supported on SBA-15.59 In that case, the reduction of Ni/SBA-15 at 350 °C generated a catalyst that totally converted CNA in 300 min of reaction with a selectivity to HCNA of 94 mol% on the whole range of conversion. It can be concluded that the very high activity of NiAl-MO sample doubled by the outstanding selectivity to HCNA endorse this sample as a high-performance catalyst in the hydrogenation of CC double bond of cinnamaldehyde.
Due to the presence of alumina in the catalyst composition, it is interesting to observe if alumina has any influence on the catalytic behavior of the samples. On the basis of the literature reports, the influence of alumina on the catalytic performance of NiAl-MO and CoAl-MO samples is in some extent debatable because, depending on the nature and amount of the metallic active sites, the preparation method as well as the reaction conditions, different behaviors were noted. For instance, Volpe et al.60 reported that alumina support manifests the same effect on the catalytic behavior as silica. Thus, copper supported on alumina and silica by impregnation showed similar selectivity to CNOL (51–73 and 48–75% for Cu/Al2O3 and Cu/SiO2, respectively at 15–30% CNA conversion; the reaction was performed at 1 MPa and iso-propanol as solvent). Lashdaf et al.61 showed that platinum catalysts supported on silica by gas-phase deposition and impregnation are more active in the hydrogenation of CNA that those on alumina, prepared by the same routes (the reaction was performed at 10 bar and iso-propanol as solvent). In a study on alumina-supported platinum catalysts, Arai et al.62 found that the catalysts, reduced with sodium tetrahydroborate at 30 °C, display high selectivity to cinnamyl alcohol (78% at 50% CNA conversion) in comparison with the catalysts reduced with hydrogen at 400 °C, that did not display selectivity to cinnamyl alcohol. Szollozi et al.63 revealed that the activity and the selectivity to cinnamyl alcohol was lower with platinum on alumina (e.g., 32 × 103 mol g−1Pt h−1 and 47% CNOL) than platinum on silica (e.g., 72 × 103 mol g−1Pt h−1 and 79% CNOL), when the catalysts are submitted to sonication under hydrogen for 10 min previous the reaction. The next example is more suitable to our situation due to the similar reaction conditions. In this case, the comparison is made between copper supported on alumina by impregnation and Cu–Al mesoporous material prepared by direct synthesis.64 The authors found that the Cu-impregnated alumina exhibited selectivity to CNOL of 30% at 35% CNA conversion while for Cu–Al mesoporous catalysts, a selectivity to CNOL of 55% at the same iso-conversion was noted. On the basis of these studies, a beneficial effect of reduction from cation incorporated into the alumina lattice to generate strong metal interaction with residual alumina support to give improved selectivities can be pointed out.
According to TPR and in situ XRD after temperature-programmed reduction, the total reduction of cobalt takes place at 700 °C, which is the reason why the hydrogenation of cinnamaldehyde on CoAl-MO sample was also carried out after reduction at 700 °C. The conversion curve and selectivity to CNOL are displayed together with those recorded after reduction at 500 °C in Fig. 8. As observed, the activity is twice the activity obtained after reduction at 500 °C suggesting the generation of a higher number of active sites by reduction at higher temperatures (i.e., 700 °C). Also, the selectivity to CNOL increased by ~15% while the selectivity to HCNA decreased suggesting that the adsorption of the carbonyl bond is further favored over the olefin bond. This improvement in chemoselectivity can be related to the modification of adsorption site density with metal particle size. It is assumed that upon reduction of the sample at 700 °C the particle size slightly increased as compared to the sample reduced at 500 °C because of particle sintering. Indeed, previous studies demonstrated that as compared with small particles, large cobalt particles are more favourable to the CNA adsorption via CO bond due to a higher proportion of dense Co (111) faces.58,65
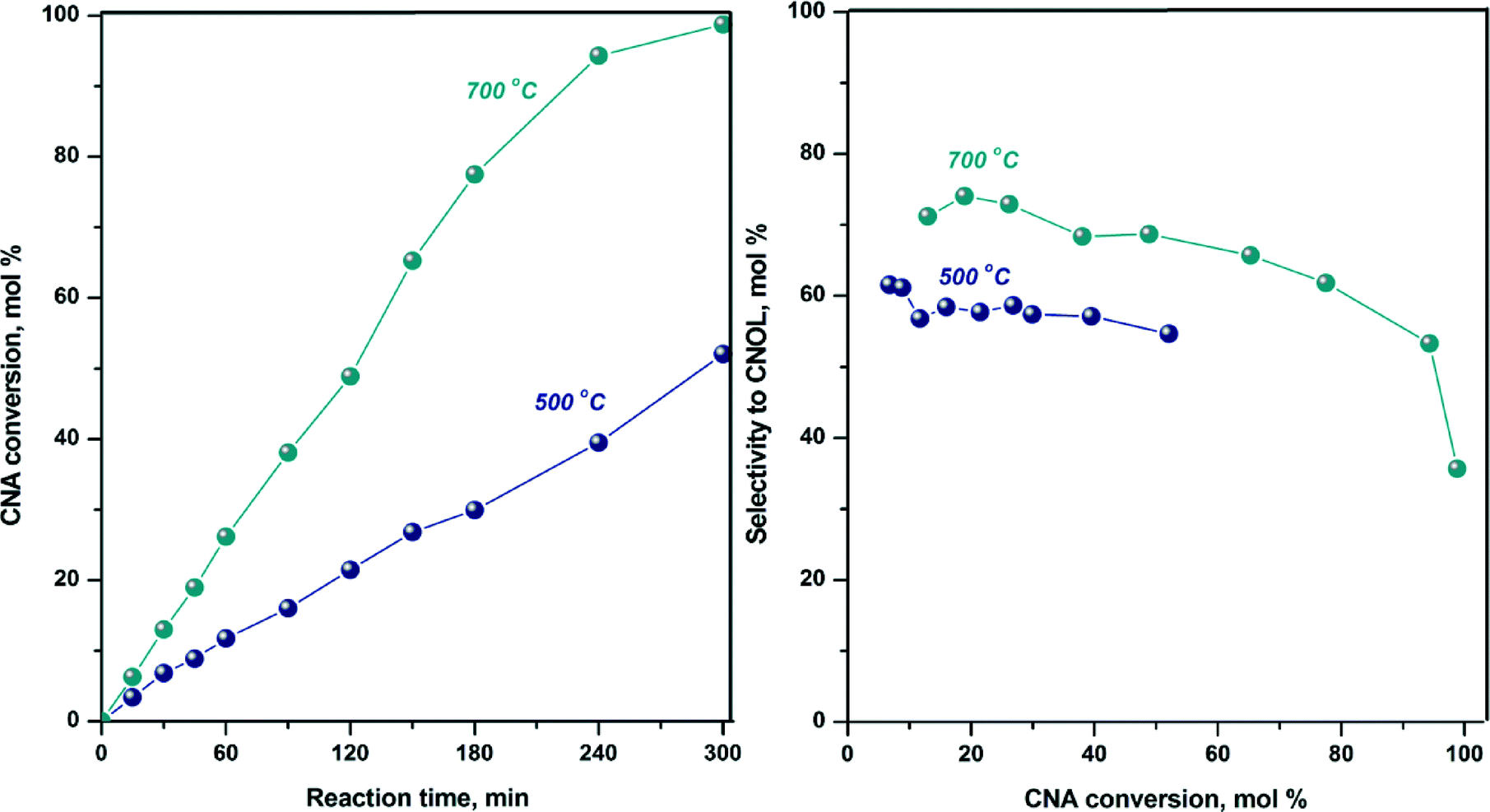 | ||
| Fig. 8 CNA conversion vs. time and selectivity to reaction products for CoAl-MO samples after reduction at 500 and 700 °C (Test conditions: Treaction = 150 °C, 0.265 g catalyst; 1 mL of CNA, 25 mL propylene carbonate as solvent, H2 flow = 1 L h−1, stirring rate = 900 rpm). | ||
It is also interesting to note that the high CNOL selectivity obtained over CoAl-MO is maintained up to high CNA conversions (Fig. 8, i.e., ~60% at 90% conversion), suggesting that during the reaction the adsorption of unsaturated alcohol on the catalyst surface is weaker than the adsorption of saturated or unsaturated aldehyde.
For a catalyst, the stability during several catalytic tests is very important for the economy of the process. Therefore, several catalytic runs with recycled catalysts were performed and the catalytic activities were evaluated (Fig. 9). It can be observed that the catalytic performances of both catalysts are not essentially changed over the three catalytic runs demonstrating their good stability and reusability.
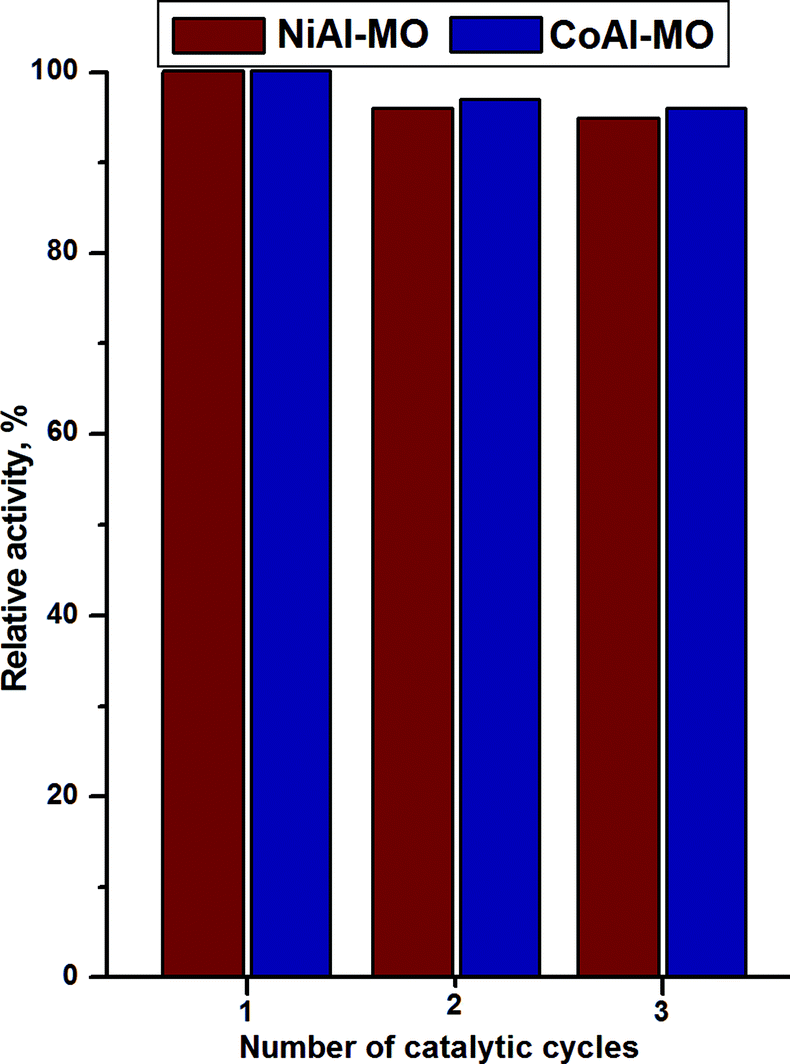 | ||
| Fig. 9 Recycling tests for NiAl-MO and CoAl-MO samples. | ||
As a conclusion, this study shows the interest to use binary precursors of NiAl and CoAl LDHs to prepare metallic catalysts with controlled and stable catalytic performances in terms of both activity and chemoselectivity.
Conclusions
Highly dispersed and thermostable nickel and cobalt nanoparticles showing high performance in cinnamaldehyde hydrogenation were prepared from NiAl takovite-like LDH precursors and related materials. The lamellar structure of the LDH precursors was confirmed by XRD while DR UV-vis indicated the incorporation of the two cations in octahedral coordination in the brucite-like sheets. The calcination of the samples generated external NiO and Co3O4 oxides phases, amorphous alumina and perhaps cobalt and nickel spinels. The TPR and in situ XRD results for the calcined NiAl sample indicated a strong interaction between nickel and alumina, which resulted in limited sintering of evolved nickel nanoparticles in the high-temperature range of 600–800 °C (i.e., the crystallite size of Ni0 nanoparticles only slightly increased with temperature from 3.6 to 5.8 nm). Likewise, a very strong interaction between cobalt and alumina was evidenced for CoAl sample, with no detectable sintering of generated cobalt nanoparticles in the high-temperature range (i.e., the crystallite size of Co0 nanoparticles was below the XRD detection limit (<3 nm) even after reduction at 800 °C). Materials derived from the studied LDH systems are found to be performance catalysts for the hydrogenation of cinnamaldehyde. Thus, at similar reduction temperature (i.e., 500 °C), NiAl catalyst presented much higher overall activity than CoAl, though the activity of CoAl can be enhanced by reduction at higher temperature (i.e., 700 °C). NiAl was highly chemoselective for the hydrogenation of CC bond (selectivity to hydrocinnamaldehyde of ~95% at complete conversion of cinnamaldehyde), whereas CoAl was chemoselective for the hydrogenation of CO bond (selectivity to cinnamyl alcohol of ~60% at ~50% conversion of cinnamaldehyde), indicative of composition-dependent chemisorption modes of CNA molecules on the catalytic active sites. Improvement in the chemoselectivity of CoAl to cinnamyl alcohol (~70% at ~50% conversion of cinnamaldehyde) was further achieved by reduction at 700 °C. Finally, it can be concluded that effective and low cost hydrogenation catalysts with chemoselective properties can be prepared from takovite-like LDH precursors and related materials.
Acknowledgements
This work was partially supported by a grant of the Romanian National Authority for Scientific Research, CNCS – UEFISCDI, project number PN-II-RU-TE-2012-3-0403. C. R. acknowledges the project CUANTUMDOC “Doctoral Studies for European Performances in Research and Innovation” ID79407 funded by the European Social Fund and Romanian Government.References
- P. Mäki-Arvela, J. Hajek, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2005, 292, 1 CrossRef PubMed.
- S. Handjani, E. Marceau, J. Blanchard, J.-M. Krafft, M. Che, P. Mäki-Arvela, N. Kumar, J. Wärn and D. Y. Murzin, J. Catal., 2011, 282, 228 CrossRef CAS PubMed.
- P. Zucca, M. Littarru, A. Rescigno and E. Sanjust, Biosci., Biotechnol., Biochem., 2009, 73, 1224 CrossRef CAS.
- US Pat. 5129951A, 1991 Search PubMed.
- X. Han, R. Zhou, B. Yue and X. Zheng, Catal. Lett., 2006, 109, 157 CrossRef CAS PubMed.
- H.-D. Jakubke and H. Jeschkeit, Concise Encyclopedia Chemistry, Walter de Gruyter & CO., Berlin, 1993, p. 506 Search PubMed.
- J. A. Goodall, J. Lyall, R. J. McBride, J. B. Murray and G. Smith, J. Clin. Pharm. Ther., 1980, 5, 323 CrossRef CAS.
- G. R. Bertolini, C. I. Cabello, M. Muñoz, M. Casella, D. Gazzoli, I. Pettiti and G. Ferraris, J. Mol. Catal. A: Chem., 2013, 366, 109 CrossRef CAS PubMed.
- V. R. Landaeta, F. López-Linares, R. Sánchez-Delgado, C. Bianchini, F. Zanobini and M. Peruzzini, J. Mol. Catal. A: Chem., 2009, 301, 1 CrossRef CAS PubMed.
- A. M. Raspolli Galletti, L. Toniolo, C. Antonetti, C. Evangelisti and C. Forte, Appl. Catal., A, 2012, 447–448, 49 CrossRef CAS PubMed.
- Y. Zhang, S. Liao, Y. Xu and D. Yu, Appl. Catal., A, 2000, 192, 247 CrossRef CAS.
- L. Zhang, J. M. Winterbottom, A. P. Boyes and S. Raymahasay, J. Chem. Technol. Biotechnol., 1998, 72, 264 CrossRef CAS.
- B. F. Machado, S. Morales-Torres, A. F. Pérez-Cadenas, F. J. Maldonado-Hódar, F. Carrasco-Marín, A. M. T. Silva, J. L. Figueiredo and J. L. Faria, Appl. Catal., A, 2012, 425–426, 161 CrossRef CAS PubMed.
- A. K. Prashar, S. Mayadevi and R. Nandini Devi, Catal. Commun., 2012, 28, 42 CrossRef CAS PubMed.
- X. Zhang, Y. C. Guo, Z. Cheng Zhang, J. S. Gao and C. M. Xu, J. Catal., 2012, 292, 213 CrossRef CAS PubMed.
- C. Milone, J. Catal., 2004, 222, 348 CrossRef CAS PubMed.
- J. Lenz, B. C. Campo, M. Alvarez and M. A. Volpe, J. Catal., 2009, 267, 50 CrossRef CAS PubMed.
- M. L. Toebes, F. F. Prinsloo, J. H. Bitter, A. Jos van Dillen and K. P. de Jong, J. Catal., 2003, 214, 78 CrossRef CAS.
- A.-M. Simion, T. Arimura and C. Simion, C. R. Chim., 2013, 16, 476 CrossRef CAS PubMed.
- B. M. Reddy, G. M. Kumar, I. Ganesh and A. Khan, J. Mol. Catal. A: Chem., 2006, 247, 80 CrossRef CAS PubMed.
- H. Li, H. Yang and H. Li, J. Catal., 2007, 251, 233 CrossRef CAS PubMed.
- W. Lin, H. Cheng, L. He, Y. Yu and F. Zhao, J. Catal., 2013, 303, 110 CrossRef CAS PubMed.
- R. S. Disselkamp, T. R. Hart, A. M. Williams, J. F. White and C. H. Peden, Ultrason. Sonochem., 2005, 12, 319 CrossRef CAS PubMed.
- K. J. A. Raj, M. G. Prakash, T. Elangovan and B. Viswanathan, Catal. Lett., 2011, 142, 87 Search PubMed.
- K.-Y. Jao, K.-W. Liu, Y.-H. Yang and A.-N. Ko, J. Chin. Chem. Soc., 2009, 56, 885 CAS.
- A. Ungureanu, D. Meloni, B. Dragoi, M. Casula, A. Chirieac, V. Solinas and E. Dumitriu, Environ. Eng. Manage. J., 2010, 4, 461 CAS ; B. Dragoi, A. Ungureanu, D. Meloni, M. Casula, A. Chirieac, A. Sasu, V. Solinas and E. Dumitriu, E. Environ. Eng. Manage. J., 2010, 9, 1203 Search PubMed; A. Chirieac, B. Dragoi, A. Ungureanu, A. Moscu, C. Rudolf, A. Sasu and E. Dumitriu, Environ. Eng. Manage. J., 2012, 11, 47 Search PubMed.
- J. Barrault, A. Derouault, G. Courtois, J. M. Maissant, J. C. Dupin, C. Guimon, H. Martinez and E. Dumitriu, Appl. Catal., A, 2004, 262, 43 CrossRef CAS PubMed.
- J. T. Kloprogge and R. L. Frost, in Layered Double Hydroxides: Present and Future, ed. V. Rives, Nova, New York, 2001, p. 164 Search PubMed.
- Ger. Pat 2 024 282, 1970 Search PubMed.
- B. Dragoi, A. Ungureanu, A. Chirieac, V. Hulea and E. Dumitriu, Acta Chim. Slov., 2010, 57, 677 CAS.
- J. M. Fernandez, C. Barriga, M. A. Ulibarri, F. M. Labajos and V. Rives, Chem. Mater., 1997, 9, 312 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Reed Educational and Professional Publishing Ltd., Oxford, 1997, pp. 1115–1148 Search PubMed.
- M. Crivello, C. Perez, E. Herrero, G. Ghione, S. Casuscelli and E. Rodriguez-Castellon, Catal. Today, 2005, 107, 215 CrossRef PubMed.
- H. T. Gomes, P. Selvam, S. E. Dapurkarc, J. L. Figueiredo and J. L. Faria, Microporous Mesoporous Mater., 2005, 86, 287 CrossRef CAS PubMed.
- M. J. Holgado, V. Rives and M. S. San Román, Appl. Catal., A, 2001, 214, 219 CrossRef CAS.
- Z. Sojka, F. Bozon-Verduraz and M. Che, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2nd edn, 2008, vol. 2, pp. 1039–1046 Search PubMed.
- L. Y. Mostovaya, T. S. Petkevich and L. A. Kupcha, J. Appl. Spectrosc., 1989, 51, 1298 CrossRef.
- M. Herrero, P. Benito, F. M. Labajos and V. Rives, J. Solid State Chem., 2007, 180, 873 CrossRef CAS PubMed.
- L. Ji, J. Lin and H. C. Zeng, J. Phys. Chem. B, 2000, 104, 1783 CrossRef CAS.
- K. Lee and W. Lee, Zeolitic Mater., 2002, 3, 8 Search PubMed.
- N. Sahli, C. Petit, A. C. Roger, A. Kiennemann, S. Libs and M. M. Bettahar, Catal. Today, 2006, 113, 187 CrossRef CAS PubMed.
- S. Velu, K. Suzuki, M. P. Kapoor, S. Tomura, F. Ohashi and T. Osaki, Chem. Mater., 2000, 12, 719 CrossRef CAS.
- V. Rives, M. A. Ulibarri and A. Montero, Appl. Clay Sci., 1995, 10, 83 CrossRef CAS.
- P. Benito, F. M. Labajos, J. Rocha and V. Rives, Microporous Mesoporous Mater., 2006, 94, 148 CrossRef CAS PubMed.
- V. Rives, in Layered Double Hydroxides: Present and Future, ed. V. Rives, Nova Science Publishers, New York, 2001, p. 243 Search PubMed.
- J. W. Lee, T. Ahn, J. H. Kim, J. M. Ko and J.-D. Kim, Electrochim. Acta, 2011, 56, 4849 CrossRef CAS PubMed.
- X. Ke, J. Cao, M. Zheng, Y. Chen, J. Liu and G. Ji, Mater. Lett., 2007, 61, 3901 CrossRef CAS PubMed.
- M. A. Ulibarri, J. M. Fernandez, F. M. Labajos and V. Rives, Chem. Mater., 1991, 3, 626 CrossRef CAS.
- Z. Wen, F. Zheng, Z. Jiang, M. Li and Y. Luo, J. Mater. Sci., 2013, 48, 342 CrossRef CAS.
- Z. Li, X. Xiang, L. Bai and F. Li, Appl. Clay Sci., 2012, 65–66, 14 CrossRef CAS PubMed.
- J. Newnham, K. Mantri, M. H. Amin, J. Tardio and S. K. Bhargava, Int. J. Hydrogen Energy, 2012, 37, 1454 CrossRef CAS PubMed.
- P. Claus, Top. Catal., 1998, 5, 51 CrossRef CAS.
- C. Mohr and P. Claus, Sci. Prog., 2001, 84, 311 CrossRef CAS.
- P. Claus and Y. Onal, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2nd edn, 2008, vol. 2, pp. 3316–3319 Search PubMed.
- F. Delbeq and P. Sautet, J. Catal., 1995, 152, 217 CrossRef.
- U. K. Singh and M. A. Vannice, J. Catal., 2001, 199, 73 CrossRef CAS.
- Y. Nitta, K. Ueno and T. Imanaka, Appl. Catal., 1989, 56, 9 CrossRef CAS.
- A. Ungureanu, B. Dragoi, A. Chirieac, C. Ciotonea, S. Royer, D. Duprez, A. S. Mamede and E. Dumitriu, ACS Appl. Mater. Interfaces, 2013, 5, 3010 CAS.
- V. Gutierrez, M. Alvarez and M. A. Volpe, Appl. Catal., A, 2012, 413–414, 358 CrossRef CAS PubMed.
- M. Lashdaf, J. Lahtinen, M. Lindbla, T. Venalainen and A. O. I. Krause, Appl. Catal., A, 2004, 276, 129 CrossRef CAS PubMed.
- M. Arai, K. Usui and Y. Nishiyama, Chem. Commun., 1993, 1853 RSC.
- G. Szollosi, B. Torok, G. Szakonyi, I. Kun and M. Bartok, Appl. Catal., A, 1998, 172, 225 CrossRef CAS.
- S. Valange, A. Derouault, J. Barrault and Z. Gabelica, J. Mol. Catal. A: Chem., 2005, 228, 255 CrossRef CAS PubMed.
- C. Ciotonea, B. Dragoi, A. Ungureanu, A. Chirieac, S. Petit, S. Royer and E. Dumitriu, Chem. Commun., 2013, 49, 7665 RSC.
| This journal is © The Royal Society of Chemistry 2014 |