Characterization and performance of electrostatically adsorbed Ru–Hbpp water oxidation catalysts†
Joan
Aguiló
a,
Laia
Francàs
b,
Hai Jie
Liu
a,
Roger
Bofill
a,
Jordi
García-Antón
a,
Jordi
Benet-Buchholz
b,
Antoni
Llobet
*ab,
Lluís
Escriche
*a and
Xavier
Sala
*a
aDepartament de Química, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, 08193, Barcelona, Spain. E-mail: lluis.escriche@uab.cat; xavier.sala@uab.cat; Fax: + 34 93 581 3101; Tel: +34 93 586 8295
bInsititute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans, 16, E-43007, Tarragona, Spain. E-mail: allobet@iciq.es; Fax: +34 977 920 222; Tel: +34 977 920 201
First published on 30th October 2013
Abstract
A new family of Ru–Hbpp dinuclear complexes containing the positively charged terpyridine derivative ligand 4-(-p-(pyridin-1-ylmethyl)phenyl)-2,2′:6′,2′-terpyridine of general formula {[Ru(L1+)]2(μ-bpp)(L-L)}m+ (L-L = μ-Cl, μ-acetato, or (H2O)2; m = 4 or 5) have been synthesized and fully characterized, both in the solid state (X-ray diffraction) and in solution (1D and 2D NMR spectroscopy, UV–vis spectroscopy and electrochemical techniques). New hybrid materials have been prepared by the electrostatic interaction of these complexes with several oxidatively rugged solid supports such as SiO2, FTO–TiO2 and FTO–Nafion®. These new hybrid materials were prepared and catalytically evaluated with regard to their capacity to chemically and electrochemically oxidize water to dioxygen.
Introduction
The use of stable redox-active catalysts such as ruthenium mono- or dinuclear complexes is an interesting strategy in order to succeed in water oxidation catalysis.1 Our group has developed several complexes of this type (mainly the so-called Ru–Hbpp family, see Chart 1) presenting a relatively good performance with regard to this challenging reaction. Furthermore, most of these oxygen-evolving species have been mechanistically investigated.2 In terms of catalyst stability, one of the common deactivation pathways operating in the above mentioned systems is the catalyst–catalyst intermolecular oxidative degradation, which generates non-active species and/or CO2 as ligand degradation products.2b,3 The immobilization of these catalysts onto conductive/semiconductive solid supports has been one of the used strategies to overcome their self-degradation. In addition, the use of conductive materials allows the electrochemical activation of the catalysts and represents a step forward towards the feasible construction of a fuel-cell for the photo-production of hydrogen.3 Our first approach towards the preparation of heterogeneous water oxidation catalysts (WOCs) dates from 2008 with the electropolymerization of a pyrrole-modified Ru–Hbpp system (Chart 1, B) onto vitreous carbon sponges (VCS) and fluorinated tin oxide electrodes (FTO).4 This system confirmed the feasibility of the solid-state approach, since it was able to perform the catalytic oxidation of water to dioxygen through electroactivation at 1.17 V vs. SSCE. However, despite the observed increase in robustness, these hybrid materials suffered from the oxidation of the polymeric matrix within the harsh catalytic conditions employed. | ||
| Chart 1 Drawing of previously reported Ru–Hbpp complexes (A, B and C) and Hbpp and trpy ligands together with their modified counterparts, including the L1+ ligand used in this work. | ||
Soon afterwards, we further modified the Ru–Hbpp catalyst through the introduction of a carboxylate functional group into the bridging ligand (Chart 1, C). That allowed its anchoring onto a more rugged, inorganic solid support such as nanoparticulated TiO2.5 The chemical (Ce(IV)) mediated activation of this system ended up with the concomitant generation of O2 and CO2, the latter due to the intermolecular ligand oxidation beginning at the benzylic position of the modified Hbpp ligand.
Taking into account all the aforementioned results presented above we envisaged a new strategy based on the electrostatic interaction between different solid supports/electrodes and positively charged Ru–Hbpp catalytic species. With this aim, herein we present the synthesis, characterization and catalytic activity of a new catalyst belonging to the Ru–Hbpp family containing extra and positively charged pyridylic rings on the trpy ligands (Chart 1, L1+). Furthermore, we report its anchoring onto TiO2, SiO2 and Nafion® surfaces, the characterization of the new hybrid materials generated and their catalytic evaluation with regard to the oxidation of water to dioxygen.
Results and discussion
Synthesis and structural characterization
The synthesis of the L1+ ligand (Chart 1, bottom-right) was carried out by slightly modifying a two-step procedure previously reported.6,7 After bromination of 4′-(4-methylphenyl)-2,2′:6′,2′-terpyridine with N-bromosuccinimide (NBS), the bromomethyl derivative obtained was subjected to a nucleophilic attack with pyridine, which generated the cationic L1+. The use of a microwave reactor in the last step allowed shorter reaction times (from 48 h to 30 min) and better yields (from 70% to 80%). After solvent evaporation and redissolution in water, the pure ligand was obtained by precipitation with NH4PF6(aq) and characterized by 1D and 2D NMR (Fig. S1, ESI†).Reaction of RuCl3·nH2O with L1+ in refluxing methanol for four hours afforded [RuCl3(L1+)](PF6), 1(PF6), as a brown solid. Further combination of the latter with the anionic tetra-N-dentate bpp− bridge (bpp− = 3,5-bis(2-pyridyl)pyrazolate) under the conditions shown in Scheme 1 leads to the binuclear ruthenium complexes here reported, 2(PF6)4, 3(PF6)4 and 45+. Under excess of acetate and the presence of stoichiometric Ag+ ions, the Cl-bridging ligand of 24+ can be easily replaced by an acetato-bridging ligand, leading to 34+. The latter is then replaced by two aqua ligands under acidic conditions to yield 45+. All of the isolated ruthenium complexes were characterized by elemental analysis and spectroscopic (UV–vis and NMR), spectrometric (MS) and electrochemical (CV, DPV) techniques.
 | ||
| Scheme 1 Synthetic strategy for the complexes described in this work. | ||
X-ray diffraction analysis was also carried out for complexes 1(PF6) and 2(PF6)4. Their crystallographic and acquisition parameters are reported in Tables S1 and S2 (ESI†), and ORTEP views of their cationic moieties are presented in Fig. 1. Single crystals of 1(PF6) were obtained by the slow evaporation of a saturated solution of 1+ in nitric acid. A selection of the more relevant bond distances and angles is reported in Table S3.† As expected, the ruthenium center is meridionally coordinated by the trpy-based ligand L1+, and three chloride anions occupy the three remaining coordination sites. The ruthenium ion adopts a distorted octahedral geometry with bond distances and angles comparable to analogous complexes reported earlier in the literature.8 The constrain imposed by the geometry of the L1+ ligand is clearly detected in the N3–Ru–N1 angle, reduced from the ideal 180° value to the observed 159°. 1+ crystallizes with one nitrate anion and one nitric acid molecule linked by a hydrogen bond. Both molecules are situated next to the pyridinium positive charge. In contrast to what was reported for the parent [RuCl3(trpy)] complex,8 almost no hydrogen bonding interactions and a less ordered packing are observed now. The unit cell of the structure is shown in Fig. S2 (ESI†) containing a total of eight 1+ cations. A head to tail orientation between the two central molecules of the unit cell is also observed. Concerning 2(PF6)4, because of the packing effect imposed by the pyridinium moiety of L1+, this complex crystallizes in an extremely large cell containing four independent complex molecules, sixteen PF6− anions and several disordered acetone and toluene molecules in the asymmetric unit (Fig. S3†). In order to avoid the disordered solvent molecules the SQUEEZE program9 was applied, leading to a refined model with a R1 value of 7.43%. The four cationic 24+ units described in the asymmetric cell display slightly different metric parameters due to the different orientation of the pyridinium moieties, and therefore only the so-called “A” complex will be described here. A selection of the more relevant bond distances and angles for all four independent molecules is reported in Table S4.† Each ruthenium atom adopts a pseudo-octahedral coordination geometry with two positions occupied by the bpp− ligand, three by the meridional L1+ ligand and the last one by a Cl-bridged ligand (Fig. 1, bottom). Bond distances and angles show no significant differences with regards to related complexes previously described in the literature.10 1D and 2D NMR spectroscopy allowed the structural characterization in solution of the isolated diamagnetic complexes (see Fig. 2, the Experimental section, and Fig. S4 and S5†). All of the resonances observed in the NMR spectra can be unambiguously assigned based on their integrals, symmetry and multiplicity. 24+ displays C2v symmetry in solution, with one symmetry plane containing the bpp− ligand, the two Ru atoms, both central terpyridine nitrogen atoms and the bridging chlorido moiety. A second plane (perpendicularly bisecting the former) passes through the chlorido bridge and the central pyrazolic carbon and bisects the N–N bond of the same pyrazolic ring, thus interconverting the two terpyridine ligands. The downfield shift of the singlet corresponding to H22 (see Fig. 2) is in accordance with the high electron-withdrawing effect of the closer pyridinium moiety, as also previously reported for related compounds.11 When the acetato-bridged complex 34+ is analyzed in solution, the NMR resonances of the external pyridyls of the L1+ ligands appear as magnetically symmetric at room temperature. In the solid state, the accommodation of the acetato bridging ligand should provoke a further distortion of the Ru pseudo-octahedral geometry, as previously reported for the parent {[RuII(trpy)]2(μ-bpp)(μ-AcO)}2+,12 (resulting in one Ru center moving above the equatorial plane together with its L1+ ligand, whereas the other metal center does the opposite). However, in solution at room temperature, these two moieties display a dynamic behavior, with the two Ru centers synchronically moving very fast below and above the equatorial plane. Consequently, the NMR resonances appear as if the complex had C2v symmetry. Furthermore, the downfield shift of the methylenic singlet with regards to 24+ (from 6.2 to 6.3 ppm) and the presence of the acetate singlet peak at 0.45 ppm (Fig. S5a†) are experimental evidences of the successful exchange of the chlorido bridge by the acetato moiety.
 | ||
| Fig. 1 ORTEP plot (ellipsoids at 50% probability) of the X-ray crystal structure of the cationic moiety of (top) 1+ and (bottom) 24+ (molecule A) and their corresponding labelling scheme. | ||
 | ||
| Fig. 2 Drawn structure with an atom labelling scheme and 1H NMR (acetone-d6) for 24+. | ||
Spectroscopic and redox properties
The UV–vis spectral features in acetone for the complexes described in this work are listed in the Experimental section, and the UV–vis spectra for 24+, 34+ and 45+ are displayed in Fig. 3. Two main regions can be distinguished: one between 350 and 550 nm, in which there are mainly broad unsymmetrical Ru(dπ)–trpyPyr/bpp(π*) metal-to-ligand charge-transfer bands; and the region above 550 nm, in which d–d transitions are observed.13 The MLCT absorption features shown in Fig. 3 nicely fit with those of previously reported chlorido, acetato and bis-aqua Ru–Hbpp dinuclear complexes.2b,14 | ||
Fig. 3 UV-spectra for 24+ (blue line) and 34+ (green line) at 26 mM concentration in acetone and 45+ (96 mM, red line) in acetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (pH = 1) (20:80). :water (pH = 1) (20:80). | ||
The redox properties of the complexes described in the present work were investigated by means of CV and DPV and are reported in Table 1, Fig. 4 and Fig. S7–S8 in the ESI.† The CV of 1+ in MeCN (Fig. S6†) exhibits a unique reversible wave at E1/2 = 0.05 V (ΔE = 71 mV), corresponding to the following process:
| [RuIIICl3(L1+)]+ + 1e− → [RuIICl3(L1+)] (0.05 V) | (1) |
| III/II | ||||
|---|---|---|---|---|
| a In acetonitrile using 0.1 M of TABH as the electrolyte. b In acetone using 0.1 M of TABH as the electrolyte. c In CH2Cl2 using 0.1 M of TABH as the electrolyte. d Aqueous solution at pH = 1 (0.1 M triflic acid). | ||||
| [RuIIICl3(trpy)]a | 0.01 | |||
| 1+ | 0.05 | |||
| III,II | III,III | IV,III | IV,IV | |
| / | / | / | / | |
| II,II | III,II | III,III | IV,III | |
| {[RuII(trpy)]2(μ-bpp)(μ-Cl)}2+c | 0.71 | 1.12 | — | — |
| 24+ | 0.79 | 1.20 | — | — |
| {[RuII(trpy)]2(μ-bpp)(μ-AcO)}2+c | 0.73 | 1.05 | — | — |
| 34+ | 0.76 | 1.09 | — | — |
| {[RuII(H2O)(trpy)]2(μ-bpp)}3+d | 0.54 | 0.61 | 0.81 | 1.10 |
| 45+ | 0.57 | 0.63 | 0.90 | 1.00 |
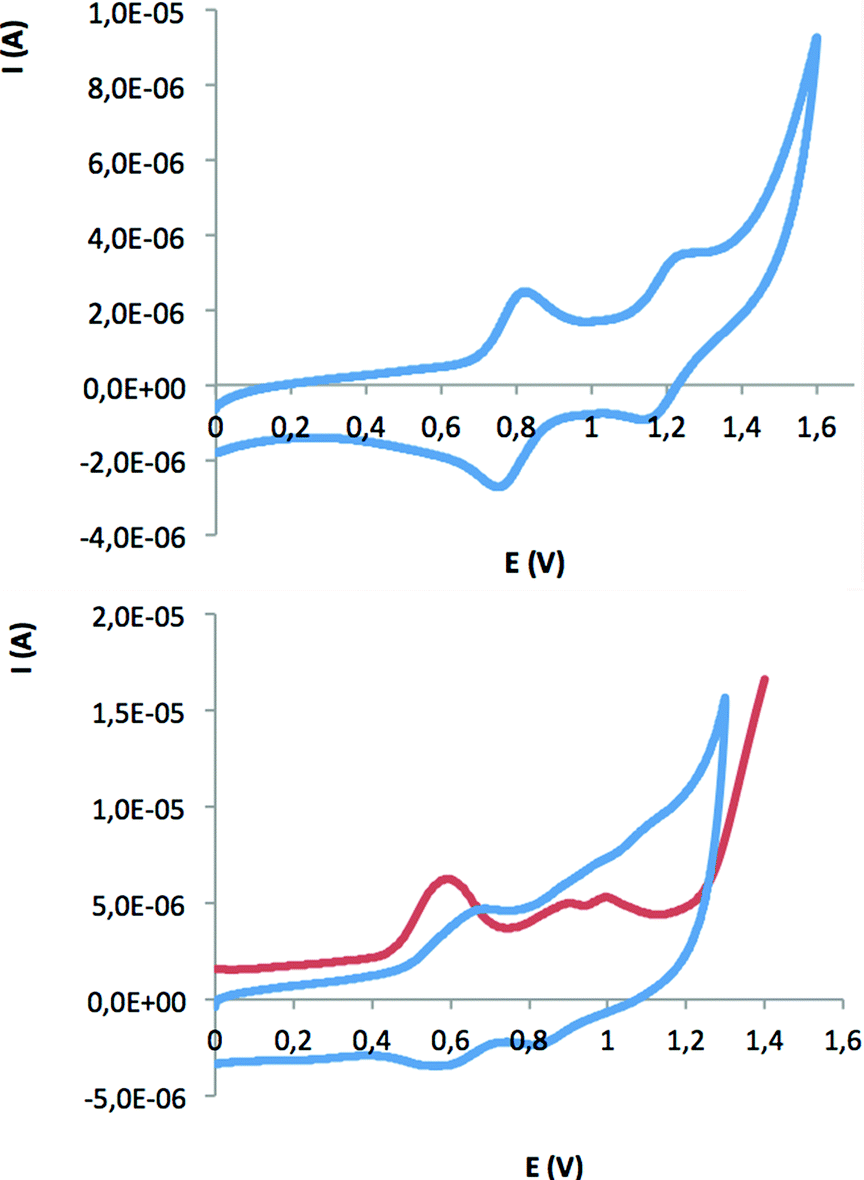 | ||
| Fig. 4 Cyclic voltammogram for the chlorido-bridged complex 24+ in 0.1 M n-Bu4NPF6 in acetone at a 100 mV s−1 scan rate (top) and differential pulse voltammogram (red line) and cyclic voltammogram (blue line) for the aqua complex 45+ at pH = 1 in a 0.1 M triflic acid aqueous solution (bottom). In both cases glassy carbon is used as the working electrode and the potential is measured vs. SSCE. | ||
Comparison with the related complex [RuIIICl3(trpy)] (Fig. S7,†E1/2 = 0.01 V, ΔE = 69 mV) revealed an up shift of the E1/2. This behavior can be assigned to the electron-withdrawing effect of the extra pyridinium group of L1+. For the dinuclear Cl− and AcO− complexes that do not contain aqua groups, the voltammograms in organic solvents show two chemically reversible and electrochemically quasi-reversible redox waves. Fig. 4, top, shows the CV of complex 24+ in acetone (see Fig. S8† for the CV of 34+). These two processes are assigned to the following electrochemical reactions (the L1+ and bpp− ligands are not shown for the sake of clarity):
| [RuIII(μ - Cl)RuII]5+ + 1e− → [RuII(μ - Cl)RuII]4+ (0.79 V) | (2) |
| [RuIII(μ - Cl)RuIII]6+ + 1e− → [RuIII(μ - Cl)RuII]5+ (1.20 V) | (3) |
The electrochemistry of 45+ has been investigated after its “in situ” generation in a pH 1 aqueous solution (0.1 M triflic acid) using 34+ as precursor (see Scheme 1). From the CV and DPV of 45+ (Fig. 4, bottom) a total of four waves are observed. These have been tentatively assigned, taking into account previous results on related complexes,10 to a total of four redox processes:
| [RuIII–RuII] + 1e− → [RuII–RuII] (0.57 V) | (4) |
| [RuIII–RuIII] + 1e− → [RuIII–RuII] (0.63 V) | (5) |
| [RuIV–RuIII] + 1e− → [RuIII–RuIII] (0.90 V) | (6) |
| [RuIV–RuIV] + 1e− → [RuIV–RuIII] (1.00 V) | (7) |
As can be observed in Table 1, in this case no significant changes are observed when comparing the potentials of 45+ with regard to those of the related Hbpp complex {[RuII2(H2O)2(trpy)2](μ-bpp)}3+, particularly when comparing the high oxidation states. When the potential is increased further up to 1.3 V a large anodic current is observed in the DPV, which can be associated with a further one electron oxidation of the complex together with the concomitant electrocatalytic oxidation of water to dioxygen in agreement with eqn (8) and (9).
{O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) RuV–RuIVO}6+ + 1e− → {ORuIV–RuIVO}5+ RuV–RuIVO}6+ + 1e− → {ORuIV–RuIVO}5+ | (8) |
| {ORuV–RuIVO}6+ + H2O → {HO–RuIV–RuIII–OOH}6+ | (9) |
The hydroperoxide intermediate is then responsible for the subsequent reactions that end up generating dioxygen. The oxidation process depicted in eqn (8) is not observed in the DPV (Fig. 4, bottom) given the concomitant and fast electrocatalytic current corresponding to the oxidation of water.
Attachment and characterization of Ru catalysts onto SiO2, FTO–TiO2 and FTO–Nafion®
The charged pyridinium rings of complexes 24+ and 34+ have been employed to electrostatically interact with several solid supports and thus generate the following new hybrid materials: SiO2–24+/34+: the attachment of 24+ and 34+ onto SiO2 was carried out by introducing a sample of SiO2 (3 g) to 0.548 mM (10 mL) acetone solutions of 24+/34+. In just 5 min, the solutions decolorized indicating the total adsorption of the complexes into SiO2, thus generating SiO2–24+ and SiO2–34+. These new materials were thoroughly washed with fresh acetone and water and finally air-dried at room temperature. The absence of UV–vis signal in the washing acetone revealed the stability of the new hybrid materials (see Fig. S9 in ESI†) and the diffuse reflectance UV–vis spectra shown in Fig. S10† confirm the structural analogy of the new hybrid materials with regards to their homogeneous counterparts.FTO–TiO2–24+: FTO–TiO2 films were prepared as usual (see the Experimental section) and then soaked overnight in a 0.3 mM acetone solution of 24+. The FTO–TiO2–24+ films were then thoroughly washed with fresh acetone and air-dried. The amount of anchored catalyst was confirmed by means of UV–vis spectroscopy. The use of films previously activated at pH = 12 (soaked overnight on a 10 mL aqueous solution at pH 12) ended up in higher amounts of anchored catalyst (see Fig. S11 in the ESI†). We propose that, under these conditions, TiO− residues are generated on the surface of the solid support, thus allowing a better support–catalyst ionic interaction. The electrochemical properties of FTO–TiO2–24+ have been also investigated by means of CV in DCM. The corresponding voltammograms at different scan rates are shown in Fig. S12 (ESI†). This new hybrid material presents similar redox potentials (0.81 V and 1.18 V vs. SSCE) to those found for complex 24+ (0.79 and 1.20 V vs. SSCE), revealing that no changes in the electrochemical (and thus structural) properties of the catalyst have occurred during the anchoring process. Later on, the stability of the FTO–TiO2–24+ films was evaluated under acidic (pH 1), neutral (pH 7) and basic (pH 12) conditions by monitoring the potential catalyst leaching via UV–vis spectroscopy. The spectra of those solutions along several hours/days are displayed in Fig. S13.† The observed absorbance increase with time of the acidic and neutral solutions containing soaked FTO–TiO2–24+ films points out the instability of the ionic interaction at these pH ranges. Surprisingly, a detachment/reattachment process of 24+ is observed when the electrode is soaked at pH 12 (Fig. S13c†). The early absorbance increase reveals an initial leaching that, after three days, is clearly reversed. The latter behavior can be due to the activation of the TiO2 surface (generation of TiO− anions at basic pH) as previously indicated.
FTO–Nafion–24+/34+: the absence of easily oxidizable –CH groups and its terminal and deprotonable sulfonate groups converts Nafion® into an excellent polymeric material to electrostatically interact with the positive residues of complexes 24+ and 34+ and thus generates rugged hybrid materials useful as catalysts for the oxidation of water. FTO–Nafion films have been prepared depositing a known volume of the Nafion® solution (Nafion 5% w/w in a mixture of water and low-weight alcohols) on a piece of FTO film, as shown in Scheme S1 in the ESI.† The films are then oven-dried at 100 °C for 30 min. After cooling at room temperature, the FTO–Nafion supports are soaked overnight into acetone solutions of 24+ or 34+. The final colorless nature of the solution after that time clearly points out to a complete attachment of the catalyst onto the Nafion® polymer. The new hybrid materials FTO–Nafion–24+ and FTO–Nafion–34+ have been electrochemically characterized by means of CV and DPV (Fig. 5) and their redox potentials have been compared with their homogeneous counterparts. The CV of FTO–Nafion–24+, shown in Fig. 5 (top), displays two chemically reversible redox waves at E1/2 = 0.64 V (ΔEp = 113 mV) and at E1/2 = 1.05 V (ΔEp = 113 mV) that are assigned to the RuIII/RuII → RuII/RuII and RuIII/RuIII → RuIII/RuII couples.
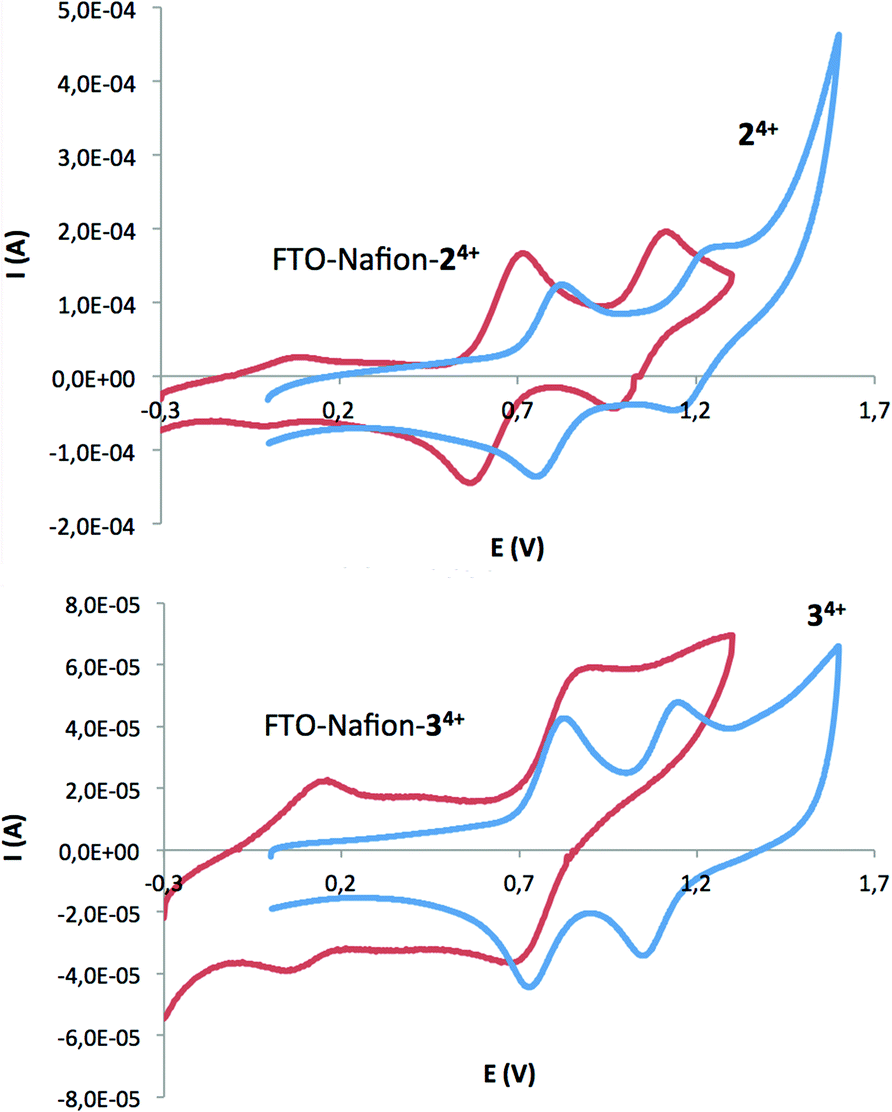 | ||
| Fig. 5 CV for the chlorido-bridged complex 24+ (blue line, signal increased 50 times for comparison purposes) and FTO–Nafion–24+ (red line) in a 0.1 M n-Bu4NPF6 acetone solution at 100 mV s−1 scan rate (top). CV for the acetato-bridged complex 34+ (blue line, signal increased 10 times for comparison purposes) and FTO–Nafion–34+ (red line) in a 0.1 M n-Bu4NPF6 acetone solution at 100 mV s−1 scan rate (bottom). A glassy carbon electrode for 24+ and 34+ and FTO for FTO–Nafion–24+ and FTO–Nafion–34+ were used as working electrodes. The potential was measured vs. SSCE. | ||
A downshift of about 100 mV in E1/2 is observed when comparing FTO–Nafion–24+ with its homogeneous counterpart, 24+. This behavior can be explained by the less electron-withdrawing effect of the pyridinium salts when its positive charge ionically interacts with the Nafion sulfonate residues (see a drawing of this interaction in Scheme S2 in the ESI†). The UV–vis spectrum of FTO–Nafion–24+ (Fig. S14, ESI†) perfectly matches that of its homogeneous counterpart (see Fig. 3 above) and further confirms the preservation of the Ru coordination sphere along the anchoring process. On the other hand, the CV of FTO–Nafion–34+ shown in Fig. 5 (bottom) displays two chemically reversible redox waves at E1/2 = 0.10 V (ΔEp = 115 mV) and at E1/2 = 0.79 V (ΔEp = 200 mV) that are assigned to the RuIII/RuII → RuII/RuII and RuIII/RuIII → RuIII/RuII couples. The clear and large downshift of both redox waves (660 mV and 300 mV, respectively) when compared with the ones observed for its homogeneous counterpart 34+ suggests changes in the first coordination sphere of the ruthenium metal ions during the anchoring process towards a significantly stronger electron-donating environment. Thus, the coordination of at least one sulfonate residue to the Ru metal center is proposed.
Chemically triggered oxidative catalysis
Complex 45+ and the hybrid materials SiO2–45+ and FTO–Nafion–45+ have been tested as WOCs upon the addition of a strong sacrificial oxidant such as Ce(IV). All these bis-aqua species have been generated “in situ” from 34+, SiO2–34+ and FTO–Nafion–24+, respectively, when poured into the acidic catalytic conditions (see the Experimental section). The total gas evolved has been manometrically measured and its composition in terms of O2:CO2 ratio analyzed by means of mass spectrometry (45+ and SiO2–45+) or a Clark electrode in solution (FTO–Nafion–45+). The overall catalytic results, together with the ones corresponding to related systems for comparison purposes, are displayed in Table 2.
:CO2 ratios for 45+, silica–45+, FTO–Nafion–45+ and other previously reported homogeneous and heterogeneous systems for comparison purposes. Catalytic conditions: Ce(IV) as oxidant. Ratio 1/100 cat./Ce(IV)
| Entry | System | [Cat.] (mM) | TN O2 | TN CO2 | [O2]/[CO2] | TN total | Eff.c | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Total volume of the reaction: 2 mL at pH = 1.0 in 0.1 M triflic acid. b Total volume of the reaction: 4 mL at pH = 1.0 in 0.1 M triflic acid. c Eff. = efficiency. d 0.350 g of SiO2–34+ (0.3%). e Total volume of the reaction: 3 mL at pH = 1.0 in 0.1 M triflic acid. f 1/200 cat./Ce(IV). tw = this work. | ||||||||
| 1 | {[Ru(H2O)(trpy)]2(μ-bpp)}3+ | 1.0a | 18 | — | — | 16 | 72.0 | 9 |
| 2 | {[RuII(H2O)(trpy)]2(μ-bpp-Bz)}3+ | 1.0a | 5.8 | 5.8 | 1.0 | 11.7 | 29.2 | 1b |
| 3 | {[RuII2(H2O)2(trpy)2]2(μ-(bpp)2-o-xyl)}6+ | 0.5af | 18.4 | 6.1 | 3.0 | 20.8 | 46.7 | 1b |
| 4 | {[RuII2(H2O)2(trpy)2]2(μ-(bpp)2-m-xyl)}6+ | 0.5af | 15.6 | 6.2 | 2.5 | 19.9 | 45.5 | 1b |
| 5 | {[RuII2(H2O)2(trpy)2]2(μ-(bpp)2-p-xyl)}6+ | 0.5af | 14.8 | 5.8 | 2.5 | 21.9 | 48.9 | 1b |
| 6 | 45+ | 1.0a | 12 | — | — | 12 | 44 | tw |
| 7 | SiO2–45+ | 0.3%ad | 1.8 | 14.2 | 0.125 | 16 | 7.2 | tw |
| 8 | TiO2–{[RuII(H2O)(trpy)]2(μ-bpp-Ra)}3+ | 0.5b | 3.5 | 3 | 1.2 | 6.5 | 55 | 4 |
| 9 | FTO–Nafion–45+ | 0.06e | 1 | — | — | 1 | 4 | tw |
When Ce(IV) is added to a pH 1 (0.1 M triflic acid) solution of 45+, a TN value of 12 is achieved (Table 2, entry 6 and Fig. S15a of the ESI†). Comparison with the parent {[RuII(H2O)(trpy)]2(μ-bpp)}3+ complex (entry 1, 18 TN)10 revealed a slight decrease in the catalytic activity of the modified complex. This decrease in the catalytic activity is in agreement with previous results attained by our research group.15 The electronic modification of Ru–Hbpp WOCs by introducing electron-withdrawing substituents into their surrounding ligands (as here is the case with the L1+ ligand) resulted in low substitution reaction rates and higher redox potentials, thus complicating the attainment of high oxidation states. Therefore, a slow down of the catalytic processes and a reduction of their efficiency are observed.15 On the other hand, no CO2 generation is detected when the catalytic process with 45+ is on-line monitored by mass spectrometry (Fig. S15b† and Table 2, entry 6), which is in sharp contrast with the significant CO2 values reported for several structurally related complexes such as {[RuII(H2O)(trpy)]2(μ-bpp-Bz)}3+ (Table 2, entry 2) and their tetranuclear {[RuII2(trpy)2(H2O)2]2(μ-(bpp)2-L-xyl)}6+ (L = ortho, meta or para) and heterogenous derivatives TiO2–{[RuII(H2O)(trpy)](μ-bpp-Ra)}3+ (Table 2, entries 3–5 and 8), all of them containing methylenic units on the modified bpp− ligands (for a drawing of several of these complexes, see Scheme S3 in the ESI†). The CO2 generation on these kinds of complexes is associated with a bimolecular reaction where oxidation of the methylenic unit of the ligands is carried out by an active Ru–O group of another molecule of the catalyst. This is the only possible pathway because Ce(IV) does not react with the free ligands at room temperature and the geometrical disposition of the Ru–O units prevents an intramolecular ligand oxidation.5 Therefore, the proximity of the positively charged pyridinium rings to the methylenic groups of the L1+ ligand of complex 45+ is key to electronically disfavor the bimolecular oxidative degradation in this particular case. Contrarily, in the SiO2–45+ system this positive charge is delocalized due to the ionic interaction with the SiO− residues of the solid support. Consequently, the methylene group in alpha position to the pyridinium nitrogen atom is here again more prone to oxidation, thus decomposing and evolving CO2 within the catalytic conditions (Fig. S15c–d,†Table 2, entry 7, and Scheme S3†). The chemically triggered catalytic activity of the bis-aqua FTO–Nafion–45+ system has been analyzed after activation of FTO–Nafion–24+ films for 24 h at pH = 1.0 in 0.1 M triflic acid (see below for the neat conversion to the bis-aqua derivative under these conditions). Surprisingly, a very low TN value of about 1 (Table 2, entry 9, and Fig. S16†) was obtained. Given the polymeric nature of the Nafion-based hybrid material, the encapsulation of the active sites within the inter-winkled polymeric chains and therefore their difficult interaction with the Ce(IV) ions could be invoked here in order to explain its poor catalytic activity when chemically activated.
Electrochemically triggered oxidative catalysis
FTO–TiO2–24+ and FTO–Nafion–24+/34+ have been investigated in terms of their electrocatalytic capacity to oxidize water to dioxygen. The instability of the FTO–TiO2–24+ films prepared under the usual catalytic conditions at low (pH = 1) or neutral (pH = 7) conditions (see above) moved us to prepare modified electrodes previously activated at pH 12. In order to test their electrocatalytic activity through bulk electrolysis, the correct potential to be applied has been electrochemically evaluated by means of DPV and estimated to be about 1.1 V vs. SSCE (Fig. S17†). The catalytic reaction has been carried out in an electrochemical cell (sketched in Scheme S4†) formed by two different compartments (A for oxidation and C for reduction) separated by a frit membrane (B). FTO–TiO2–24+ was used as working electrode in the above-mentioned electrochemical cell applying a potential of 1.1 V vs. SSCE for 8 h (Fig. S18†). On-line O2 measurement on the headspace of the cell compartment using a Clark electrode revealed no oxygen evolution (Fig. S19†). The electrochemical characterization of the electrode after the CPE experiment revealed leaching of 45+ from the FTO–TiO2–45+ material to the basic aqueous solution. Effectively, Fig. S17b† displays the electrode CV after CPE, where the total disappearance of the redox waves can be appreciated. In addition, the UV–vis spectra of the solution after CPE (Fig. S20†) displayed the typical bands for 45+, further confirming this hypothesis.FTO–Nafion–24+ and FTO–Nafion–34+ films have been investigated in terms of their potential electrocatalytic capacity to oxidize water to dioxygen at pH = 1.0 in a 0.1 M triflic acid aqueous solution, as displayed in Fig. 6. The DPV of FTO–Nafion–24+ (Fig. 6, bottom) shows the “activation” of the film, characterized by the growing of an electrocatalytic wave at around 1.4 V, assignable to an electrochemically triggered water oxidation reaction. These results are in agreement with the exchange of the chlorido-bridged ion for two water molecules, as shown in Scheme S5 in the ESI.† As reported here, the “in situ” generation of Ru–OH2 species from their Ru–Cl counterparts in acidic media usually provokes a decrease of the intensity of their redox waves and the convergence of several oxidation states in a narrow potential range, as well as the appearance of intense electrocatalytic currents due to WO catalysis. These phenomena have been extensively studied for several related systems.2c,16,17 In contrast, the DPV of FTO–Nafion–34+ (Fig. 6, top) shows neither a shift of the redox waves nor an increase of the electrocatalytic current. The latter is in agreement with the above-proposed coordination of at least one sulfonate group to the Ru metal center and the consequent blocking of its putative catalytic activity (see above in the Electrochemical Characterization section).
 | ||
| Fig. 6 DPV of FTO–Nafion–24+ (bottom) and FTO–Nafion–34+ (top) in aqueous solutions at pH = 1.0 in 0.1 M triflic acid. FTO was used as working electrode and the potential measured vs. SSCE. | ||
Conclusions
Ru–Hbpp complexes constitute a wide family of efficient WOCs extraordinarily well studied both from a structural and a mechanistic approach.2b–e,10,12,15–17 The accurate understanding of the limitations and consequences of anchoring such family of WOCs onto solid surfaces is a must when attempting to include them into photoelectrochemical cells (PECs).18 Therefore, research in order to explore and optimize the different types of catalyst–support interactions is a mandatory step. With this leit motif in mind, our group has explored several anchoring strategies for the Ru–Hbpp family (covalent anchoring, electropolymerization, etc.) but so far none of them has been proven to be fully satisfactory.4,5 In this work, the ionic interaction between Ru–Hbpp WOCs and several rugged solid supports has been analyzed. For this purpose a new family of Ru–Hbpp complexes (24+ and 34+) containing the positively charged ligand 4′-(-p-(pyridin-1-ylmethyl)phenyl)-2,2′:6′,2′-terpyridine L1+ has been synthesized and fully characterized, both in the solid state and in solution. Furthermore, their in situ conversion (aqueous media, pH = 1) into its bis-aqua derivative 45+ has been electrochemically demonstrated. The anchoring of the new catalysts onto TiO2, SiO2 and Nafion® has been achieved and the new hybrid materials prepared have been characterized by means of electrochemical and spectroscopic techniques.The catalytic evaluation of 45+ with Ce(IV) leads to a stable catalyst that evolved only O2 (and no CO2) as reaction product, potentially due to the electron-withdrawing effect of the positive pyridinium groups of the L1+ ligands close to the easily oxidizable –CH2 groups. However, the catalytic evaluation of SiO2–45+ has shown high CO2:O2 ratios, thus revealing the oxidative degradation of the catalyst under the harsh reaction conditions. This result, compared with the clean O2 evolution observed for the homogenous 45+ counterpart, points out that the activation of the methylene groups in alpha position to the pyridinium residues happens when the positive charge of the ligand interacts with the SiO− residues.
On the other hand, the interaction of the positively charged pyridines of 24+ with TiO2 in FTO–TiO2–24+ seems to be weak, since clear leaching of the catalyst was observed when a CPE of the electrode was performed. Finally, despite the poor performance of FTO–Nafion–45+ as a chemically-driven WOC, yielding only stoichiometric amounts of O2, the evaluation of the FTO–Nafion–45+ by DPV has revealed its clear electrocatalytic activity and, therefore, its potential use as a modified electrode for the oxidation of water. In conclusion, the work herein presented demonstrates the feasibility of using ionic interactions to generate stable and active Ru–Hbpp hybrid materials, such as FTO–Nafion–45+, able to electrochemically oxidize water to dioxygen.
Experimental
Materials
All reagents used in the present work were obtained from Aldrich Chemical Co. and were used without further purification. Reagent-grade organic solvents were obtained from Scharlab. RuCl3·3H2O was supplied by Alfa Aesar and was used as received. Titanium dioxide paste (100% anatase) was supplied by Solaronix. Both the silica and the Nafion polymer were provided by Sigma Aldrich. The starting ligands bis-(2-pyridyl)pyrazole (from now on Hbpp)19 and 4′-(-p-(bromomethyl)phenyl)-2,2′:6′,2′-terpyridine20 were prepared as described in the literature. All synthetic manipulations were routinely performed under nitrogen atmosphere using Schlenk tubes and vacuum-line techniques.Instrumentation and measurements
UV–Vis spectroscopy was performed using a HP8453 spectrometer using 1 cm quartz cells. NMR spectroscopy was performed on a Bruker DPX 250 MHz, DPX 360 MHz or a DPX 400 MHz spectrometer. Samples were run in CD3CN or acetone-d6 with internal references. Elemental analyses were performed using a Carlo Erba CHMS EA-1108 instrument provided by the Chemical Analysis Service of the Universitat Autònoma de Barcelona (CAS-UAB). Electrospray Ionization Mass Spectrometry (ESI–MS) measurements were carried out on a HP298s gas chromatography (GC–MS) system from the CAS-UAB. UV–vis absorption spectra of the solid samples were recorded by diffuse reflectance measurement with a 8 cm integrating sphere (ISR-240 A) using BaSO4 as reference in a Shimadzu UV-2401 PC spectrophotometer. Cyclic voltammetry and differential pulse voltammetry experiments were performed on an Ij-Cambria HI-660 potentiostat using a three-electrode cell. A glassy carbon electrode (2 mm diameter) was used as a working electrode, platinum wire as an auxiliary electrode and a SSCE as a reference electrode. Working electrodes were polished with 0.05 micron alumina paste washed with distillated water and acetone before each measurement. The complexes were dissolved in acetone containing the necessary amount of n-Bu4NPF6 (TABH) as a supporting electrolyte to yield 0.1 M ionic strength. E1/2 values reported in this work were estimated from CV experiments as the average of the oxidative and reductive peak potentials (Ep,a + Ep,c)/2. On-line manometry measurements were carried out on a Testo 521 differential pressure manometer with an operating range of 1–100 hPa and accuracy within 0.5% of the measurement, coupled to thermostatted reaction vessels for dynamic monitoring of the headspace pressure above each reaction. The secondary ports of the manometers were connected to thermostatically controlled reaction vessels that contained the same solvents and headspace volumes as the sample vials. On-line monitoring of the gas evolution was performed on a Pfeiffer Omnistar GSD 301C mass spectrometer. Typically, 16.04 mL degassed vials containing a suspension of the catalysts in 0.1 M triflic acid (1.5 mL) were connected to the apparatus capillary tubing. Subsequently, the previously degassed solution of Ce(IV) (0.5 mL) at pH = 1 (triflic acid, 200 equiv.) was introduced using a Hamilton gastight syringe, and the reaction was dynamically monitored. A response ratio of 1:2 was observed when equal concentrations of dioxygen and carbon dioxide, respectively, were injected, and was then used for calculation of their relative concentrations.
X-ray crystal structure determination
Crystal structure determination for 1+ was carried out using a Bruker-Nonius diffractometer equipped with an APPEX II 4K CCD area detector, a FR591 rotating anode with Mo Kα radiation, Montel mirrors as a monochromatic and a Kryoflex low-temperature device (T = −173°C). Crystal structure determination for 24+ was carried out using a Apex DUO Kappa 4-axis goniometer equipped with an APPEX 2 4K CCD area detector, a microfocus source E025 IuS using MoKα radiation, Quazar MX multilayer optics as monochromator and an Oxford Cryosystems low temperature device Cryostream 700 plus (T = −173 °C). Full-sphere data collection was used with w and f scans. Programs used: data collection APPEX II,21 data reduction, Bruker Saint V/.60A,22 absorption corrections, SADABS.23 CCDC 949999 & 950000 contains supplementary data for this paper.Structure and refinement
The SHELXTL24 software was used. The crystal data parameters are listed in Tables S1 and S2.† For the structure of {[RuII(L1+)]2(μ-bpp)(μ-Cl)}(PF6)4 (2(PF6)4), the program SQUEEZE9 implemented in Platon was used in order to avoid the highly disordered solvent molecules (8 acetone and 13 toluene molecules could be detected). Applying the program SQUEEZE the R1 value could be lowered from 11.45% to 7.43%.Synthetic preparations
4′-(-p-(Pyridin-1-ylmethyl)phenyl)-2,2′:6′,2′-terpyridine (L1(PF6)): 4′-(-p-(bromomethyl)phenyl)-2,2′:6′,2′-terpyridine (0.200 g, 0.497 mmol) was dissolved in pyridine (30 mL). The mixture was allowed to react in a microwave reactor at 150 °C and 300 W for 30 minutes. The volume was reduced until dryness and the solid residue was redissolved in water (30 mL). The final suspension was filtered through celite and a saturated aqueous NH4PF6 solution (1 mL) was added to the colorless solution to obtain a white precipitate. The mixture was filtered after stirring for two hours to yield 0.2 g (85%) of the desired powder. 1H NMR (400 MHz, acetonitrile-d3): δ = 8,79 (d, 2H23-27, J23-24 = 6,40 Hz) 8,73 (s, 2H9-7), 8,73 (d, 2H1-15), 8,69 (d, 2H4-12, J3-4 = 8,30 Hz), 8,55 (t, 1H25, J24-25 = 8,10 Hz), 8,06 (t, 2H24-26, J24-25 = 8,10 Hz, J23-24 = 6,40 Hz ), 7,99 (ddd, 2H3-13, J3-4 = 8,30 Hz, J3-2 = 7,60 Hz, J1-3 = 1,46 Hz), 7,99 (d, 2H17-21, J17-18 = 8,10 Hz), 7,63 (d, 2H18-20, J17-18 = 8,10 Hz), 7,47 (t, 2H2-14, J2-3 = 7,60 Hz, J1-2 = 6,0 Hz), 5,81 (s, 2H22). 13C{1H} NMR (100 MHz, acetonitrile-d3): δ = 157,45 (C6-10), 156,65 (C5-11), 150,56 (C1-15), 150,47 (C8), 147,78 (C25), 145,99 (C23-27), 141,17 (C16), 138,89 (C3-13), 135,16 (C19), 131,47 (C18-20), 130,09 (C24-26), 129,57 (C17-21), 125,86 (C2-14), 122,54 (C4-12), 119,94 (C9-7), 65,40 (C22).[RuCl3(L1+)](PF6) (1(PF6)): L1(PF6) (0.915 g, 1.617 mmol) and RuCl3·3H2O (0.422 g, 1.617 mmol) were dissolved in dry MeOH (130 mL). The mixture was stirred and heated at reflux temperature for 4 h. Then the solution was kept cool until a brown precipitate appeared. The solid was filtered, washed with cold water (3 × 5 mL) and diethyl ether (3 × 5 mL) and finally dried under vacuum to afford complex 1(PF6) (0.949 g, 77%). ESI–MS (MeOH): m/z = 610 ([M−PF6]+). Elemental analysis (%) found: C, 43.02; H, 2.81; N, 7,43. Calcd. for C27H21Cl3F6N4PRu: C, 42.89; H, 2.96; N, 7.22.
{[RuII(L1+)]2(μ-bpp)(μ-Cl)}(PF6)4 (2(PF6)4): 1(PF6) (0.942 g, 1.77 mmol) and LiCl (0.113 g, 2.65 mmol) were dissolved in a solution of NEt3 (492 mL, 3.54 mmol) and dry MeOH (180 mL). The mixture was stirred at room temperature for 20 min, and then Hbpp (0.197 g, 0884 mmol) and 0.6684 M MeONa (1.32 mL, 0.844 mmol) in dry MeOH (20 mL) were added. The resulting solution was heated for 4 h and then stirred in the presence of a 100 W tungsten lamp for 8 h. The reaction mixture was filtered and then a saturated aqueous NH4PF6 solution (1 mL) was added to obtain a brown precipitate. The solid was collected, washed with cold water (3 × 5 mL) and diethyl ether (3 × 5 mL) and finally dried under vacuum to afford complex 2(PF6)4 (0.860 g, 75%). 1H NMR (400 MHz, acetone-d6): δ = 9.34 (d, 4H, J23-24 = 6.08 Hz; H23), 9.01 (s, 4H; H7), 8.80 (t, 2H, J25-24 = 7.80 Hz; H25), 8.72 (d, 4H, J3-4 = 8.00 Hz; H4), 8.54 (s, 1H; H8), 8.41 (d, 4H , J1-2 = 5.76 Hz; H1), 8.35 (t, 4H, J24-25 = 7.80 Hz; H24), 8.31 (m, 6H; H28-17), 7.97 (t, 4H, J3-4,2 = 8.00 Hz; H3), 7.89 (d, 4H, J17-18 = 8.20 Hz; H18), 7.83 (t, 2H, J29-28,30 = 8.00 Hz; H29), 7.64 (t, 4H, J2-1,3 = 6.57 Hz; H2), 7.50 (d, 2H, J30-31 = 5.90 Hz; H31), 6.82 (t, 2H, J30-31 = 5.90 Hz; H30), 6.20 (s, 4H, H22). 13C{1H} NMR (100 MHz, acetone-d6): δ = 159.39 (C6), 159.05 (C5), 158.78 (C32), 153.89 (C31), 153.56 (C1), 148.48 (C33), 146.57 (C25), 145.06 (C23), 145.00 (C8), 138.44 (C16), 137.07 (C3), 136.92 (C29), 135.15 (C19), 130.14 (C18), 128.95 (C24), 128.57 (C17), 127.39 (C2), 123.87 (C4), 122.19 (C30), 120.47 (C28), 120.20 (C7), 103.29 (C34), 64.16 (C22). UV–vis (acetone): lmax (ε) = 366 (26775), 480 (19187), 509 (20235). ESI–MS (MeOH): m/z = 1697.2 ([M−PF6]+). Elemental analysis (%) found: C, 43.60; H, 2.91; N, 9.00. Calcd. for C67H51ClF24N12P4Ru2: C, 43.70; H, 2.79; N, 9.13.
{[RuII(L1+)]2(μ-bpp)(μ-O2CMe)}(PF6)4 (3(PF6)4): a sample of 2(PF6)4 (0.225 g, 0.120 mmol), sodium acetate (0.054 g, 0.660 mmol) and AgBF4 (0.023 g, 0.120 mmol) was dissolved in acetone–water (3:1, 40 mL), and the solution was heated at reflux overnight in the dark. The resulting solution was filtered, and a saturated aqueous NH4PF6 solution (1 mL) was added. A solid precipitated appeared upon reducing the volume. The solid was collected and washed with cold water (3 × 5 mL) and diethyl ether (3 × 5 mL) and finally dried under vacuum to afford complex 3(PF6)4 (0.166 g, 73%). 1H NMR (400 MHz, acetone-d6): δ = 9.35 (d, 4H, J23-24 = 6.90 Hz; H23), 9.10 (s, 4H; H7), 8.82 (m, 6H; H25-4), 8.56 (s, 1H; H34), 8.47 (d, 4H , J1-2 = 5.70 Hz; H1), 8.37 (t, 4H, J24-25,23 = 6.90 Hz; H24), 8.35 (d, 4H, J17-18 = 8.15 Hz; H17), 8.22 (d, 2H, J28-29 = 7.97 Hz; H28), 8.05 (t, 4H, J3-4,2 = 7.85 Hz; H3), 7.94 (d, 4H, J17-18 = 8.15 Hz; H18), 7.76 (t, 2H, J29-28,30 = 7.90 Hz; H29), 7.55 (t, 4H, J2-1,3 = 6.50 Hz; H2), 7.45 (d, 2H, J30-31 = 5.70 Hz; H31), 6.83 (t, 2H, J30-31 = 7.73 Hz; H30), 6.30 (s, 4H, H22), 0.45 (s, 3H, H36). 13C{1H} NMR (100 MHz, acetone-d6): δ = 191.59 (C35), 160.34 (C6), 159.77 (C5), 156.47 (C32), 153.81 (C1), 152.80 (C31), 151.89 (C33), 146.59 (C25), 145.02 (C23), 144.77 (C8), 138.83 (C16), 137.32 (C3), 135.99 (C29), 135.17 (C19), 130.19 (C18), 128.95 (C24), 128.59 (C17), 127.46 (C2), 123.89 (C4), 122.24 (C30), 120.36 (C7), 119.61 (C28), 103.89 (C34), 64.20 (C22), 25.25 (C36). UV–vis (acetone): lmax (ε) = 367 (27615), 497 (17119), 525 (15923). ESI–MS (MeOH): m/z = 788.09 ([M−2PF6]2+). Elemental analysis (%) found: C, 43.92; H, 2.75; N, 8.95. Calcd. for C69H55F24N12O2P4Ru2: C, 44.21; H, 2.97; N, 9.01.
Preparation of FTO–TiO2–24+: on clean FTO films, anatase TiO2 paste was spread uniformly. Then the films were heated for 10 min at 100 °C in order to reduce the surface irregularities. The films were calcinated following the appropriated temperature ramps (see Fig. S26†). The anchoring process was carried out by soaking overnight every film into 5 mL of an acetone solution (0.305 mM) of 24+.
Preparation of FTO–Nafion–X4+ (X = 24+ or 34+): on clean FTO films, Nafion 5% w/w in water and low weight alcohols (50 mL) was uniformly deposited. Then the films were heated for 30 min at 100 °C in order to remove water and low weight alcohols. After cooling down until room temperature, the films were soaked overnight into an acetone solution of X4+ (10 mL, 0.0201 mM).
Preparation of silica–X4+ (X = 24+ or 34+): silica (3 g) was poured into 10 mL of a solution of 24+ or 34+ (0.548 mM) in acetone. The mixture was stirred for some minutes until the solution became colorless. Then the pink solids were washed several times with acetone and diethyl ether and were finally air-dried.
Acknowledgements
Support from MINECO (CTQ2010-21497, CTQ2010-21532-C02-02 and CTQ2011-26440) and ICIQ is gratefully acknowledged. JA is grateful for a PIF pre-doctoral grant from UAB.Notes and references
- (a) X. Sala, L. Escriche and A. Llobet, in Molecular Solar Fuels, 1st edn, ed. T. J. Wydrzynski and W. Hillier, Royal Society of Chemistry, 2012, ch. 10, pp. 273–288 Search PubMed; (b) L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296–3313 RSC.
- (a) X. Sala, M. Z. Ertem, L. Vigara, T. K. Todorova, W. Chen, R. C. Rocha, F. Aquilante, C. J. Cramer, L. Gagliardi and A. Llobet, Angew. Chem., Int. Ed., 2010, 49, 7745–7747 CrossRef CAS PubMed; (b) L. Francàs, X. Sala, E. Escudero-Adan, J. Benet-Buchholz, L. Escriche and A. Llobet, Inorg. Chem., 2011, 50, 2771–2781 CrossRef PubMed; (c) J. García-Antón, R. Bofill, L. Escriche, A. Llobet and X. Sala, Eur. J. Inorg. Chem., 2012, 4775 CrossRef; (d) S. Romain, F. Bozoglian, X. Sala and A. Llobet, J. Am. Chem. Soc., 2009, 131, 2768–2769 CrossRef CAS PubMed; (e) J. Mola, C. Dinoi, X. Sala, M. Rodriguez, I. Romero, T. Parella, X. Fontrodona and A. Llobet, Dalton Trans., 2011, 40, 3640–3646 RSC.
- (a) L. Li, L. Duan, Y. Xu, M. Gorlov, A. Hagfeldtab and L. Sun, Chem. Commun., 2010, 46, 7307–7309 RSC; (b) F. Li, B. Zhang, X. Li, Y. Jiang, L. Chen, Y. Li and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 12276–12279 CrossRef CAS PubMed; (c) X. Sala, M. Rodríguez, I. Romero, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842–2852 CrossRef CAS PubMed; (d) S. Roeser, P. Farras, F. Bozoglian, M. Martinez-Belmonte, J. Benet-Buccholz and A. Llobet, ChemSusChem, 2011, 4, 197–207 CrossRef CAS PubMed; (e) L. Duan, Y. Xu, M. Gorlov, L. Tong, S. Andersson and L. Sun, Chem.–Eur. J., 2010, 16, 4659–4668 CrossRef CAS PubMed.
- J. Mola, E. Mas-Marza, X. Sala, I. Romero, I. M. Rodríguez, C. Viñas, T. Parella and A. Llobet, Angew. Chem., Int. Ed., 2008, 47, 5830–5832 CrossRef CAS PubMed.
- L. Francas, X. Sala, J. Benet-Buchholz, L. Escriche and A. Llobet, ChemSusChem, 2009, 2, 321–329 CrossRef CAS PubMed.
- W. Spahni and G. Calzaferri, Helv. Chim. Acta, 1984, 67, 450–454 CrossRef CAS.
- S. Chakraborty, T. J. Wadas, H. Hester, C. Flaschenreim, R. Schmehl and R. Eisenberg, Inorg. Chem., 2005, 44, 6284–6293 CrossRef CAS PubMed.
- F. Laurent, E. Plantalech, B. Donnadieu, B. A. Jimenez, F. Hernández, M. Martínez-Ripoll, M. Biner and A. Llobet, Polyhedron, 1999, 18, 3321–3331 CrossRef CAS.
- A. L. Spek, SQUEEZE implemented in Platon, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. Sens, I. Romero, M. Rodríguez, A. Llobet, A. T. Parella and J. Benet-Buchholz, J. Am. Chem. Soc., 2004, 126, 7798–7799 CrossRef CAS PubMed.
- F. Dumur, C. R. Mayer, K. Hoang-Thi, I. Ledoux-Rak, F. Miomandre, G. Clavier, E. Dumas, E. R. Méaallet-Renault, M. Frigoli, J. Zyss and F. Sécheresse, Inorg. Chem., 2009, 48, 8120–8133 CrossRef CAS PubMed.
- N. Planas, G. J. Christian, E. Mas-Marzà, X. Sala, X. Fontrodona, F. Maseras and A. Llobet, Chem.–Eur. J., 2010, 16, 7965–7968 CrossRef CAS PubMed.
- (a) M. Haga, E. S. Dodsworth and A. B. P. Lever, Inorg. Chem., 1986, 25, 447–453 CrossRef CAS; (b) S. C. Rasmussen, S. E. Ronco, D. A. Mlsna, M. A. Billadeau, W. T. Pennington, J. Kolis and J. D. Petersen, Inorg. Chem., 1995, 34, 821–829 CrossRef CAS; (c) O. Kohle, S. Ruile and M. Gratzel, Inorg. Chem., 1996, 35, 4779–4787 CrossRef CAS; (d) A. Ben Altabel, S. B. Ribotta, R. de Gallo, M. E. Folquer and N. E. Katz, Inorg. Chim. Acta, 1991, 188, 67–70 CrossRef; (e) P. E. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, F. G. Strouse, N. C. Thomas, T. A. Treadway and T. A. White, Inorg. Chem., 1995, 34, 6145–6157 CrossRef CAS; (f) K. R. Barqawi, A. Llobet and T. J. Meyer, J. Am. Chem. Soc., 1988, 110, 7751–7759 CrossRef.
- F. Bozoglian, S. Romain, M. Z. Ertem, T. K. Todorova, C. Sens, J. Mola, M. Rodríguez, I. Romero, J. Benet-Buchholz, X. Fontrodona, C. J. Cramer, L. Gagliardi and A. Llobet, J. Am. Chem. Soc., 2009, 131, 15176–15187 CrossRef CAS PubMed.
- S. Roeser, Dissertation, Univ. Rovira i Virgili, 2011 Search PubMed.
- S. Roeser, M. Z. Ertem, C. Cady, R. Lomoth, J. Benet-Buchholz, L. Hammarström, B. Sarkar, W. Kaim, C. J. Cramer and A. Llobet, Inorg. Chem., 2012, 51, 320–327 CrossRef CAS PubMed.
- L. Francàs, Dissertation, Univ. Autònoma of Barcelona, 2011 Search PubMed.
- J. Liu, G. Cao, Z. Yang, D. Wang, D. Dubois, X. Zhou, G. L. Graff, L. R. Pederson and J.-G. Zhang, ChemSusChem, 2008, 1, 676–697 CrossRef CAS PubMed.
- J. Pons, F. J. Sánchez, X. López, F. Teixidor and J. Casabó, Polyhedron, 1990, 9, 2839–2845 CrossRef CAS.
- J. M. Haider, M. Chavarot, S. Weidner, I. Sadler, R. M. Williams, L. De Cola and Z. Pikramenou, Inorg. Chem., 2001, 40, 3912–3921 CrossRef CAS PubMed.
- Data collection with: Apex II, version v2009.1-02; Bruker AXS Inc., Madison, WI, 2007 Search PubMed.
- Data reduction with: SAINT, version V7.60A; Bruker AXS Inc., Madison, WI, 2007 Search PubMed.
- SADABS, version V2008/1 (2001), Bruker AXS Inc., Madison, WI, 2008; CrossRef; R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed (SHELXTL, version V6.14).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 949999 & 950000. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cy00643c |
| This journal is © The Royal Society of Chemistry 2014 |