 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneous catalysts based on supported Rh–NHC complexes: synthesis of high molecular weight poly(silyl ether)s by catalytic hydrosilylation†‡
Guillermo
Lázaro
a,
Francisco J.
Fernández-Alvarez
*a,
Manuel
Iglesias
a,
Cristina
Horna
a,
Eugenio
Vispe
a,
Rodrigo
Sancho
a,
Fernando J.
Lahoz
a,
Marta
Iglesias
b,
Jesús J.
Pérez-Torrente
a and
Luis A.
Oro
*a
aDepartamento de Química Inorgánica-ISQCH; Universidad de Zaragoza-CSIC, Facultad de Ciencias 50009, Zaragoza, Spain. E-mail: oro@unizar.es; paco@unizar.es
bICMM-CSIC; C/Sor Juana Inés de la Cruz 3, Cantoblanco 28049, Madrid, Spain
First published on 23rd September 2013
Abstract
The new rhodium(I) complexes [Rh(Cl)(COD)(R-NHC-(CH2)3Si(OiPr3)3)] (R = 2,6-diisopropylphenyl (2a); n-butyl (2b)) have been synthesised and fully characterised. The study of their application as ketone hydrosilylation catalysts showed a clear N-substituent effect, 2a being the most active catalyst precursor. Complex 2a has been immobilised in the mesoporous materials MCM-41 and KIT-6. The new hybrid materials have been fully characterised and used as catalyst precursors for the preparation of poly(silyl ether)s by catalytic hydrosilylation. The heterogeneous catalytic systems based on the materials 2a–MCM-41 and 2a–KIT-6 afford polymers with high average molecular weight (Mw) Mw = 2.61 × 106 g mol−1 (2a–MCM-41) and Mw = 4.43 × 105 g mol−1 (2a–KIT-6).
1. Introduction
Transition-metal catalysed hydrosilylation has been demonstrated to be a convenient route for the synthesis of new functional materials.1,2 In this context, it is remarkable that the most efficient hydrosilylation catalysts are based on platinum or rhodium complexes.1 Such metals are particularly expensive and scarce. Consequently, there is an increasing interest in the development of heterogeneous and therefore easily recyclable hydrosilylation catalytic systems. The immobilisation of homogenous catalysts onto supports has been a methodology commonly used to produce heterogeneous catalysts maintaining or improving the catalytic performance.3–9During the last decades N-heterocyclic carbenes (NHCs) have been widely used as ligands in homogeneous catalysis.10,11 This could be attributed to the interesting electronic properties of NHC ligands. Indeed, they are strong σ-donor ligands with a very poor π-acceptor character.12 On the other hand, Si(OR)3-functionalised transition metal–NHC complexes are species of great interest since they have the potential for immobilisation on mineral supports by the reaction between the Si–OR functionalities and the Si–OH active sites present on the support surface.13–17
Here we report on the synthesis and characterisation of new Rh(I)–NHC complexes functionalized with a triisopropoxysilyl tail group for their immobilisation on silica-based inorganic supports. Fine tuning of the homogeneous catalyst by modification of the N-substituent on the NHC ligand has allowed for the synthesis of active ketone hydrosilylation catalysts. Moreover, the supported catalysts have found application in the synthesis of high molecular weight poly(silyl ether)s by rhodium catalysed hydrosilylation.
2. Experimental
General information
All manipulations were performed with rigorous exclusion of air at an argon/vacuum manifold using standard Schlenk-tube techniques or in a dry-box (MB-UNILAB). Solvents were dried by the usual procedures and distilled under argon prior to use or taken under argon from a Solvent Purification System (SPS). The reagents 1,1,1,3,5,5,5-heptamethyltrisiloxane, 1,1,3,3,5,5-hexamethyltrisiloxane, terephthalaldehyde and the solids MCM-41 and KIT-6 were purchased from commercial sources and used without further purification. The rhodium(I) [Rh(μ-Cl)(COD)]2 complex18 and I(CH2)3(iPrO)319 were prepared according to methods reported in the literature. NMR spectra were recorded on a Varian Gemini 2000, Bruker ARX 300, Bruker Avance 300 MHz or Bruker Avance 400 MHz instrument. Chemical shifts (expressed in parts per million) are referenced to residual solvent peaks (1H, 13C{1H}). Coupling constants, J, are given in hertz. C, H, and N analyses were carried out using a Perkin-Elmer 2400 CHNS/O analyzer. Mass spectrometry was performed on an Esquire 3000+ with an ion trap detector interfaced to an Agilent 1100 series HPLC system. FT-IR spectra were recorded on a Nicolet Nexus 5700 FT spectrophotometer equipped with a Nicolet Smart Collector diffuse reflectance accessory. TEM microscopy images were collected with an INCA 200 X-Sight from Oxford Instruments with an energy resolution between 136 eV and 5.9 keV. Gas chromatography analyses were performed using an Agilent 6890N with a FID detector equipped with an HP-1 column from J&W Scientific (30 m, 0.25 mm i.d.). Parameters were as follows: initial temperature 50 °C, initial time 5 min, ramp 30 °C min−1, final temperature 250 °C, final time 5 min, injector temperature 250 °C, and detector temperature 300 °C. Isotherms were obtained on a Quantachrome AUTOSORB by measuring the volume of N2 absorbed at relative pressures between 0.05 and 0.99 at 77.3 K after drying the sample at 120 °C in vacuo. Absolute molecular weights of polymers were determined by GPC-MALS. Before injection all samples were filtered through 0.45 mm PTFE membranes. The column oven temperature was maintained at 35 °C during all the experiments. The measurements were carried out on a Waters 2695 autosampler equipped with three in line PLGel Mixed C (7.8 × 300 mm) columns and a Wyatt three detector setup (Minidaw TREOS® (MALS), Optilab Rex® (DRI) and VIscostarII® Viscometer). The samples were eluted with THF at a rate of 1 mL min−1. Tg of the polymer was determined by DSC on a TA instruments DSC Q1000 with a liquid nitrogen cooling system. The temperature program for the analysis was begun at −120 °C and the temperature was increased at a rate of 10 °C min−1 to 25 °C.Preparation of 1-(3-triisopropoxysilylpropyl)-3-(2,6-diisopropylphenyl)-imidazolium iodide (1a)
To an acetonitrile (30 mL) solution of 1-(2,6-diisopropylphenyl)imidazole (465 mg, 2.00 mmol), I(CH2)3Si(iPrO)3 (1.12 g, 3.00 mmol) was added. The reaction mixture was stirred at 90 °C for 24 h. The resulting solution was filtered through Celite and the solvent was removed in vacuo. The residue thus obtained was washed with hexane (2 × 20 mL) to give a brown solid. Yield 1.07 g (89%). Anal. calcd. for C27H47IN2O3Si: C, 53.81, H, 7.86, N, 4.65. Found: C, 54.03, H, 7.92, N, 4.53. 1H NMR (300 MHz, CDCl3, 293 K) plus COSY: δ 9.95 (s, 1H, NCHN), 7.91 (t, 1H, JH–H = 2 Hz, CHimd), 7.54 (m, 1H, CH), 7.30 (m, 2H, CH), 7.23 (t, 1H, JH–H = 2 Hz, CHimd), 4.79 (t, 2H, JH–H = 7 Hz, CH2N), 4.20 (spt, 3H, JH–H = 6 Hz, CH–iPrO), 2.30 (spt, 2H, JH–H = 7 Hz, CH–iPr), 2.11 (m, 2H, –CH2–), 1.23 (d, 6H, JH–H = 7 Hz, CH3–iPr), 1.17 (d, 18H, JH–H = 6 Hz, CH3–iPrO), 1.14 (d, 6H, JH–H = 7 Hz, CH3–iPr), 0.58 (m, 2H, CH2Si). 13C{1H} NMR plus HSQC (75.46 MHz, CDCl3, 293 K): δ 145.5 (Cipso, 2C), 138.0 (NCHN), 132.2 (CH), 130.1 (Cipso), 124.9 (CH, 2C), 124.3 (CHimd), 123.5 (CHimd), 65.5 (CH–iPrO), 52.5 (CH2N), 28.9 (CH–iPr), 25.7 (CH3–iPrO), 24.9 (–CH2–), 24.6 (CH3–iPr), 24.3 (CH3–iPr), 8.4 (CH2Si). 29Si NMR (64.52, CDCl3, 293 K): δ −51.6 (CH2Si). Mass spectrometry (ESI+): m/z 475.3 (M+–I).Preparation of 1-(3-triisopropoxysilylpropyl)-3-(n-butyl)-imidazolium iodide (1b)
To an acetonitrile (30 mL) solution of 1-(butyl)imidazole (124 mg, 1.00 mmol), I(CH2)3Si(iPrO)3 (486 mg, 1.30 mmol) was added. The reaction mixture was stirred at 90 °C for 24 h. The resulting solution was filtered through Celite and the solvent was removed in vacuo. The residue thus obtained was washed with hexane (2 × 20 mL) to give a white solid. Yield 459 mg (92%). Anal. calcd. for C19H39IN2O3Si: C, 45.78, H, 7.89, N, 5.62. Found: C, 45.96, H, 7.51, N, 5.81. 1H NMR (300 MHz, CDCl3, 293 K) plus COSY: δ 10.0 (s, 1H, NCHN), 7.62 (t, 1H, JH–H = 2 Hz, CHimd), 7.36 (t, 1H, JH–H = 2 Hz, CHimd), 4.33 (m, 2H, CH2N(3)), 4.29 (m, 2H, CH2N(1)) 4.12 (spt, 3H, JH–H = 6 Hz, CH–iPrO), 1.94 (m, 2H, CH2CH2CH2Si), 1.85 (m, 2H, CH2CH2CH2but), 1.32 (m, 2H, CH3CH2), 1.10 (d, 18H, JH–H = 6 Hz, CH3–iPrO), 0.89 (t, 3H, JH–H = 7 Hz, CH3CH2), 0.47 (m, 2H, CH2Si). 13C{1H} NMR plus HSQC (75.46 MHz, CDCl3, 293 K): δ 136.0 (NCHN), 122.6 (CHimd), 122.0 (CHimd), 65.2 (CH–iPrO), 51.7 (CH2N(1)), 49.8 (CH2N(3)), 32.1 (CH2CH2CH2), 25.5 (CH3–iPrO), 24.6 (CH2CH3), 19.4 (CH2CH2CH2Si), 13.4 (CH2CH3), 8.5 (CH2Si). 29Si NMR (64.52 MHz, CDCl3, 293 K): δ −50.0 (CH2Si). Mass spectrometry (ESI+): m/z 371.3 (M+–I).Preparation of 2a
Dichloromethane (15 mL) was added to a mixture of the imidazolium salt 1a (602 mg, 1.00 mmol) and Ag2O (232 mg, 1.00 mmol). The reaction mixture was stirred in the absence of light at room temperature for 4 h. The resulting mixture was filtered and the solvent was removed in vacuo to yield a brown residue of the silver carbene. A THF (15 mL) solution of [RhCl(COD)]2 (247 mg, 0.500 mmol) was added to the residue and the mixture was stirred at room temperature for 16 h. The solvent was removed in vacuo and the residue thus obtained was extracted with hexane (3 × 20 mL) to afford a yellow crystalline solid. Yield 635 mg (88%). Anal. calcd. for C35H58ClN2O3RhSi: C, 58.30, H, 8.11, N, 3.88. Found: C, 58.09, H, 8.40, N, 4.07. 1H NMR plus COSY (300 MHz, C6D6, 293 K): δ 7.31 (dd, 1H, JH–H = 8 Hz, JH–H = 2 Hz, CH), 7.25 (t, 1H, JH–H = 8 Hz, CH), 6.97 (dd, 1H, JH–H = 8 Hz, JH–H = 2 Hz, CH), 6.43 (d, 1H, JH–H = 2 Hz, CHimd), 6.36 (d, 1H, JH–H = 2 Hz, CHimd), 5.38 (m, 2H, CHCOD, 1H, CH2N), 4.34 (m, 1H, CH2N), 4.25 (spt, 3H, JH–H = 6 Hz, CH–iPrO), 4.06 (spt, 1H, JH–H = 7 Hz, CH–iPr), 3.60 (m, 1H, CHCOD), 3.04 (m, 1H, CHCOD), 2.51 (m, 1H, CH2COD), 2.32 (m, 1H, –CH2–), 2.23–2.05 (m, 1H, –CH2–, 2H, CH2COD), 2.00 (spt, 1H, JH–H = 7 Hz, CH–iPr), 1.89 (m, 1H, CH2COD), 1.83 (d, 3H, JH–H = 7 Hz, CH3–iPr), 1.71 (m, 1H, CH2COD), 1.58 (m, 2H, CH2COD), 1.45 (s, 1H, CH2COD), 1.22 (d, 18H, JH–H = 6 Hz, CH3–iPrO), 1.06 (d, 3H, JH–H = 7 Hz, CH3–iPr), 1.03 (d, 3H, JH–H = 7 Hz, CH3–iPr), 0.92 (d, 3H, JH–H = 7 Hz, CH3–iPr), 0.76 (m, 2H, CH2Si). 13C {1H} NMR plus HSQC (75 MHz, C6D6, 293 K): δ 183.5 (d, JRh–C = 52 Hz, RhCcarbene), 148.3 (Cipso), 145.1 (Cipso), 136.2 (Cipso), 129.6 (CH), 124.6 (CH), 123.8 (CHimd), 122.8 (CH), 120.1 (CHimd), 96.9 (d, JRh–C = 7 Hz, CHCOD), 96.5 (d, JRh–C = 7 Hz, CHCOD), 67.1 (d, JRh–C = 14 Hz, CHCOD), 66.9 (d, JRh–C = 14 Hz, CHCOD), 64.9 (CH–iPrO), 54.4 (CH2N), 34.5 (CH2COD), 31.5 (CH2COD), 29.4 (CH2COD), 28.4 (CH–iPr), 28.0 (CH–iPr), 27.9 (CH2COD), 26.0 (CH3–iPr), 25.8 (CH3–iPr), 25.5 (CH3–iPrO), 24.9 (–CH2–), 23.8 (CH3–iPr), 22.6 (CH3–iPr), 9.6 (CH2Si). 29Si NMR (64.52 MHz, CDCl3, 293 K): δ −49.9 (CH2Si). Mass spectrometry (ESI+): m/z 685.2 (M+–Cl).Preparation of 2b
CH2Cl2 (15 mL) was added to a mixture of the imidazolium salt 1b (498 mg, 1.00 mmol) and Ag2O (232 mg, 1.00 mmol). The reaction mixture was stirred in the absence of light at room temperature for 4 h. The resulting mixture was filtered and the solvent was removed in vacuo to yield an off-white residue of the silver carbene. A THF (15 mL) solution of [RhCl(COD)]2 (247 mg, 0.500 mmol) was added to the residue and the mixture was stirred at room temperature for 16 h. The solvent was removed in vacuo and the residue thus obtained was extracted with hexane (3 × 20 mL) to afford a yellow crystalline solid. Yield 0.55 g (89%). Anal. calcd. for C27H50ClN2O3RhSi: C, 52.55, H, 8.17, N, 4.54. Found: C, 51.97, H, 8.40, N, 4.27. 1H NMR plus COSY (300 MHz, C6D6, 293 K): δ 6.29 (d, 1H, JH–H = 2 Hz, CHimd), 6.12 (d, 1H, JH–H = 2 Hz, CHimd), 5.45 (m, 2H, CHCOD), 4.60 (m, 1H, CH2N(3)), 4.43 (m, 1H, CH2N(1)), 4.28 (spt, 3H, JH–H = 6 Hz, CH–iPrO; m, 1H, CH2N(3)), 4.08 (m, 1H, CH2N(1)), 3.32 (m, 1H, CHCOD), 3.25 (m, 1H, CHCOD), 2.42 (m, 2H, CH2COD), 2.26 (m, 3H, CH2COD; 1H, CH2CH3), 2.11 (m, 1H, CH2CH3), 1.80 (m, 3H, CH2COD), 1.57 (m, 2H, CH2CH2CH2But), 1.31 (m, 2H, CH2CH2CH2But), 1.23 (dvd, 18H, JH–H = 6 Hz, N = 6.5 Hz, CH3–iPrO), 0.88 (t, 3H, CH2CH3), 0.78 (m, 2H, CH2Si). 13C{1H} NMR plus HSQC (75 MHz, C6D6, 293 K): δ 183.3 (d, JRh–C = 51 Hz, RhCcarbene), 120.2 (CHimd), 120.0 (CHimd), 98.2 (d, JRh–C = 7 Hz, CHCOD), 97.9 (d, JRh–C = 7 Hz, CHCOD), 67.4 (d, JRh–C = 14 Hz, CHCOD), 67.0 (d, JRh–C = 14 Hz, CHCOD), 65.3 (CH–iPrO), 53.6 (CH2N(3)), 50.7 (CH2N(1)), 33.7 (CH2COD), 33.3 (CH2COD), 33.1 (CH2CH2CH2But) 29.6 (CH2COD), 29.2 (CH2COD), 25.9 (CH3–iPrO), 25.4 (CH2CH3), 20.3 (CH2CH2CH2Si), 14.0 (CH2CH3), 9.9 (CH2Si). 29Si NMR (64.52 MHz, CDCl3, 293 K): δ −49.9 (CH2Si). Mass spectrometry (ESI+): m/z 581.7 (M+–Cl).Crystal structure determination of compound 2a
Crystals of 2a suitable for X-ray analysis were obtained by the slow evaporation of a solution of the complex in hexane. X-ray diffraction data were collected at 100(2) K with graphite-monochromated MoKα radiation (λ = 0.71073 Å), using narrow ω rotation (0.3°) on a Bruker APEX DUO diffractometer. Intensities were integrated with the SAINT+ program and corrected for the absorption effect with SADABS, integrated in the APEX2 package. The structures were solved by direct methods with SHELXS-97.20 Refinement, by full-matrix least-squares on F2, was performed with SHELXL-97.20 Hydrogen atoms were included in calculated positions and defined with displacement and positional riding parameters. A typical disorder for the oxygen atoms of the alkoxy groups was observed and modelled with two oxygens having complementary occupancy factors (0.839 and 0.161(6)). Half a molecule of hexane, highly disordered, was also observed in the asymmetric unit of the crystal; six residual peaks were introduced in the final refinement to take account of this electron density. Crystal data for 2a: C35H58ClN2O3RhSi 0.5 C6H14, M = 764.37; yellow prism, 0.211 × 0.211 × 0.141 mm3; triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; a = 10.2529(12), b = 12.6020(15), c = 16.390(2) Å, α = 73.082(2), β = 87.303(2), γ = 89.419(2)°; Z = 2; V = 2023.8(4) Å3; Dc = 1.254 g cm−3; μ = 0.553 mm−1, min. and max. correction factors 0.675 and 0.926; 2θmax = 61.0°; 19
; a = 10.2529(12), b = 12.6020(15), c = 16.390(2) Å, α = 73.082(2), β = 87.303(2), γ = 89.419(2)°; Z = 2; V = 2023.8(4) Å3; Dc = 1.254 g cm−3; μ = 0.553 mm−1, min. and max. correction factors 0.675 and 0.926; 2θmax = 61.0°; 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 266 collected reflections, 10794 unique [Rint = 0.0440]; number of data/restraints/parameters 10794/7/442; final GoF 1.012; R1 = 0.0545 [8226 reflections, I > 2σ(I)]; wR2 = 0.1460 for all data; largest difference peak: 1.71 e Å−3 close to the metal atom. CCDC 945443 (2a) contains the supplementary crystallographic data for this paper.
266 collected reflections, 10794 unique [Rint = 0.0440]; number of data/restraints/parameters 10794/7/442; final GoF 1.012; R1 = 0.0545 [8226 reflections, I > 2σ(I)]; wR2 = 0.1460 for all data; largest difference peak: 1.71 e Å−3 close to the metal atom. CCDC 945443 (2a) contains the supplementary crystallographic data for this paper.
Immobilisation of 2a on inorganic supports
To a suspension of the corresponding inorganic support (0.50 g) in wet toluene (10.0 mL) a solution of 2a (50 mg) in toluene (5.0 mL) was added. The reaction slurry was stirred for one hour at r.t. and after that refluxed for 24 h. The resulting mixture was cooled down to r.t. The solid was separated by decantation and washed with toluene (10 mL), dichloromethane (2 × 10 mL) and diethylether (2 × 10 mL) and dried in vacuo at 60 °C to afford an off-white solid.
2a–MCM-41 yield 0.465 g (93.0%). Found: Rh, 0.96% (9.57 mg g−1). FT-IR: 1629 cm−1 (br.), ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and ν(CC), 1241 cm−1, 1043 cm−1, 800 cm−1ν(Si–O–Si). 13C-NMR CP-MAS: δ 147.5 (CH), 137.2 (CH), 130.8 (CH), 124.8 (CHimd), 69.4 (CHCOD), 53.7 (CH2N), 32.4–19.4 (CH2COD and –CH2–), 8.5 (CH2Si). 29Si-NMR CP-MAS: δ −92.4, −101.1, −110.7 (MCM-41), −70.2 to −51.4 (br, CH2Si).
N) and ν(CC), 1241 cm−1, 1043 cm−1, 800 cm−1ν(Si–O–Si). 13C-NMR CP-MAS: δ 147.5 (CH), 137.2 (CH), 130.8 (CH), 124.8 (CHimd), 69.4 (CHCOD), 53.7 (CH2N), 32.4–19.4 (CH2COD and –CH2–), 8.5 (CH2Si). 29Si-NMR CP-MAS: δ −92.4, −101.1, −110.7 (MCM-41), −70.2 to −51.4 (br, CH2Si).
2a–KIT-6 yield 0.484 g (96.8%). Found: Rh, 0.55% (5.53 mg g−1). FT-IR: 1636 cm−1 (br.), ν(CN) and ν(CC), 1241 cm−1, 1048 cm−1, 800 cm−1ν(Si–O–Si). 13C-NMR CP-MAS: δ 147.5 (CH), 136.9 (CH), 130.4 (CH), 123.1 (CHimd), 96.2 (CHCOD), 67.6 (CHCOD), 53.9 (CH2N), 28.1–22.8 (CH2COD and –CH2–), 8.4 (CH2Si). 29Si-NMR CP-MAS: δ −92.5, −100.9, −110.0 (KIT-6), −66.3 to −52.6 (br, CH2Si).
Hydrosilylation of acetophenone
A mixture of 1,1,1,3,5,5,5-heptamethylsiloxane (2.0 mL), the corresponding catalyst (14.4 mg (2a), 12.3 mg (2b), 0.020 mmol), acetophenone (0.234 mL, 2.00 mmol) and mesitylene (0.234 mL, 2.00 mmol, internal standard) was stirred at T = 80 °C. Samples were taken at regular time intervals and analysed by gas chromatography.Co-polymerization by homogeneous catalytic hydrosilylation (2.0 mol% catalyst)
4-Dioxane (2.0 mL) was added to a mixture of 1,1,3,3,5,5-hexamethylsiloxane (0.254 mL, 1.00 mmol) terephthalaldehyde (134 mg, 1.00 mmol) and 2a (14.4 mg, 0.020 mmol). The reaction mixture was stirred at 110 °C for 4 days. The solvent was removed in vacuo and the residue was extracted with THF (5 mL) to afford an orange oil.Co-polymerization by heterogeneous catalytic hydrosilylation (2.0 mol% catalyst based on Rh content)
1,4-Dioxane (2.0 mL) was added to a mixture of 1,1,3,3,5,5-hexamethylsiloxane (0.025 mL, 0.100 mmol,), terephthalaldehyde (13.4 mg, 0.100 mmol) and the corresponding heterogeneous catalysts (0.002 mmol). The reaction mixture was stirred at 110 °C for 4 days. The heterogeneous catalyst was removed by decantation. The resulting mixture was dried in vacuo and the residue was extracted with THF (5 mL) to afford an orange oil.Co-polymerization by heterogeneous catalytic hydrosilylation (0.2 mol% catalyst based on Rh content)
1,4-Dioxane (2.0 mL) was added to a mixture of 1,1,3,3,5,5-hexamethylsiloxane (0.254 mL, 1.00 mmol), terephthalaldehyde (134 mg, 1.00 mmol) and the corresponding heterogeneous catalysts (0.002 mmol). The reaction mixture was stirred at 110 °C for 1 day. The heterogeneous catalyst was removed by decantation. The resulting mixture was dried in vacuo and the residue was extracted with THF (5 mL) to afford an amber gelatinous compound.Recycling of the heterogeneous catalyst
The reaction mixture was centrifuged and the corresponding heterogeneous catalyst was separated from the product by decantation, washed with CH2Cl2 (3× 3.0 mL), Et2O (3× 3.0 mL), dried in vacuo and reused without further purification.3. Results and discussion
3.1. Synthesis and characterisation of the catalyst precursors
[Rh(X)(COD)(NHC)] (X = Cl, Br) complexes have been revealed as efficient precatalysts in homogeneously catalysed hydrosilylation processes.10 In particular, Si(OR)3-functionalized rhodium–NHC complexes are species of great interest due to their potential for immobilisation on mineral supports.13,21–23The preparation of Si(OR)3-functionalised rhodium–NHC complexes can be accomplished using imidazolium salts as NHC ligand precursors in order to obtain silver(I)–NHC compounds, which are widely used as reagents for transmetallation reactions.24 The imidazolium iodide salts used as ligand precursors in this work were synthesized in good yield by the reaction of the corresponding 1-substituted imidazole and I(CH2)3Si(iPrO)3 in acetonitrile at 90 °C (Scheme 1). The imidazolium salts 1a and 1b have been fully characterised by elemental analysis, mass spectrometry (ESI+), 1H, 13C{1H} and 29Si NMR spectroscopy (see Experimental).
 | ||
| Scheme 1 | ||
The rhodium complexes 2a and 2b were synthesised by Lin's method of transmetallation from the intermediate silver(I) complexes prepared in situ by reaction of the corresponding imidazolium salt with Ag2O (Scheme 2).24 Complexes 2a and 2b were isolated, after recrystallisation, as pure yellow crystalline solids in high yield and characterised by elemental analysis, mass spectrometry (ESI+) and 1H and 13C{1H} NMR spectroscopy. The 1H NMR spectra of complexes 2a and 2b provided evidence for the absence of the NCHN resonance of the parent imidazolium salts and showed the characteristic set of resonances for both the 3-triisopropoxysilylpropyl and the 2,6-diisopropylphenyl (2a) and n-butyl (2b) substituents on the NHC ligands. The 13C{1H} NMR spectra exhibit a doublet resonance at δ 183.5 ppm (1JRh–C = 52 Hz) (2a) and 183.3 ppm (1JRh–C = 51 Hz) (2b), which confirms the coordination of the carbene ligand to the rhodium centre.23,25,26
 | ||
| Scheme 2 | ||
The structure of complex 2a was determined by X-ray crystallography. A view of the molecular geometry of this species is shown in Fig. 1. The rhodium atom displays a slightly distorted square planar coordination. The COD ligand bonds to the metal through its two olefinic bonds in a chelate mode, showing a typical tub conformation; the carbon atom C(9) of the NHC ligand and the chlorido ligand complete the metal coordination environment. The NHC ligand contains two different N-substituents, namely, 2,6-diisopropylphenyl at N(1) and triisopropoxysilylpropyl at N(2). The geometry of the fragment [Rh(Cl)(COD)(NHC)] compares well with those reported for the complex [Rh(Cl)(COD)(2-methoxyethyl-NHC-(CH2)3Si(OiPr3)3)] (2c)23 and related compounds.25,26 The most striking feature of the structure concerns the high structural trans effect showed by the NHC ligand compared with that of the chloride, making the two Rh–olefin bond distances clearly different (Rh–M(1) 1.989(3) Å trans to Cl, and Rh–M(2) 2.084(3) Å trans to NHC ligand; see Fig. 1 caption).
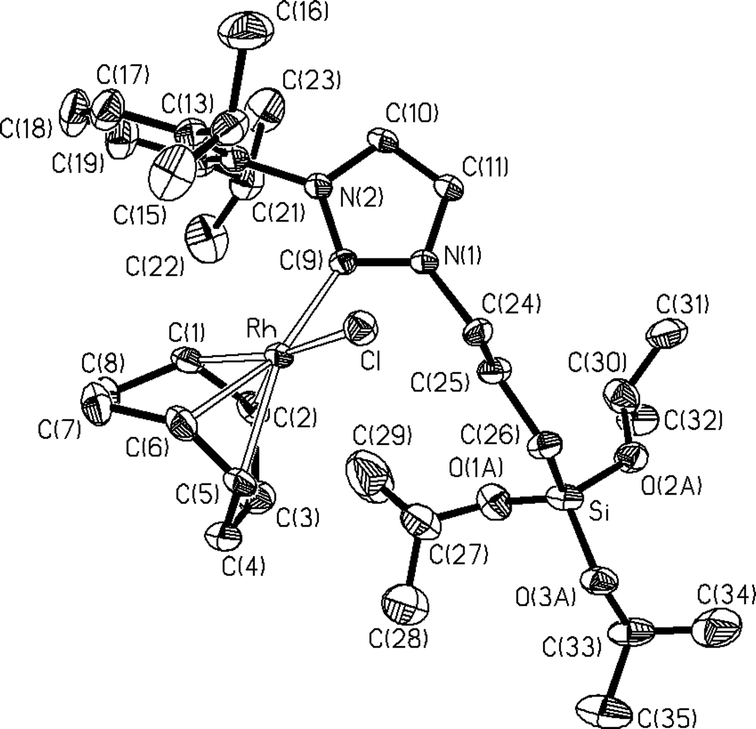 | ||
| Fig. 1 Molecular diagram of complex 2a. Selected bond lengths (Å) and angles (°): Rh–Cl 2.3913(9), Rh–C(1) 2.111(3), Rh–C(2) 2.105(3), Rh–C(5) 2.203(3), Rh–C(6) 2.188(3), Rh–C(9) 2.043(3), C(1)–C(2) 1.400(5), C(5)–C(6) 1.380(5); Cl–Rh–C(9) 87.79(9), Cl–Rh–M(1) 175.82(8), Cl–Rh–M(2) 90.53(8), C(9)–Rh–M(1) 94.39(11), C(9)–Rh–M(2) 87.36(10), N(1)–C(9)–N(2) 104.6(2). M(1) and M(2) represent the midpoints of the olefinic bonds C(1)–C(2) and C(5)–C(6). | ||
3.2. Homogeneous catalytic hydrosilylation of acetophenone: influence of the N-substituent
The rhodium-catalyzed hydrosilylation of acetophenone with 1,1,1,3,5,5,5-heptamethyltrisiloxane (HepTMS) to produce PhMeCH–O–SiMe(OSiMe3)2 (Scheme 3) has been demonstrated to be an accurate activity test bench, whose results could be extrapolated to the synthetic methodology established for the preparation of poly(silyl ether)s by catalytic hydrosilylation.23 The new compounds 2a and 2b and the previously reported 2c, having a 2-methoxyethyl substituent of hemilabile character, were used as catalyst precursors for the catalytic hydrosilylation of acetophenone with HepTMS (Scheme 3).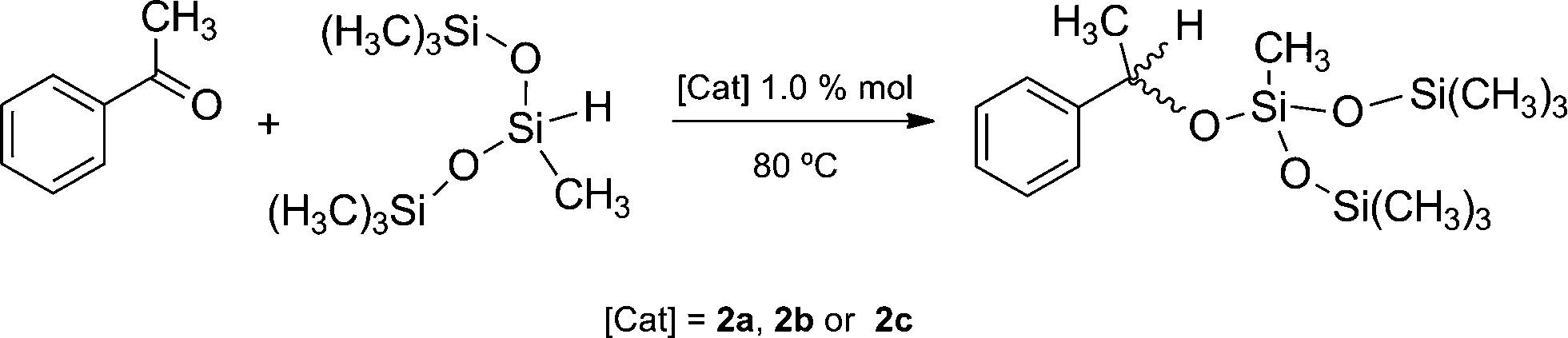 | ||
| Scheme 3 | ||
The catalytic reactions of acetophenone with HeptMTS using the rhodium(I)-catalyst 2a, 2b or 2c were monitored by GC and 1H NMR. As can be seen in Fig. 2, the N-substituent on the NHC ligand strongly influences the catalytic activity. Complex 2a, which contains the bulky 2,6-diisopropylphenyl substituent, exhibited the highest catalytic activity with conversion of 95% in less than 2 h and a TOF1/2 of 73 h−1. Complexes 2b and 2c, which contain a straight-chain flexible wingtip group of approximately the same length, were considerably less active, exhibiting TOF1/2 values of 36 and 24 h−1, respectively. Nevertheless, full acetophenone conversion was observed at longer reaction times with both catalyst precursors.
 | ||
| Fig. 2 Conversion (%) versus time (min) for catalytic hydrosilylation of acetophenone with HeptMTS using 2a, 2b or 2c (1.0 mol%). Turnover frequency (TOF1/2) recorded at 50% conversion. | ||
On the other hand, the inferior catalytic activity of 2c compared to 2b points to a negative influence of the potentially hemilabile 2-methoxyethyl substituent. This observation is in sharp contrast with the positive effect of this group on the hydrogen transfer catalytic activity of the related complex [Ir(Cl)(COD)(Me-NHC-(CH2)2OCH3], which has been substantiated by DFT calculations.26 The superior catalytic activity of 2a could be ascribed to the steric protection of the catalytic active species introduced by the bulky substituent on the NHC ligand.
1H NMR studies of the reaction of acetophenone with HepMTS in C6D6, using 2a (10 mol%) as catalyst confirm that a pre-activation step is required as no reaction was observed below 60 °C. Initially, we observed the appearance of resonances corresponding to the hydrosilylation product, PhMeCH–O–SiMe(OSiMe3)2,23 together with a doublet resonance at δ −20.62 ppm (1JRh–H = 40 Hz) due to a Rh–H intermediate species. After 30 minutes at 60 °C, the 1H NMR spectra showed an increase of the resonances due to PhMeCH–O–SiMe(OSiMe3)2 and the formation of a mixture of unidentified rhodium hydride complexes. On the other hand, solutions of 2a in C6D6 and wet C6D6 (5% H2O) were stable for 24 h at 80 °C and no traces of any decomposition product nor iPrOH were observed. These observations suggest a hydride-mediated mechanism resulting from the silane activation, in full agreement with the established rhodium-catalysed ketones hydrosilylation mechanisms.27–30
3.3. Immobilisation of 2a on inorganic supports
The results described above clearly evidenced the superior catalytic performance of precursor 2a. These results prompted us to select complex 2a as precursor for the preparation of different heterogeneous catalysts. Thus, 2a–MCM-41 and 2a–KIT-6 were prepared by refluxing a mixture of 2a and the corresponding inorganic support in wet toluene for 24 h (Scheme 4). | ||
| Scheme 4 | ||
The new materials were isolated as off-white solids that were characterised by ICP-MS, FT-IR, 13C and 29Si CP-MAS NMR and TEM. We have determined a rhodium loading of 9.57 mg g−1 and 5.53 mg g−1 for 2a–MCM-41 and 2a–KIT-6, respectively. The FT-IR spectra of the new materials show the stretching vibration modes of the mesoporous framework (Si–O–Si) at around 1241 cm−1, 1043 cm−1 and 800 cm−1.13 The FT-IR spectra of the grafted materials exhibit an additional broad absorption corresponding to the ν(CN) and ν(CC) stretching modes of the ligand at around 1630 cm−1.21,22
The resonances observed in the 13C CP-MAS solid state NMR spectra of 2a–MCM-41 and 2a–KIT-6 compare well with those observed in the 13C{1H} NMR spectra of the parent complex 2a. The most prominent resonances in the 13C CP-MAS solid state NMR spectra of the solids are those due to the CHimd [δ 124.8−123.1 ppm], CH2N [δ 53.9−52.6 ppm] and CH2Si [δ 8.5−8.3 ppm] carbon atoms. The 29Si CP-MAS solid state NMR spectra of these new materials exhibit resonances of great intensity corresponding to the silicon atoms of the silica (at around −92.0, −100 and −110 ppm) and less intense broad resonances in between δ −51.4 and −70.2 ppm (2a–MCM-41) and δ −52.6 and −66.3 ppm (2a–KIT-6) assigned to T1, T2 and T3 environments of the CH2Si silicon atoms.31
The results of N2-adsorption/desorption studies of MCM-41, 2a–MCM-41, KIT-6 and 2a–KIT-6, including BET surface area, total pore volume, and Barret–Joyner–Halenda (BJH) pore size are shown in Table 1. These results showed that the surface area, pore volume and pore diameter of 2a–MCM-41 and 2a–KIT-6 decreased, which suggests the inclusion of the rhodium species 2a inside the channels of the mesoporous materials MCM-41 and KIT-6. As a whole, the above data confirm that complex 2a has been effectively immobilised on MCM-41 and KIT-6 (Fig. 3).
 | ||
| Fig. 3 Transmission electron microscopy (TEM) of 2a–MCM-41 (a-left 100 nm; a-right 20 nm) and 2a–KIT-6 (b-left 200 nm; b-right 50 nm). | ||
3.4. Synthesis of high molecular weight poly(silyl ether)s
Poly(silyl ether)s represent a family of promising polymeric materials.32–34 The backbones of poly(silyl ether)s contain hydrolytically reactive Si–O–C bonds,35 which make them attractive in many applications, such as biodegradable materials or controlled release of drugs.36–38 Weber's group reported in 1998 the first example of synthesis of poly(silyl ether)s by transition metal-catalysed hydrosilylation using [Ru(CO)H2(PPh3)3] as precatalyst.39 This method allowed the synthesis of a number of poly(silyl ether)s with average Mw between 10000 and 150000 g mol−1.40,41 Following our interest in the chemistry and catalytic applications of Rh(I)–NHC (NHC = N-heterocyclic carbene) species,25,26,42–44 we have recently found that both homogeneous and heterogeneous catalytic systems based on the species 2c, which contain a potentially hemilabile 2-methoxyethyl substituent, are also effective for the synthesis of poly(silyl ether)s.23 However, such catalytic systems afford polymers with a relatively low average Mw up to 94000 g mol−1, and the recycled heterogeneous catalyst showed a drastic decrease in the average Mw. Thus, the design and development of effective synthetic methodologies that permit the synthesis of high molecular weight poly(silyl ether)s and recycling of heterogeneous catalysts still remain a challenge.
In this work, we have studied the reaction of terephthalaldehyde with 1,1,3,3,5,5-hexamethyltrisiloxane (HexMTS) in 1,4-dioxane at 110 °C using the rhodium chlorido species 2a, or the materials 2a–MCM-41 and 2a–KIT-6 as catalyst precursors. These catalytic reactions proceed quantitatively, affording orange oils or amber gelatinous compounds (depending on the Mw) which have been characterised as the poly(silyl ether) 3 by comparison of their 1H, 13C{1H} and 29Si{1H} NMR spectra with reported data (Scheme 5).23 It is worth noting that GPC analyses of 3 showed UV absorption at 254 nm due to the aromatic rings and all along the polymer peaks, in agreement with the copolymeric nature of the samples.
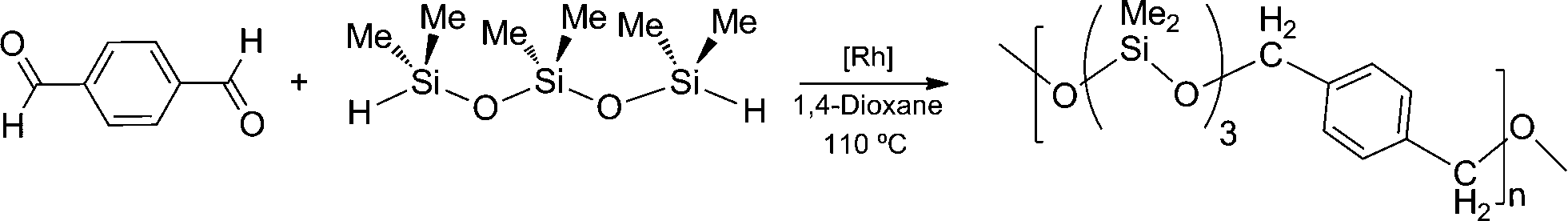 | ||
| Scheme 5 | ||
It is notable that glass transition temperature (Tg) and temperature of thermic destruction (Td) values are related to the average Mw (Table 2). The Tg of these polymers are in the range of −86.3 °C and −89.4 °C. The polymers with high Mw (Table 2, entry 2) have higher Tg values. These Tg values compare well with those reported for analogous poly(silyl ether)s23 and are lower than those reported for polymers obtained with the ruthenium catalytic system.39,40 Thermogravimetric analysis (TGA) studies showed complete destruction of the polymers in the range of 456 °C and 510 °C with a weight loss of around 96%. In this case, the average Mw also influences the temperature of thermic destruction which is higher for polymers with high average Mw (Table 2). Thus, an increase in the average Mw produces polymers with higher thermic resistance.
The homogenous catalyst precursor 2a gave a polymer with an average Mw = 34000 g mol−1, much higher than that obtained with 2c as catalyst precursor (Mw = 5200 g mol−1) under the same reaction conditions (2.0 mol% of catalyst and 110 °C in 1,4-dioxane).23 The solvent has an impact on the molecular weight of the polymer (Table 3). Interestingly, the copolymerization reaction also occurs under solvent-free conditions (Table 3, entry 3). However, polymers with higher average Mw were always observed when 1,4-dioxane was used as solvent (Table 3, entry 2).
| Entry | Solvent | M w (g mol−1) | PDI |
|---|---|---|---|
| a 4 days at 110 °C using 2.0 mol% of 2a as catalyst precursor, 100% of conversion. | |||
| 1 | Toluene | 2300 | 2.4 |
| 2 | 1,4-Dioxane | 34000 |
2.2 |
| 3 | HexMTS | 7000 | 1.4 |
The hybrid catalyst 2a–MCM-41 (2.0 mol% of catalyst and 4 days at 110 °C in 1,4-dioxane) efficiently catalysed the copolymerisation of terephthalaldehyde with 1,1,3,3,5,5-hexamethyltrisiloxane (HexMTS), affording polymers also with bimodal distribution. However, the polymer with the lowest average molecular weight fraction, Mw = 28100 g mol−1 and PDI = 1.2, represents 81% of the total mass, whereas the high average molecular weight, Mw = 378000 g mol-1 and PDI = 1.9, is 19% (Table 4, entry 1).
| Entry | Catalyst | Catalyst loading (mol%) | M w e (g mol−1) | PDI |
|---|---|---|---|---|
| a 100% conversion. b 4 days at 110 °C. c 1 day at 110 °C. d 2 days at 110 °C. e Polymers with a bimodal distribution (mass % wt). | ||||
| 1 | 2a–MCM-41b | 2.0 | 3.78 × 105/(19%) | 1.9 |
| 2.81 × 104/(81%) | 1.2 | |||
| 2 | 2a–MCM-41c | 0.2 | 2.61 × 106/(35%) | 2.1 |
| 1.79 × 105/(65%) | 1.2 | |||
| 3 | 2a–MCM-41d | 0.2 | 2.78 × 106/(34%) | 1.6 |
| 6.48 × 105/(66%) | 1.2 | |||
| 4 | 2a–KIT-6b | 2.0 | 1.34 × 105/(13%) | 1.8 |
| 3.8 × 103/(87%) | 1.4 | |||
| 5 | 2a–KIT-6c | 0.2 | 4.43 × 105/(21%) | 2.3 |
| 2.37 × 104/(79%) | 1.2 | |||
| 6 | 2a–KIT-6d | 0.2 | 5.30 × 105/(19%) | 2.1 |
| 2.66 × 104/(81%) | 1.2 | |||
On the other hand, the heterogeneous catalyst 2a–KIT-6 produced polymers of lower average molecular weight exhibiting also a bimodal distribution (Table 4, entry 4). The low molecular weight polymers represent the most abundant fraction (≈90%) with average Mw lower than 4000 g mol−1 and PDI values in the range of 1.3–1.4. However, the average Mw of the high molecular weight fraction obtained using the heterogeneous catalyst 2a–KIT-6 (Table 4, entry 4), Mw = 134000 g mol−1 is considerably bigger.
Interestingly, upon reducing the catalyst loading and reaction time (0.2 mol% catalyst and 1 day of reaction at 110 °C) and using the heterogeneous catalysts 2a–MCM-41 and 2a–KIT-6, we observed an extraordinary enhancement of the average Mw of the obtained polymers. This is remarkable in the case of the catalytic systems based on the material 2a–MCM-41 which allow for the preparation of polymers with an average Mw = 2.61 × 106 g mol−1 (Table 4, entry 2). Additionally, we have proved that increasing the reaction time from 1 day to 2 days did not produce a noticeable change (Table 4, entries 3 and 6). This is an exciting result and represents the first synthetic route to poly(silyl ether)s with high average molecular weight.
The results described above provide evidence that the inorganic support strongly influences the average Mw of the polymers. Thus, polymers with higher average Mw were obtained using 2a–MCM-41 and 2a–KIT-6 as catalysts (Table 4). The best catalytic performance, in terms of average Mw, was achieved with 2a–MCM-41. The polymers obtained using the hybrid heterogeneous catalysts have a bimodal distribution This fact could be attributed to a confinement effect exerted by the support.
GC monitoring of the copolymerisation reactions of terephthalaldehyde with 1,1,3,3,5,5-hexamethyltrisiloxane (HexMTS) revealed the total consumption of the co-monomers after 5 min, using both the homogeneous catalyst precursor 2a and the heterogeneous catalyst 2a–MCM-41. Additionally, studies on the variation of Mw with reaction time provide evidence that in both homo- and heterogeneously catalysed processes, polymerisation proceeds in a stepwise manner with the Mw of the polymer continuously increasing with time. These results are in agreement with step-growth polymerisation.45
Recycling studies using 2a–MCM-41 and 2a–KIT-6 as catalysts showed that in both cases and independent of the catalyst loading, the average molecular weight of the obtained polymers decreased after three uses, being dramatic in the case of 2a–MCM-41. We have studied the possibility of leaching of active species from the heterogeneous material; however, the solutions obtained after decantation of the heterogeneous catalysts showed no activity in the hydrosilylation of acetophenone. Furthermore, in order to discard the leaching of active species during reaction, we heated a suspension of 2a–MCM-41 in 1,4-dioxane for 24 hours at 110 °C. The 1,4-dioxane solution was filtered, and acetophenone and HepTMS were added. The mixture was heated for 16 hours and no trace of the hydrosilylation product was observed. An important detail observed is the slight enhancement of the weight of the recycled heterogeneous catalyst. Although other reasons cannot be excluded, this fact is compatible with the presence of polymer chains blocking the channels of the heterogeneous catalyst, which could be an explanation for diminishing Mw after each cycle.
4. Conclusions
The new rhodium(I) complexes [Rh(Cl)(COD)(R-NHC-(CH2)3Si(OiPr3)3)] (R = 2,6-diisopropylphenyl (2a); n-butyl (2b)) have been synthesised and fully characterised. The study of their application as ketone hydrosilylation catalysts showed a clear N-substituent effect, with 2a being the most active catalyst precursor. Complex 2a has been immobilised in the mesoporous materials MCM-41 and KIT-6. The new hybrid materials have been fully characterised and used as catalyst precursors for the preparation of poly(silyl ether)s. The heterogeneous catalytic systems based on the materials 2a–MCM-41 and 2a–KIT-6 afford polymers with higher average molecular weight (Mw) than the homogeneous catalyst 2a. Remarkably, upon reducing the catalyst loading from 2.0 mol% to 0.2 mol% and using the most active heterogeneous catalytic systems, it was possible to obtain polymers with high average molecular weight Mw = 2.61 × 106 g mol−1 (2a–MCM-41) and Mw = 4.43 × 105 g mol−1 (2a–KIT-6).In summary, it has been demonstrated that by using bulky N-substituents on the NHC ligand and by employing mesoporous materials as catalyst supports, it is possible to obtain copolymers with high average Mw. A comprehensive study including a variety of co-monomers is in progress.
Acknowledgements
Financial support from the Spanish “Ministerio de Economia y Competividad” projects, CONSOLIDER INGENIO CSD2006-0015 and CSD2009-00050, CTQ2011-27593 and CTQ2012-35665, “Juan de la Cierva” program (M. I.) and “Diputación General de Aragón-Fondo Social Europeo” group E07 is acknowledged.Notes and references
- A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef.
- B. Marciniec, K. H. Maciejewski, C. Pietraszuk and P. Pawluć, in Hydrosilylation: A Comprehensive Review on Recent Advances, ed. B. Marciniec, Springer, London, 2008 Search PubMed.
- I. Chorkendorff and J. W. Niemantsverdriet, in Concepts of Modern Catalysis and Kinetics, Wiley-VCH, Weinheim, 2003 Search PubMed.
- G. Rothenberg, in Catalysis: Concepts and Green Applications, Wiley-VCH, Weinheim, 2008 Search PubMed.
- B. Marciniec, K. Szubert, M. J. Potrzebowski, I. Kownacki and H. Maciejewski, ChemCatChem, 2009, 1, 304–310 CrossRef CAS.
- C. E. Song and S. Lee, Chem. Rev., 2002, 102, 3495–3524 CrossRef CAS PubMed.
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108–122 RSC.
- M. Tada and Y. Iwasawa, Coord. Chem. Rev., 2007, 251, 2702–2716 CrossRef CAS PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- F. E. Hahn, ChemCatChem, 2013, 5, 419–430 CrossRef CAS.
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- K. H. Park, S. Kim and Y. K. Chung, Bull. Korean Chem. Soc., 2008, 29, 2057–2060 CrossRef CAS.
- S. Dastgir, K. S. Coleman and M. L. H. Green, Dalton Trans., 2011, 40, 661–672 RSC.
- C. S. J. Cazin, M. Veith, P. Braunstein and R. B. Bedford, Synthesis, 2005, 622–626 CAS.
- A. Monge-Marcet, R. Pleixats, X. Cattoën and M. Wong Chi Man, Catal. Sci. Technol., 2011, 1, 1544–1563 CAS.
- S. Berardi, M. Carraro, M. Iglesias, A. Sartorel, G. Scorrano, M. Albrecht and M. Bonchio, Chem.–Eur. J., 2010, 16, 10662–10666 CrossRef CAS PubMed.
- R. Uson, L. A. Oro and J. A. Cabeza, Inorg. Synth., 1985, 23, 126–130 CrossRef CAS.
- E. Besson, A. Mehdi, D. A. Lerner, C. Reyé and R. J. P. Corriu, J. Mater. Chem., 2005, 15, 803–809 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. del Pozo, A. Corma, M. Iglesias and F. Sánchez, Organometallics, 2010, 29, 4491–4498 CrossRef CAS.
- G. Villaverde, A. Corma, M. Iglesias and F. Sánchez, ChemCatChem, 2011, 3, 1320–1328 CrossRef CAS.
- G. Lázaro, M. Iglesias, F. J. Fernández-Alvarez, P. J. Sanz Miguel, J. J. Pérez-Torrente and L. A. Oro, ChemCatChem, 2013, 5, 1133–1141 CrossRef.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, V. Gierz, F. J. Lahoz and L. A. Oro, Organometallics, 2008, 27, 224–234 CrossRef.
- M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, F. J. Modrego, S. Winterle, C. Cunchillos, F. J. Lahoz and L. A. Oro, Organometallics, 2011, 30, 5493–5508 CrossRef.
- I. Ojima, M. Nihonyanagi, T. Kogure, M. Kumagai, S. Horiuchi, K Nakatsugawa and Y. Nagai, J. Organomet. Chem., 1975, 94, 449–461 CrossRef CAS.
- G. Z. Zheng and T. H. Chan, Organometallics, 1995, 14, 70–79 CrossRef CAS.
- N. Schneider, M. Finger, C. Haferkemper, S. Bellemin-Laponnaz, P. Hofmann and L. H. Gade, Chem.–Eur. J., 2009, 15, 11515–11529 CrossRef CAS PubMed.
- D. Imao, M. Hayama, K. Ishikawa, T. Ohta and Y. Ito, Chem. Lett., 2007, 36, 366–367 CrossRef CAS.
- M. Jia, A. Seifert and W. R. Thiel, Chem. Mater., 2003, 15, 2174–2180 CrossRef CAS.
- Y. Li, M. Seino and Y. Kawakami, Macromolecules, 2000, 33, 5311–5314 CrossRef CAS.
- Y. Li and Y. Kawakami, Macromolecules, 1999, 32, 6871–6873 CrossRef CAS.
- T. Nishikubo, A. Kameyama, Y. Kimura and T. Nakamura, Macromolecules, 1996, 29, 5529–5534 CrossRef CAS.
- M. G. Voronkov, V. P. Mileshkevich and Y. A. Yuzhelevskii, in The Siloxane Bond, Consultants Bureau, New York, 1978, Si–O–Si, pp. 146–149, Si–O–C, pp. 323–340 Search PubMed.
- Y. Nagasaki, F. Matsukura, M. Kato, H. Aoki and T. Tokuda, Macromolecules, 1996, 29, 5859–5863 CrossRef CAS.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- A. M. Issam and M. Haris, J. Inorg. Organomet. Polym., 2009, 19, 454–458 CrossRef CAS.
- J. K. Paulasaari and W. P. Weber, Macromolecules, 1998, 31, 7105–7107 CrossRef CAS.
- J. M. Mabry, J. K. Paulasaari and W. P. Weber, Polymer, 2000, 41, 4423–4428 CrossRef CAS.
- J. M. Mabry, M. K. Runyon and W. P. Weber, Macromolecules, 2002, 35, 2207–2211 CrossRef CAS.
- A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente, F. J. Lahoz, V. Polo and L. A. Oro, Angew. Chem., Int. Ed., 2011, 50, 3938–3942 CrossRef CAS PubMed.
- A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente, M. Crucianelli, V. Polo, R. Sancho, F. J. Lahoz and L. A. Oro, J. Am. Chem. Soc., 2012, 134, 8171–8183 CrossRef CAS PubMed.
- R. Azpiroz, A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente and L. A. Oro, Chem.–Eur. J., 2013, 19, 3812–3816 CrossRef CAS PubMed.
- G. Odian, in Principles of Polymerisation, Wiley-Interscience, New Jersey, 2004, 4th edn Search PubMed.
Footnotes |
| † CCDC 945443. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cy00598d |
| ‡ Dedicated to the memory of Prof. Maria Pilar Garcia Clemente. |
| This journal is © The Royal Society of Chemistry 2014 |