
The influence of the electrolyte on chemical and morphological modifications of an iron sulfide thin film negative electrode
Feng
Liao
,
Jolanta
Światowska
*,
Vincent
Maurice
,
Antoine
Seyeux
,
Lorena H.
Klein
,
Sandrine
Zanna
and
Philippe
Marcus
PSL Research University, Chimie ParisTech - CNRS, Institut de Recherche de Chimie Paris, 11 rue Pierre et Marie Curie, 75005 Paris, France. E-mail: jolanta.swiatowska@chimie-paristech.fr; Tel: +33(0)144278026
First published on 10th November 2014
Abstract
The chemical and morphological modifications of FeS thin film as anode material for LiBs have been studied in detail in two classical electrolytes usually used in Li-ion batteries: 1 M LiClO4-PC and 1 M LiPF6-EC/DMC. The X-ray photoelectron spectroscopic (XPS) analysis evidenced the formation of a solid electrolyte interphase (SEI) that contains a more significant amount of inorganic salt residues formed in LiPF6-EC/DMC than in LiClO4-PC, which is likely to increase the ionic resistivity of the SEI, thus impeding the lithiation–delithiation in the first cycles while improving its reversibility. Ion depth profiles performed by time-of-flight secondary ion mass spectrometry (ToF-SIMS) show volume expansion–shrinkage of the thin film leading to cracking and pulverization of the electrode material, which is also confirmed by scanning electron microscopy (SEM) analysis. The prolonged cycling results in penetration and accumulation of the electrolyte in a bulk electrode with accumulation of the inorganic species in the inner part of the SEI enhanced in a fluoride-containing electrolyte. Cycling in these two different electrolytes leads also to formation of two different electrode morphologies: with a compact electrode structure formed in LiClO4-PC and a foam-like, porous structure in LiPF6-EC/DMC. A model of this conversion-type thin film electrode modification based on these thorough spectroscopic and microscopic analyses induced by cycling in two different electrolytes is proposed.
1. Introduction
Understanding the surface chemistry and the lithiation-induced bulk modifications of electrode materials is crucial for improving the electrochemical performance of Li-ion batteries (LiBs). The formation and stability of the so-called solid electrolyte interphase (SEI), a layer formed on the electrode surface by reductive decomposition of the electrolyte, is one of the most significant factors influencing the performance and electrochemical efficiency of LiBs.1 The SEI is thought to be mainly composed of a compact inorganic inner layer and a porous organic outer layer and it can be strongly influenced by the electrolyte chemical composition and the electrode material.1–6Today the most conventional organic liquid electrolytes used in LiBs are based on propylene carbonate (PC), or a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC), or diethylcarbonate (DEC) solvents. On negative electrode surfaces, these solvents undergo reduction to form SEIs principally composed of lithium carbonate (Li2CO3), lithium alkyl carbonate (ROCO2Li), lithium alkoxides (RCH2OLi) and lithium alkyls (LiR).7–20 Furthermore, due to oligomerization, polymers can be formed initially and decompose during subsequent lithiation cycles.21
Salts in the electrolytes play an important role in the formation and composition of the SEI. LiClO4 can be reduced to Li2O, LiCl and/or LiClO4−n (n = 1, 2, 3), and LiPF6 to LiF and LixPFy.12 Strong Lewis acids, like PF5 and BF3 (originating from LiPF6 and LiBF4 salts, respectively) can promote polymerization by ring-opening.16 If water traces are present in the electrolyte, the moisture-sensitive ROCO2Li can convert to Li2CO3 instantly13,22 and PF5 can be further decomposed to POF3 and/or POF2(OH).23
Ultimately, the SEI should form a passivating layer avoiding further electrolyte decomposition and protecting the anode material from cracking and exfoliation. The relationship between electrolyte, interphase chemistry and morphological modification of negative electrodes has been thoroughly studied on intercalation-type and alloy-type materials, like graphite and Sn-based alloy, respectively,24–26 but not on conversion-type materials. Principally, the electrochemical studies performed previously on conversion-type FeS electrode materials prepared by different methods, such as, electrolytic deposition, thermal sulfidation, mechanical milling or sol–gel combined with casting slurry coating27–30 show a good lithiation–delithiation reversibility. Our previous extensive research studies on FeS (troilite) using a thin film electrode prepared by thermal sulfidation of a pure Fe substrate were essentially related to the conversion31 and aging mechanisms.32 Here, we report on the influence of electrolyte (LiClO4-PC and LiPF6-EC/DMC) on the chemical and morphological modifications of FeS thin film electrodes induced by cycling. The combined spectroscopic and microscopic studies performed by means of X-ray Photoelectron Spectroscopy (XPS), Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) and Scanning Electron Microscopy (SEM), respectively, were applied to investigate the interphase chemistry (including SEI) and the morphological and bulk chemical modifications of this conversion electrode material.
2. Experimental
The FeS thin film samples were prepared by thermal sulfidation of iron foil substrates (99.5% purity, Goodfellow) as described previously in detail.32 As prepared, the chemical formula was FeS1−x (x ∼ 0.07) and the thickness was about 210 nm. The sulfidized foil substrates were transferred under vacuum to an Ar filled glove box and then cut into several samples.All electrochemical measurements were performed in Teflon Swagelok half-cells controlled by a VMP3 Biologic multi-channel potentiostat/galvanostat and with metallic Li foil (Aldrich) as a reference/counter electrode and dried filter paper as a separator. The electrolytes were 1 MLiClO4-PC (Aldrich) and 1 M LiPF6-EC/DMC (1/1 wt%, Aldrich and Alfa Aesar). Galvanostatic lithiation–delithiation was performed between 2.9 to 1 V and vice versa at a constant current of 9.43 μA and 9.71 μA (∼1/4 C) in PC and EC/DMC-based electrolytes, respectively. Lithiation–delithiation by cyclic voltammetry (CV) was carried out at a scan rate of 0.5 mV s−1 between 0.25 and 3 V starting from the open circuit potential (OCP) to the negative potential direction. After electrochemical (de)lithiation, the specimens were washed with non-aqueous acetonitrile, dried by Ar flow and then transferred directly under anhydrous and anaerobic conditions using a direct transfer from glove box to the XPS32 or ToF-SIMS analysis chamber. For SEM analysis, the samples were exposed to ambient air for less than 5 min.
The XPS spectrometer (VG ESCALAB 250, Thermo Electron Corporation) was operated without using the magnetic lens to avoid sample magnetization. An Al Kα monochromatized X-ray source (hν = 1486.6 eV) was employed. Survey spectra were recorded with a 100 eV pass energy, and high resolution spectra (i.e. C1s, F1s, P2p) with a 20 eV pass energy. The photoelectron take-off angle was 90°. Peak fitting and decomposition was performed with the Avantage software provided by Thermo Electron, using a Shirley type background and Gaussian/Lorentzian peak shapes with a fixed ratio of 70/30.33 Binding energies (BEs) were calibrated using clean Cu, Ag, and Au samples. The C1s and O1s BE's were corrected from charging effects by setting the carbon peak for –CH2–CH2– bonds at 285.0 eV.
The ToF-SIMS spectrometer (ToF-SIMS 5, IonTof Gmbh) was operated in negative ions polarity. For depth profiling, a pulsed 25 keV Bi+ primary ion source delivering 1 μA over 100 × 100 μm2 was used for analysis and interlaced with a 1 keV Cs+ ion gun delivering 70 nA over 300 × 300 μm2 for sputtering. Analysis conditions were identical for all samples in order to allow direct comparison. Data acquisition and post-processing were performed using the Ion-Spec software.
Microstructural analysis was performed using a LEO 1530 VP Gemini Field Emission Scanning Electron Microscope (FESEM) operated at an acceleration voltage of 3 keV. The surface fraction occupied by cracks was measured using the Image-Pro plus 6.0 software.
3. Results and discussion
3.1. Electrochemical performance
The cyclic voltammetry (CV) performed on the FeS thin film (Fig. 1) shows one main cathodic/anodic peak (marked respectively C/A) in both electrolytes (LiClO4-PC and LiPF6-EC/DMC), corresponding to the conversion–deconversion reaction:27,32| FeS + 2Li+ + 2e− ↔ Fe + Li2S | (1) |
During the 2nd cycle, a small positive potentials shift of the cathodic/anodic peaks and lower peak current densities can be observed in both electrolytes, which can be related to surface modifications i.e. formation of a passive layer. Further cycles do not show any peak displacement, which suggests that the conversion–deconversion process is quasi-reversible.
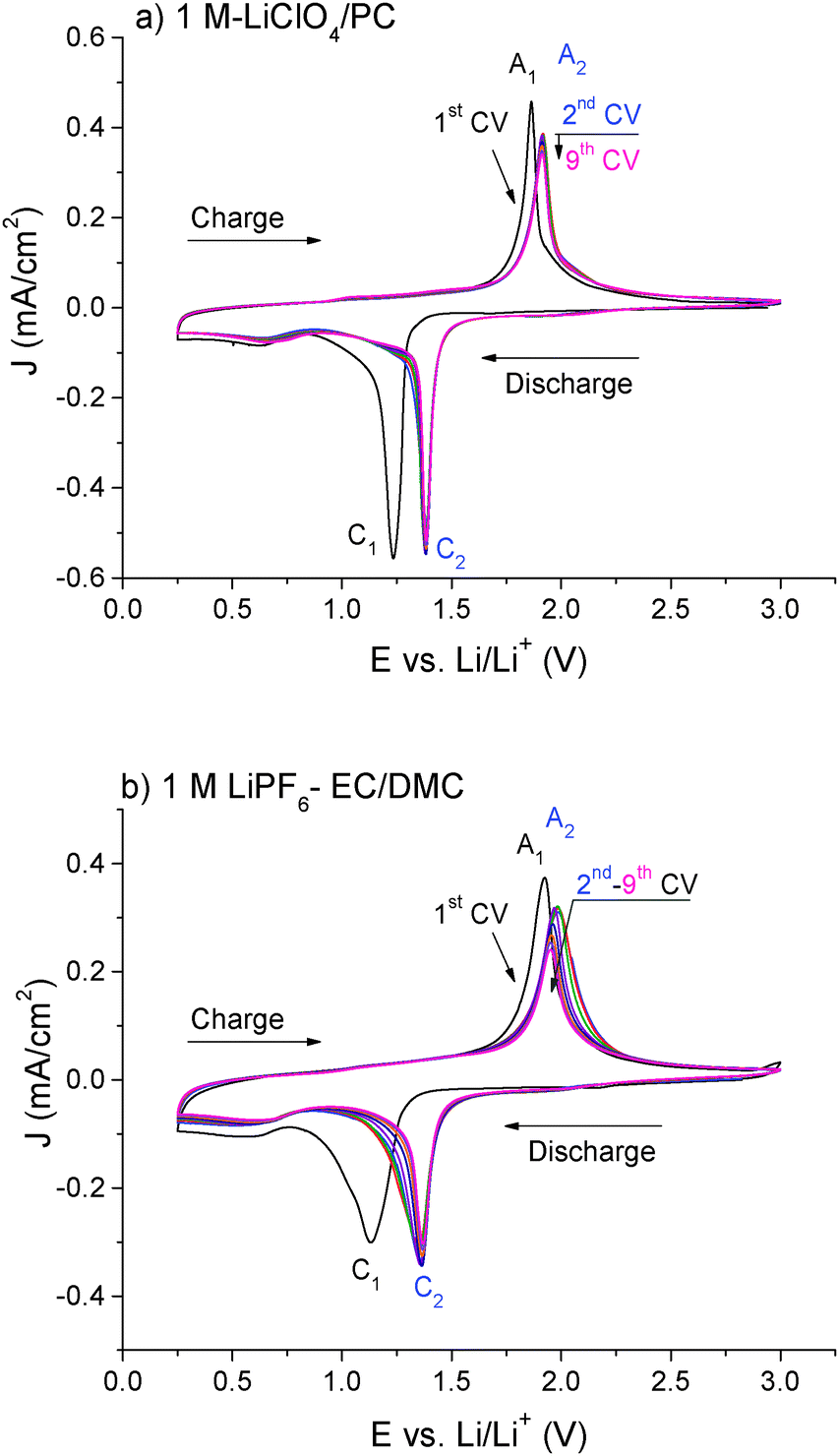 | ||
| Fig. 1 First nine cyclic voltammograms (CVs) on FeS thin film (Vscan = 0.5 mV s−1) in (a) 1 M LiClO4-PC or (b) 1 M LiPF6-EC/DMC. | ||
The galvanostatic discharge–charge curves (Fig. 2a) performed on a FeS thin film electrode display plateaus corresponding to the lithiation–delithiation (conversion–deconversion) process. The discharge plateau is displaced from 1.37 V to more positive 1.48 V and the charge plateau from ∼1.3 to ∼1.45 V in the 2nd cycle in agreement with the displacement of the cathodic/anodic peak in CV measurements. In the 60th cycle, the discharge–charge plateaus (at ∼1.5 and ∼1.8 V, respectively) are drastically reduced in width and conversion and deconversion occur more continuously. These changes in the galvanostatic curves can be a result of significant interfacial and bulk modifications of the electrode occurring in the 1st cycle and being amplified in the following cycles.
 | ||
| Fig. 2 (a) Charge–discharge curves of the FeS thin film electrodes in 1 M LiClO4-PC (solid line) and 1 M LiPF6-EC/DMC (dashed line) between 1.0 and 3.0 V at rates of about 1/4 C. (b) Charge–discharge capacity (left axis) and coulombic efficiency (right axis) versus cycle number. | ||
Higher than FeS theoretical capacity (609 mA h g−1) observed in both electrolytes (742 mA h g−1 inLiClO4-PC and 687 mA h g−1 in LiPF6-EC/DMC, Fig. 2b) is due to SEI layer formation by reductive decomposition of the electrolyte as shown hereafter. Lower initial (first 17 cycles) discharge capacity in LiPF6-EC/DMC can result from less extensive electrolyte decomposition occurring in EC/DMC than in PC-based electrolyte, as already reported,21 but also possibly from higher ionic resistivity of the formed layer hindering the ionic transport.34,35 Higher resistivity of the SEI layer formed in LiPF6-EC/DMC would be caused by the presence of lithium fluorides instead of lithium carbonates, oxides, and hydroxides components.36,37 In the following cycles (after 17 cycles), the discharge capacity becomes higher in LiPF6-EC/DMC and reaches 532 mA h g−1versus 490 mA h g−1 in LiClO4-PC after 60 cycles, possibly because of different evolutions of the SEI layers and electrode morphology as discussed below.
The columbic efficiency increases during the first 10 cycles and it is lower in LiClO4-PC (with 94%) than in LiPF6-EC/DMC (where it reaches 99%) as shown in Fig. 2b. During cycling the columbic efficiency remains relatively stable especially in LiClO4-PC. Small decrease of columbic efficiency can be observed in a case of LiPF6-EC/DMC showing some surface modification of i.e. lower SEI stability and/or its different properties.
3.2. Surface chemical modifications
The differences of the surface layers formed on the FeS thin film electrode in these two electrolytes have been studied by electronic spectroscopy (XPS) and ionic mass spectrometry (ToF-SIMS). Here, we merely present the XP core level spectra corresponding to constituents of the SEI layer (i.e. C1s, F1s, and P2p) (Fig. 3). Due to formation of a thick SEI layer and complete attenuation of the iron and sulfur core level peaks corresponding to electrode material, the Fe2p and S2p peaks are not present here.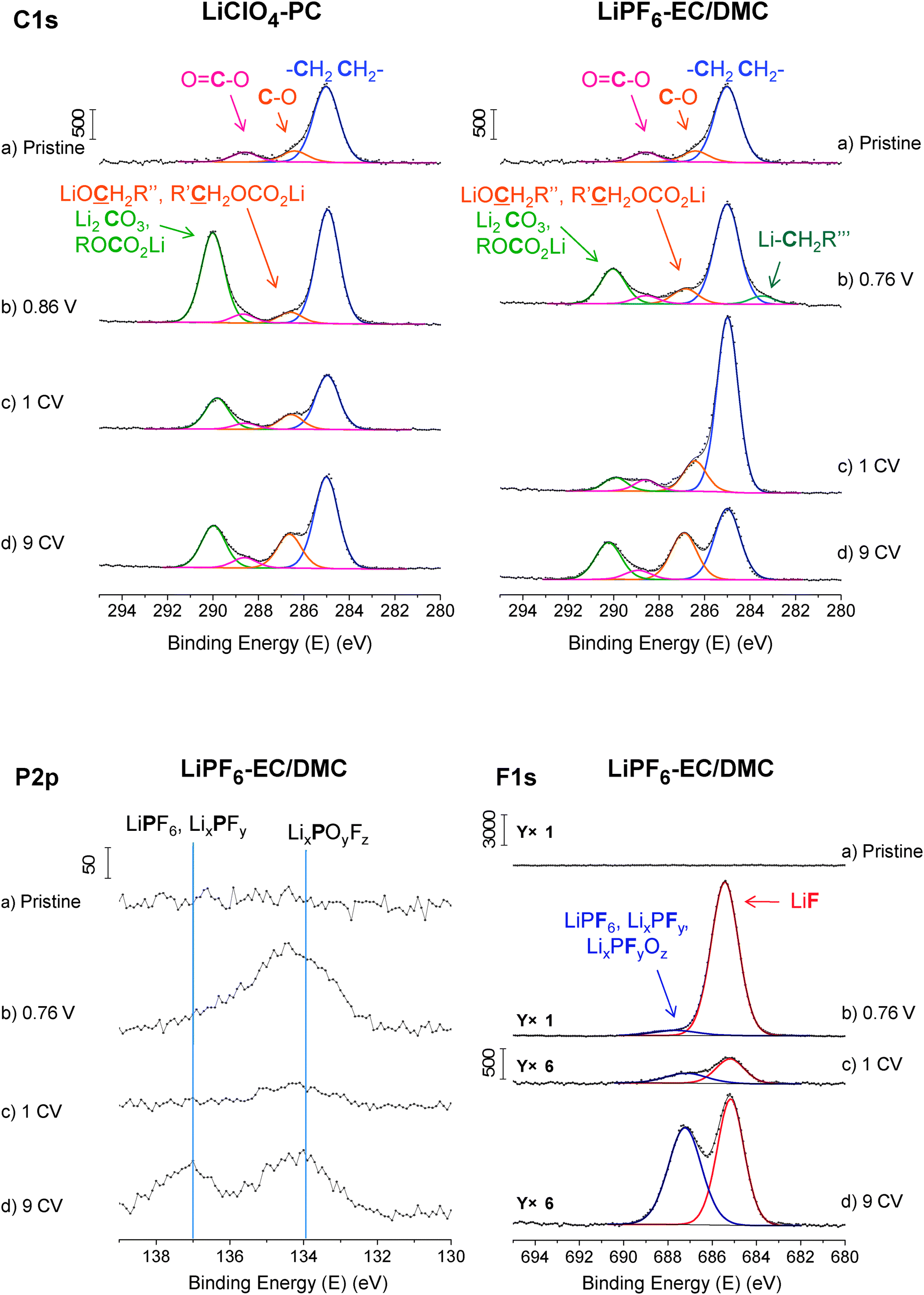 | ||
| Fig. 3 XP C1s, F1s and P2p core level spectra for FeS thin film electrodes before (a) and after lithiation at (b) 0.86 V (0.76 V) in 1 M LiClO4-PC (1 M LiPF6-EC/DMC) and (c) 1 and (d) 9 lithiation–delithiation cycles (1 and 9 CVs). | ||
The cycling in both electrolytes clearly evidences the formation of the SEI layer, principally by appearance of an additional carbon peak C1sD at 290.0 ± 0.2 eV (Fig. 3), assigned to Li2CO3 and/or ROCO2Li (R = alkyl group).19,28,31,38 In LiClO4-PC, carbonates are the dominant components of the SEI and remain so after cycling as shown by the higher relative intensity of the C1sD peak. The C1sB peak at 287.7.0 ± 0.2 eV, assigned to R'CH2OCO2Li and/or LiOCH2R”, increases in relative intensity with multi cycling showing an increasing amount of single-bonded carbon in the SEI. In LiPF6-EC/DMC, double-bonded carbon from carbonates also initially dominates the SEI composition but the lower intensity of the C1sD peak indicates less extensive electrolyte decomposition than in PC. Single-bonded carbon becomes dominant already after 1 cycle in LiPF6-EC/DMC. A minor C1sE peak at 283.5 eV, corresponding to the Li–C bond in LiCH2CH2OCO2Li,17,39,40 was observed only in the lithiated state in LiPF6-EC/DMC (Fig. 3b).
The presence and modification of the inorganic component of the SEI layer is studied by using the F1s and P2p (for the electrode cycled in LiPF6-EC/DMC) and the Cl2p (for the electrode cycled in LiClO4-PC) and ToF-SIMS mass spectra. For the sample cycled in LiPF6-EC/DMC, the F1s spectra show a major peak at 685.3 ± 0.1 eV, attributed to LiF,41,42 and a minor peak at 687.4 ± 0.3 eV, attributed to LiPF6, LixPFy, or LixPOyFz,41,43,44 the latter growing in relative intensity with multi cycling (Fig. 3). LiF, LixPFy or LixPOyFz can be decomposition products of the LiPF6 salt, accumulating in the SEI.17,45,46 ToF-SIMS mass spectra (Fig. 4) confirm the presence of fluorine (19F−) and phosphorous (31P−, 47PO−, 63PO2−, 79PO3−) originating from salt (LiPF6) decomposition and/or hydrolysis products in the SEI. The presence on the surface of some salt residues, not completely removed from the electrode surface by rinsing, cannot be excluded. The presence of a very small quantity of the hydrolysis product LixPOyFz is confirmed by the XP P2p3/2 peak observed at ∼133.9 eV (Fig. 3).46,47 Another XP P2p3/2 peak at ∼137.0 eV evidences the presence of LixPFy and/or LiPF6.46,47
 | ||
| Fig. 4 Relative elemental surface composition (at%) of the FeS thin film electrodes before and after cycling in 1 M LiClO4-PC and 1 M LiPF6-EC/DMC. | ||
For the sample cycled in LiClO4-PC, no Cl2p signal (not shown here) was observed either in the lithiated or delithiated states. It can be then concluded that the quantity of Cl-compound is well below the detection limit of XPS (∼0.5 at%) and/or the LiClO4 salt shows much lower activity than LiPF6 salt.34 However the much more sensitive ToF-SIMS technique indicated the presence of traces of chlorides in the SEI by measurable 35Cl− ions and 42LiCl− ions (not shown here).
To have a better overview of different SEI layer components as a function of cycling in these two different electrolytes, Fig. 4 summarizes the elemental surface compositions (Li1s, P2p, F1s, O1s and C1s) calculated from XPS. More Li is found in the SEI formed in LiPF6-EC/DMC than in LiClO4-PC, however, upon cycling the Li decreases in both electrolytes. The SEI layer formed in LiPF6-EC/DMC includes measurable amounts of inorganic products (containing Li, P and F) in contrast to that formed in LiClO4-PC for which only the organic constituents are observed. A similar composition of the SEI layer, enriched in inorganic products after cycling in LiPF6-EC/DMC but not after cycling in LiClO4-PC has been already observed in a case of Si negative electrodes.42 Assuming higher ionic resistivity of the SEI layer rich in inorganic components, the SEI initially formed in LiPF6-EC/DMC could impede the conversion reaction, which would contribute to the lower initial discharge capacity but higher reaction reversibility in LiPF6-EC/DMC. After 60 cycles, the surface composition of the SEI layers is very similar in both electrolytes. It is suggested that with multi cycling the organic components accumulate in the outer part of the SEI and the inorganic salt products in the inner part, thus forming a duplex structure whose inner part would not be measured by XPS due to intensity attenuation of the photoelectrons by the organic outer part. Such a duplex-like structure of the SEI layer has been already previously proposed by Peled for carbonaceous electrodes1,2 and confirmed for alloy-type and intercalation-type materials.3–5,42 It was evidenced for conversion-type sulfide31 and oxide48 materials only recently and it is further supported by the present data.
The compositional chemical differences on the surface of electrodes cycled in two different electrolytes can be also observed from surface analysis by means of ToF-SIMS negative ion mass spectra in the 40.96–41.08 and 70.96–71.08 m/z ranges (Fig. 5). C2HO− ions (m/z = 41.01) and C3H5− ions (m/z = 41.03) are present on the electrode cycled in LiClO4-PC but not in LiPF6-EC/DMC, which is contrary to the ions with m/z = 71.03, assigned to a mixture of C3H3O2− and C4H7O− (exact m/z values of 71.0133 and 71.0497, respectively49), appearing on the samples cycled in LiPF6-EC/DMC but not in LiClO4-PC. These latter ions are assigned to fragments of larger, polymer-type molecules like LiOCO2C2H4OCO2Li, LiOCO2C4H8OCO2Li, and (CH2CH2O)x/(CH2CH2OCO2)y (ref. 50) which can be formed in the electrolyte based on the EC/DMC solvent.8,14,15,21 This polymer-like character of the SEI layer can have an influence on the slight decrease of columbic efficiency during cycling. According to previous studies,12,21,51 the reduction of the PC solvent may also result in the formation of large molecules like LiOCO2C3H6OCO2Li, LiOCO2C6H12OCO2Li, and (CH2CH2O)x/(CH2CH2OCO2)y. However, here in this study no significant amount of hydrocarbon oxide CxHyOz− ions, for example C3H5O− and C6H11O− (exact m/z values of 57.0340 and 99.0810, respectively49), could be observed by ToF-SIMS on the electrodes cycled in LiClO4-PC. This is in good agreement with the XPS data that showed that Li carbonates, as a major component of the SEI layer, are more dominant in the SEI formed in LiClO4-PC than that formed in LiPF6-EC/DMC.
 | ||
| Fig. 5 ToF-SIMS negative ion mass spectra in the regions of 41 and 71 m/z for the pristine FeS thin film electrodes and after lithiation at 0.86 V (0.76 V) in 1 M LiClO4-PC (1 M LiPF6-EC/DMC) and after 1 cycle (1 CV). The spectra were recorded from the extreme surface down to 30 s of sputtering. | ||
Thus, it can be clearly concluded that the EC/DMC electrolyte containing fluorinated species favors the formation of inorganic compounds, whereas, the PC electrolyte containing LiClO4 salt leads to formation of carbonates. Moreover, the ToF-SIMS mass spectra allowed detection of polymer-type compounds in the SEI layer formed in the electrolyte made of EC/DMC. The differences in the chemical composition of the SEI layer formed in both electrolytes are redressed after 60 cycles, where only small quantity of inorganic components can be observed on the extreme surface of the electrode cycled in LiPF6-EC/DMC.
3.3. Bulk chemical modifications
To observe the bulk modifications of thin film FeS electrodes induced by the first lithiation and during the first 9 cycles the ToF-SIMS depth profiles of the LiS− and C− ions were collected and presented in Fig. 6. The LiS− ion profiles were used together with the Fe2− ion profiles (not shown) to mark the interface limits of the FeS thin film with the SEI and substrate interfaces. In the lithiated state, the LiS− ion profile shows a more uniform in-depth distribution in LiPF6-EC/DMC than in LiClO4-PC, which can be explained by a homogenous in-depth distribution of converted material. After 1 CV, two peaks are observed at the SEI/FeS and the FeS/Fe interfaces. Assuming no matrix effect, more lithium sulfide (i.e. Li2S) converted products seem to be trapped at the SEI/FeS interface in LiPF6-EC/DMC, which appears consistent with a higher ionic resistivity of the SEI owing to the presence of inorganic products as discussed above.34–37 After 9 CVs, the LiS− ion profiles markedly increase in intensity due to accumulation and trapping of Li sulfide converted products in the bulk of the thin film electrode. | ||
| Fig. 6 ToF-SIMS depth profiles of the LiS− and C− ions for the pristine FeS thin film electrodes and after lithiation at 0.86 V (0.76 V) in 1 M LiClO4-PC (1 M LiPF6-EC/DMC) and 1 and 9 cycle (1 and 9 CVs). | ||
In the first seconds of sputtering, the C− ion depth profiles all show a peak more intense on the treated samples than on the pristine sample, confirming the presence of the SEI on the surface of the cycled electrodes (Fig. 6). In the lithiated state, the peak is more intense on the sample treated in LiClO4-PC, in line with more extensive decomposition of the PC-based electrolyte as discussed above. One notices a similar width of this peak in the two electrolytes, indicating no major difference in the thickness of the SEI layers. The displacement of the FeS/Fe interface towards longer sputtering time (with lithiation and cycling) is consistent with volume expansion of the FeS thin film electrode material, which is more significant in LiPF6-EC/DMC (∼50%) than in LiClO4-PC (∼33%). However, after the first delithiation (1 CV), the sputtering time decreases, but it remains higher (∼50% in both electrolytes) than on the pristine sample. The further cycling (9 CV) leads to continuous increase of sputtering time, which can be attributed to larger volume expansion of the thin film electrodes. The volume expansion reaches 150% for the electrode cycled in LiPF6-EC/DMC and almost 100% for the electrode cycled in LiClO4-PC. After 1 and 9 cycles, the C− ion profiles are markedly changed in the bulk thin films and at the FeS/Fe interface as a result of the penetration of the electrolyte in the bulk thin film electrode. The accumulation of the C-containing products at the FeS/Fe interface after 1 and 9 CVs is consistent with the formation of cracks penetrating the thin film electrode material until the iron substrate, as observed in the SEM data presented below. The C− peak at the FeS/Fe interface becomes markedly broadened after 9 CVs in both electrolytes, suggesting formation of a more defective, thick FeS thin film electrode, more permeable to electrolyte.
Hereafter, the ToF-SIMS negative ion depth profiles for the FeS thin film electrodes obtained after 60 galvanostatic cycles in both electrolytes were compared with depth profiles made on the pristine electrode (Fig. 7). For the pristine film (Fig. 7a), the FeS bulk region can be clearly distinguished by the high intensity plateau of the FeS− ions. The thickness (∼210 nm) of the pristine FeS film corresponds to about 650 s of sputtering. C, F and Cl initial contamination predominates at the film surface and at the interface with the Fe substrate. After 60 cycles (Fig. 7b and c), the new peaks at the beginning of the LiCO2−, CH2− and LiO− ion profiles show SEI formation. A shifted FeS bulk region to higher sputtering times and the broader C− peak observed for the sample cycled in LiPF6-EC/DMC (∼240 s) than in LiClO4-PC (∼130 s), indicate formation of a thicker SEI layer in EC/DMC than in PC-based electrolyte. Consistently, the longer and markedly higher F− plateau observed for the electrode cycled in LiPF6-EC/DMC than the Cl− plateau for the LiClO4-PC-treated electrode indicates higher accumulation of inorganic products of electrolyte decomposition in EC/DMC-based than in PC-based electrolytes.
 | ||
| Fig. 7 ToF-SIMS negative ion depth profiles of FeS thin film electrodes (a) before (pristine) and after 60 cycles (galvanostatic) in (b) 1 M LiClO4-PC and (c) 1 M LiPF6-EC/DMC. | ||
A more significant volume expansion of thin film electrode cycled in LiPF6-EC/DMC than in LiClO4-PC can be deduced from higher sputtering time ∼850 vs. ∼930 s, respectively. Taking into account the sputtering time of the SEI layer and the FeS regions, the volume expansion is 120% and 170% for the thin film cycled in PC- and EC/DMC-based electrolytes, respectively. However, it must be emphasized that the SEI layer taken into consideration in the calculation of volume changes is an important contributor as it can be seen from the interfaces marked in the Fig. 7b and c. The high intensities of C− and CH2− ion profiles observed in the FeS bulk region are consistent with the penetration of the electrolyte. The large humps of C− and CH2− observed at the FeS/Fe interface for both electrolytes may account for an increased electrode roughness and/or formation of defects.
The more significant volume expansion observed for thin film iron sulfide electrodes after galvanostatic cycling in EC/DMC than in the PC-based electrolyte can be explained by:
– better and more homogenous distribution of LiS-like compounds in the bulk electrode cycled in EC/DMC-based electrolyte, deduced from a high intensity LiS− ion profile,
– more significant quantity of inorganic compounds accumulated in the bulk thin film electrode cycled in EC/DMC-based electrolyte confirmed by a high intensity of the F− ion profile,
– higher accumulation of organic products containing polymer-type species observed from the C− ion profiles and confirmed by mass spectra.
3.4. Morphology modifications
The SEM analysis of the FeS thin film electrodes before and after electrochemical lithiation–delithiation is presented in Fig. 8. The pristine sample (Fig. 8a) is constituted of homogeneously distributed grains with an average lateral size of ∼90 nm. The average grain size increases to ∼120 and ∼160 nm after the 1st lithiation in LiClO4-PC (Fig. 8b) and in LiPF6-EC/DMC (Fig. 8c), respectively, which is consistent with the volume expansion of thin film electrode observed by ToF-SIMS. One also notices (see insets) the grain pulverization to be more marked in LiPF6-EC/DMC of initially homogeneous pristine film. | ||
| Fig. 8 SEM images of FeS thin film before (a) and after (b) 1st lithiation at 0.86 V, (d) 1 CV and (f) 60 galvanostatic cycles in 1 M LiClO4-PC, and after (c) first lithiation at 0.76 V, (e) 1 CV and (g) 60 galvanostatic cycles in 1 M LiPF6-EC/DMC. | ||
The first lithiation–delithiation cycle performed in both electrolytes introduces already the irreversible morphological alteration like cracks combined with pulverization (Fig. 8d and e). This confirms that volume expansion–shrinkage upon lithiation–delithiation (conversion–deconversion) generates stress in the material independent of the electrolyte. The surface fraction occupied by cracks is measured to be ∼12% in both electrolytes after one lithiation–delithiation cycle. The grains crumbling and formation of subparticles is confirmed at this stage for both electrolytes (see insets), the effect still being more marked in LiPF6-EC/DMC. These modifications are expected to facilitate electrolyte transport and thus lithium insertion into the electrode, as also observed by ToF-SIMS. This is proposed to be at the origin of the positive potential shift of the 2nd lithiation and the improved reaction reversibility observed electrochemically.
After 60 cycles (Fig. 8f and g), further cracking and grain pulverization is confirmed in both electrolytes (see insets). The grain division appears finer in LiPF6-EC/DMC than in LiClO4-PC leading to the formation of a more homogeneous morphology of the thin film outer surface. Cracks have enlarged which reveals the bulk morphology below the thin film surface. The cracked surface fraction is ∼15% in LiClO4-PC and ∼21% in LiPF6-EC/DMC, indicating larger morphological damage at least to the surface of the electrode material in EC/DMC than in PC-based electrolytes after 60 cycles.
Fig. 9 compares SEM images of particles from the thin film electrodes after 60 cycles. In LiClO4-PC, the thickness of the peeled-off particle ranges between 260 and 350 nm (Fig. 9a). Clearly, this is thicker than the pristine film (∼210 nm) and confirms that multi cycling-induced thickening of the thin film electrode, as observed by ToF-SIMS. This also shows that exfoliation has taken place due to deadhesion at the FeS/Fe interface. Besides, this cross-sectional view reveals a compact bulk structure of the thin film multi cycled in LiClO4-PC. In LiPF6-EC/DMC, the cross-sectional view of an exfoliated particle (circled in red in Fig. 9b) indicates a thickness of ∼140 nm. This is smaller than for the thickness of initial film electrode and indicates deadhesion by fracture in the bulk of the thin film and not at the FeS/Fe interface as observed in LiClO4-PC. Moreover, a foam-like, porous structure (magnified in Fig. 9c) is revealed underneath in the bulk of the thin film electrode.
 | ||
| Fig. 9 SEM images of FeS thin film particles after 60 lithiation–delithiation cycles in 1 M LiClO4-PC (a) and in 1 M LiPF6-EC/DMC (b and c). | ||
3.5. Model of electrode modifications
Based on the combined analysis by XPS, ToF-SIMS and SEM of the FeS thin film electrodes cycled in the LiClO4-PC and LiPF6-EC/DMC-based electrolytes, a model of the electrode modifications taking into account the effect of the electrolyte is proposed (Fig. 10). The pristine FeS thin film electrode has a compact granular morphology and presents a flat and homogeneous surface, slightly oxidized,28 and covered by a thin, organic contamination layer. | ||
| Fig. 10 Model of the FeS thin film electrode modifications induced by cycling in 1 M LiClO4-PC and 1 M LiPF6-EC/DMC. | ||
Lithiation-induced conversion causes a large volume expansion of the thin film electrode material, leading to significant roughness increase31 and enlargement of the converted grains. These modifications are more pronounced in LiPF6-EC/DMC. This process is accompanied by the reductive decomposition of the electrolyte leading to formation of a Li carbonate SEI in both electrolytes. The SEI layer thickness is similar in both electrolytes but much more salt residues enter its composition in LiPF6-EC/DMC suggesting more pronounced influence of the salt than the solvent. It is possibly less dense in LiPF6-EC/DMC. Deconversion causes volume shrinkage of the material and thickness decrease of the SEI.31 Expansion–shrinkage of the material causes mechanical stress that triggers the formation of cracks (possibly voids) and grain pulverization. These microstructural modifications open new pathways for the electrolyte penetration in the bulk electrode, promoting the conversion process already in the second discharge and formation of the SEI in the bulk material.
Cycling promotes SEI thickening more significantly in LiPF6-EC/DMC, with formation of a duplex structure with the inner and outer parts composed mainly of inorganic and organic compounds, respectively. Moreover, cycling amplifies the morphology modifications which results in penetration of the electrolyte into the bulk electrode and leads to crack enlargement and exfoliation of the electrode material. Owing to the inactivity of metallic Fe towards lithiation, the volume variations accompanying the conversion–deconversion process and the concomitant SEI penetration generate stress not only in the bulk of the thin film but also at the FeS/Fe interface. Cracks propagate in depth in the compact structure of the thin film cycled in LiClO4-PC, facilitating the electrolyte transport up to the current collector (Fe substrate) and leading to deadhesion at the FeS/Fe interface with loss of contact of the electrode material with current collector and capacity fading. The formation of a foam-like, porous structure with a polymerized SEI in LiPF6-EC/DMC, obviously better accommodates the stress generated by the volume variations and concomitant SEI formation. This impedes in-depth crack propagation and exfoliation is limited to the outer part of the electrode material, which results in capacity retention.
4. Conclusions
The influence of two different electrolytes (1 M LiClO4-PC or 1 M LiPF6-EC/DMC) on the surface and bulk chemical and morphological modifications of conversion-type FeS electrodes have been studied by XPS, ToF-SIMS and FESEM and indicate the following.• Formation of a lithium carbonate-rich SEI layer in both electrolytes with polymer-type compounds preferentially occurs in LiPF6-EC/DMC. Cycling in LiPF6 leads also to enrichment of SEI in inorganic salt residues with high F composition (up to 24 F%).
• Grain pulverization and thin film cracking occur to a larger extent in LiPF6-EC/DMC than in LiClO4-PC.
• Continuous uptake of the SEI layer is particularly observed in LiPF6-EC/DMC with the inorganic residues concentrated in the inner part of the SEI.
• Morphology modifications lead to formation of a compact structure in LiClO4-PC and a foam-like, porous structure in LiPF6-EC/DMC, which results in easier electrode degradation in PC than in EC/DMC-based electrolytes and better stress accommodation and/or electrolyte transport in EC/DMC than in PC-based electrolytes.
The observed aforementioned electrode modifications induced by cycling in these two different electrolytes allowed us to conclude about enhanced capacity retention and better reaction reversibility in LiPF6-EC/DMC than in LiClO4-PC.
Moreover, the combination of spectroscopic and microscopic analytical techniques applied here allowed for better understanding of fine chemical and morphological differences in this electrode material. The methodology used here in this work can be transposed to many other high capacity electrode materials widely tested for application in Li-ion batteries, which suffer volume modifications induced by cycling.
References
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS PubMed.
- E. Peled, D. Golodnitsky and G. Ardel, J. Electrochem. Soc., 1997, 144, L208 CrossRef CAS PubMed.
- K. Kanamura, H. Tamura, S. Shiraishi and Z.-I. Takehara, J. Electroanal. Chem., 1995, 394, 49 CrossRef.
- J.-T. Li, J. Swiatowska, V. Maurice, A. Seyeux, L. Huang, S.-G. Sun and P. Marcus, J. Phys. Chem. C, 2011, 115, 7012 CAS.
- D. Bar-Tow, E. Peled and L. Burstein, J. Electrochem. Soc., 1999, 146, 824 CrossRef CAS PubMed.
- E. Nanini-Maury, J. Światowska, A. Chagnes, S. Zanna, P. Tran-Van, P. Marcus and M. Cassir, Electrochim. Acta, 2014, 115, 223 CrossRef CAS PubMed.
- Y. Matsumura, S. Wang and J. Mondori, J. Electrochem. Soc., 1995, 142, 2914 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS.
- K. Xu and A. V. Cresce, J. Mater. Chem., 2011, 21, 9849 RSC.
- D. Aurbach, M. L. Daroux, P. W. Faguy and E. Yeager, J. Electrochem. Soc., 1987, 134, 1611 CrossRef CAS PubMed.
- K. Xu, J. Electrochem. Soc., 2009, 156, A751 CrossRef CAS PubMed.
- D. Aurbach and Y. S. Cohen, in Lithium-Ion Batteries: Solid-Electrolyte Interphase, ed. B. Balbuena and Y. Wang, Imperial College Press, 2004, p. 70 Search PubMed.
- A. Naji, J. Ghanbaja, B. Humbert, P. Willmann and D. Billaud, J. Power Sources, 1996, 63, 33 CrossRef CAS.
- Y. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123, 11708 CrossRef CAS PubMed.
- R. Dedryvère, S. Laruelle, S. Grugeon, L. Gireaud, J.-M. Tarascon and D. Gonbeau, J. Electrochem. Soc., 2005, 152, A689 CrossRef PubMed.
- A. M. Andersson and K. Edström, J. Electrochem. Soc., 2001, 148, A1100 CrossRef CAS PubMed.
- K. W. Schroder, H. Celio, L. J. Webb and K. J. Stevenson, J. Phys. Chem. C, 2012, 116, 19737 CAS.
- K. Tasaki, J. Phys. Chem. B, 2005, 109, 2920 CrossRef CAS PubMed.
- R. Dedryvère, L. Gireaud, S. Grugeon, S. Laruelle, J.-M. Tarascon and D. Gonbeau, J. Phys. Chem. B, 2005, 109, 15868 CrossRef PubMed.
- G. V. Zhuang, H. Yang, P. N. Ross, Jr., K. Xu and T. R. Jow, Electrochem. Solid-State Lett., 2006, 9, A64 CrossRef CAS PubMed.
- H. Tavassol, J. W. Buthker, G. A. Ferguson, L. A. Curtiss and A. A. Gewirth, J. Electrochem. Soc., 2012, 159, A730 CrossRef CAS PubMed.
- K. Xu, G. V. Zhuang, J. L. Allen, U. Lee, S. S. Zhang, P. N. Ross, Jr. and T. R. Jow, J. Phys. Chem. B, 2006, 110, 7708 CrossRef CAS PubMed.
- T. Kawamura, S. Okadaa and J.-I. Yamaki, J. Power Sources, 2006, 156, 547 CrossRef CAS PubMed.
- J. O. Besenhard, M. Winter, J. Yang and W. Biberacher, J. Power Sources, 1995, 54, 228 CrossRef CAS.
- S. Bhattacharya, A. R. Riahi and A. T. Alpas, J. Power Sources, 2011, 196, 8719 CrossRef CAS PubMed.
- M. Winter, W. K. Appel, B. Evers, T. Hodal, K.-C. Möller, I. Schneider, M. Wachtler, M. R. Wagner, G. H. Wrodnigg and J. O. Besenhard, Monatsh. Chem., 2001, 132, 473 CrossRef CAS.
- R. D. Apostolova, L. I. Neduzhko and E. M. Shembel, Russ. J. Appl. Chem., 2009, 82, 1939 CrossRef CAS.
- F. Liao, J. Światowska, V. Maurice, A. Seyeux, L. H. Klein, S. Zanna and P. Marcus, Electrochim. Acta, 2014, 120, 359 CrossRef CAS PubMed.
- Y. Kim and J. B. Goodenough, J. Phys. Chem. C, 2008, 112, 15060 CAS.
- L. Fei, Q. Lin, B. Yuan, G. Chen, P. Xie, Y. Li, Y. Xu, S. Deng, S. Smirnov and H. Luo, ACS Appl. Mater. Interfaces, 2013, 5, 5330 CAS.
- F. Liao, J. Światowska, V. Maurice, A. Seyeux, L. H. Klein, S. Zanna and P. Marcus, Appl. Surf. Sci., 2013, 283, 888 CrossRef CAS PubMed.
- J. Światowska-Mrowiecka, V. Maurice, S. Zanna, L. Klein and P. Marcus, Electrochim. Acta, 2007, 52, 5644 CrossRef PubMed.
- J. T. Li, J. Swiatowska, A. Seyeux, L. Huang, V. Maurice, S. G. Sun and P. Marcus, J. Power Sources, 2010, 195, 8251 CrossRef CAS PubMed.
- D. Aurbach, I. Weissman, A. Zaban and O. Chusid, Electrochim. Acta, 1994, 39, 51 CrossRef CAS.
- D. Aurbach, B. Markovsky, A. Schechter, Y. Ein-Eli and H. Cohen, J. Electrochem. Soc., 1996, 143, 3809 CrossRef CAS PubMed.
- D. Aurbach and A. Zaban, J. Electrochem. Soc., 1995, 142, L108 CrossRef CAS PubMed.
- D. Aurbach, M. Moshkovich, Y. Cohen and A. Schechter, Langmuir, 1999, 15, 2947 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332 CrossRef CAS PubMed.
- D. Aurbach, I. Weissman, A. Schechter and H. Cohen, Langmuir, 1996, 12, 3991 CrossRef CAS.
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423 CrossRef CAS.
- C. K. Chan, R. Ruffo, S. S. Hong and Y. Cui, J. Power Sources, 2009, 189, 1132 CrossRef CAS PubMed.
- C. Pereira-Nabais, J. Światowska, A. Chagnes, F. Ozanam, A. Gohier, P. Tran-Van, C.-S. Cojocaru, M. Cassir and P. Marcus, Appl. Surf. Sci., 2013, 266, 5 CrossRef CAS PubMed.
- H. Bryngelsson, M. Sjterndahl, T. Gustafsson and K. Edström, J. Power Sources, 2007, 174, 970 CrossRef CAS PubMed.
- W. Li and B. L. Lucht, J. Electrochem. Soc., 2006, 153, A1617 CrossRef CAS PubMed.
- S. Leroy, H. Martinez, R. Dedryvère, D. Lemordant and D. Gonbeau, Appl. Surf. Sci., 2007, 253, 4895 CrossRef CAS PubMed.
- A. M. Andersson, D. P. Abraham, R. Haasch, S. MacLaren, J. Liu and K. Amine, J. Electrochem. Soc., 2002, 149, A1358 CrossRef CAS PubMed.
- M. Herstedt, D. P. Abraham, J. B. Kerr and K. Edström, Electrochim. Acta, 2004, 49, 5097 CrossRef CAS PubMed.
- L. Martin, H. Martinez, D. Poinot, B. Pecquenard and F. Le Cras, J. Power Sources, 2014, 248, 861 CrossRef CAS PubMed.
- G. Audi, A. H. Wapstra and C. Thibault, Nucl. Phys. A, 2003, 729, 337 CrossRef PubMed.
- A. M. Lesson, M. R. Alexander, R. D. Short, D. Briggs and M. J. Hearn, Surf. Interface Anal., 1997, 25, 261 CrossRef.
- O. Chusid, E. Ein-Eli, D. Aurbach, M. Babai and Y. Carmeli, J. Power Sources, 1993, 43, 47 CrossRef CAS.
| This journal is © the Owner Societies 2015 |