 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct measurements of the total rate constant of the reaction NCN + H and implications for the product branching ratio and the enthalpy of formation of NCN
Nancy
Faßheber
,
Johannes
Dammeier
and
Gernot
Friedrichs
*
Institut für Physikalische Chemie, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Str. 1, 24118 Kiel, Germany. E-mail: friedrichs@phc.uni-kiel.de; Fax: +49 431 880 7743; Tel: +49 431 880 7742
First published on 29th April 2014
Abstract
The overall rate constant of the reaction (2), NCN + H, which plays a key role in prompt-NO formation in flames, has been directly measured at temperatures 962 K < T < 2425 K behind shock waves. NCN radicals and H atoms were generated by the thermal decomposition of NCN3 and C2H5I, respectively. NCN concentration-time profiles were measured by sensitive narrow-line-width laser absorption at a wavelength of λ = 329.1302 nm. The obtained rate constants are best represented by the combination of two Arrhenius expressions, k2/(cm3 mol−1 s−1) = 3.49 × 1014 exp(−33.3 kJ mol−1/RT) + 1.07 × 1013 exp(+10.0 kJ mol−1/RT), with a small uncertainty of ±20% at T = 1600 K and ±30% at the upper and lower experimental temperature limits.The two Arrhenius terms basically can be attributed to the contributions of reaction channel (2a) yielding CH + N2 and channel (2b) yielding HCN + N as the products. A more refined analysis taking into account experimental and theoretical literature data provided a consistent rate constant set for k2a, its reverse reaction k1a (CH + N2 → NCN + H), k2b as well as a value for the controversial enthalpy of formation of NCN, ΔfHo298K = 450 kJ mol−1. The analysis verifies the expected strong temperature dependence of the branching fraction ϕ = k2b/k2 with reaction channel (2b) dominating at the experimental high-temperature limit. In contrast, reaction (2a) dominates at the low-temperature limit with a possible minor contribution of the HNCN forming recombination channel (2d) at T < 1150 K.
1 Introduction
Nitrogen oxides, NO and NO2 (NOx), are major atmospheric pollutants formed by different reaction mechanisms in combustion processes. Especially under fuel rich combustion conditions, the so-called prompt-NO formation pathway becomes significant. According to Fenimore,1 prompt-NO formation is initiated by the reaction of small hydrocarbon radicals with molecular nitrogen from the combustion air. Although it has been proven both theoretically2–4 and experimentally5,6 that the key initiation reaction CH + N2 yields the spin-allowed products H + NCN,| CH(2Π) + N2(1Σ+) → H(2S) + NCN(3Σg−), | (1a) |
| CH(2Π) + N2(1Σ+) → N(4S) + HCN(1Σ+), | (1b) |
| NCN + H → CH + N2, | (2a) |
| NCN + H → HCN + N. | (2b) |
Depending on the reaction condidtions, two additional minor reaction channels forming HNC + N and HNCN are accessible (see Discussion section). The rate of reaction (2) and its exact branching ratio turned out to be crucial factors for modelling the fate of NCN in hydrocarbon flames.7,8,19 On the one hand reaction (2a) constitutes the reverse of the prompt-NO initiation reaction (1a) and can be calculated from k1avia the thermochemical equilibrium constant K(CH + N2 ⇌ NCN + H) = k1a/k2a. On the other hand the products of reaction (2b) are the same as the products of the formerly assumed spin-forbidden reaction (1b), which brings the new NCN chemistry back to the old Fenimore NOx formation route.
Reported rate constant values for reaction NCN + H have been included in the Arrhenius diagram shown in Fig. 1. An early rate constant estimate of Glarborg et al.20 assumed reaction (2b) to proceeds with a temperature independent rate constant close to the collisional rate, k2b = 1 × 1014 cm3 mol−1 s−1 (upper solid line). Shortly after, the reaction NCN + H has been theoretically analyzed by Moskaleva and Lin.2 As a side note in their paper on the overall rate constant of the reaction CH + N2 → NCN + H, but unfortunately without giving much details on the underlying theoretical model, they reported a pressure independent rate constant expression of k2b = 1.89 × 1014 × exp(−35.3 kJ mol−1/RT) cm3 mol−1 s−1 revealing that the reaction takes place over a sizable barrier (lower dash-dotted line). Hence, with k2b = 1.1 × 1013 cm3 mol−1 s−1 at T = 1500 K, the reaction is one order of magnitude slower than the initial estimate. Experimentally, Vasudevan et al.6 indirectly determined the rate constant of reaction (2b) in the temperature range of 2378 K < T < 2492 K by measuring absorption-time profiles of NCN in ethane–N2 mixtures behind shock waves (triangles with error bars). Following fast NCN generation by reaction (1a), the observed slow decays of the NCN radical concentration profiles were found to be consistent with k2b values close to the ones reported by Moskaleva and Lin.
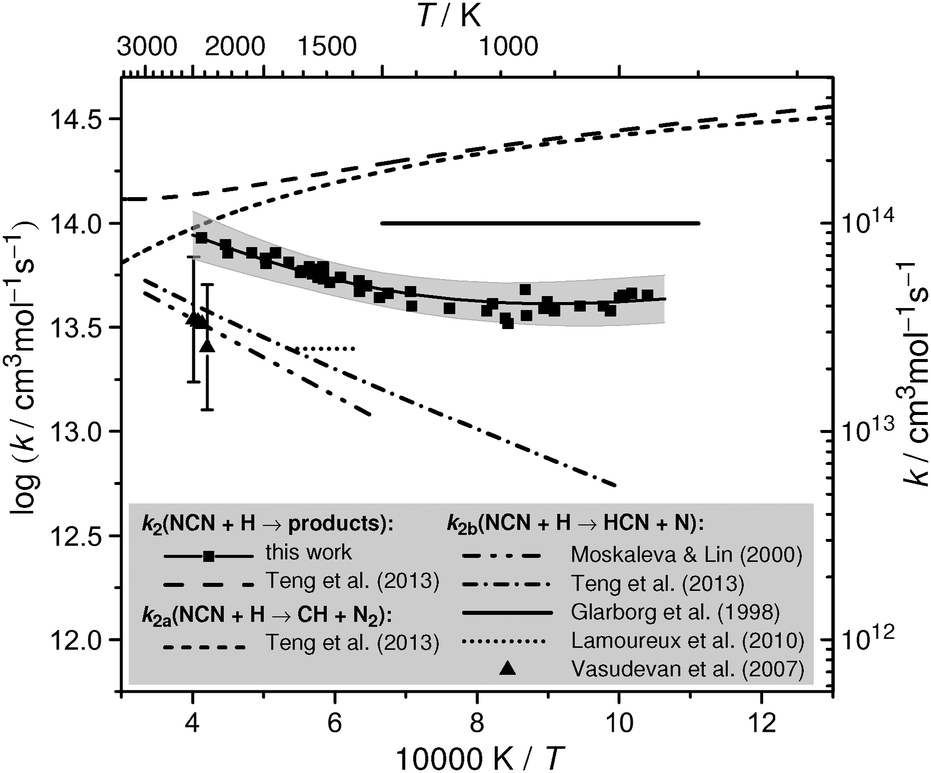 | ||
| Fig. 1 Experimental and theoretical literature rate constant data for the reaction NCN + H in comparison with the results of this work. Estimate of Glarborg et al.,20 shock tube data of Vasudevan et al.,6 flame data of Lamoureux et al.,8 and theoretical predictions of Teng et al.4 and Moskaleva and Lin2 are illustrated as outlined in the legend. The squares depict the experimental data of this work; the shaded area and the thick solid curve correspond to the range of uncertainty and a fit of the experimental data, respectively. | ||
In an indirect experimental and numerical study of the role of NCN formation in low pressure flames, Lamoureux et al.8 reported the value k2b = 2.5 × 1013 cm3 mol−1 s−1 (dotted line), which was also adopted for the GDFkin3.0_NCN mechanism. Essentially, this value had been chosen to match the experimentally measured NCN and NO concentration profiles in methane and acetylene flames with peak flame temperatures of T = 1600–1835 K. Therefore, this reported value is highly dependent on the value of the assumed absorption cross section for NCN, which is subject to ongoing discussion.21 Furthermore, the assumed enthalpy of formation of NCN, here ΔfHo298K = 452 kJ mol−1, is a critical quantity as k2a is calculated from k1avia thermochemical equilibrium in their simulations. The crucial role of NCN thermochemistry for modelling prompt-NO formation in flames has been recently highlighted in a paper by Goos et al.22 They nicely demonstrated that the modelled branching ratio of the overall reaction (2) heavily depends on the assumed enthalpy of formation value for NCN. For example, by switching the enthalpy of formation from the low value ΔfHo298K = 444.5 kJ mol−1 (representative for theoretical estimates based on single-reference computations)16,23 to the high value 466.5 kJ mol−1 (experimental result based on photodissociation experiments)24 both the simulated NCN peak mole fraction and final NO yield varied by a factor of about 3 for a fuel rich low-pressure CH4–O2–N2 flame. In view of this large variation it becomes clear that also the indirect k2b value of Lamoureux et al. is uncertain.
Very recently the M. C. Lin group4 updated their theoretical prediction of the rate constants of the reactions CH + N2 and NCN + H based on (i) high-level ab initio calculation (CCSD(T) with complete basis set limit) of the underlying quartet and doublet potential energy surfaces and (ii) by correcting a previous coding error in a program used in their original paper2 from the year 2000. Teng et al.4 clearly showed that reaction (2b) is a spin-allowed process predominantly taking place on a quartet surface, in contrast to reaction (2a) taking place only on a doublet surface. In comparison with their previous work, they now recommend the rate expression k2b = 4.96 × 1012 × T0.41 × exp(−22.8 kJ mol−1/RT) cm3 mol−1 s−1 yielding 20% to 40% higher k2b values at temperatures from 1500 K to 2000 K (upper dash-dotted line in Fig. 1). Another important finding was that an alternative reaction channel yielding HNC + N is minor and that the recombination reaction yielding HNCN, which dominates at room temperature, contributes to less than 5% at combustion relevant temperatures of T > 1000 K at 1 bar total pressure. Remarkably high total rate constant values with a shallow minimum of k2 ≈ 1.3 × 1014 cm3 mol−1 s−1 at T = 3180 K have been reported in their work as well (long-dashed curve), which we consider unfeasible. As it turns out in this work, the recommendation of Teng et al. for reaction channel (2a) alone is already up to 6 times higher (short-dashed curve) than our experimentally determined total rate constant k2 (see also Section 4).
From this short overview of existing literature data we conclude that a reliable modeling of NCN chemistry in flames is not possible so far. Clearly, experimental data on the rate constant of the reaction NCN + H are needed to constrain the rate constant uncertainties and to advance current prompt-NO formation models.
2 Experimental
Shock tube apparatus
All experiments were carried out in an electropolished stainless steel shock tube with inner diameter of 81 mm. A more detailed description can be found elsewhere.25 Briefly, hydrogen or mixtures of hydrogen and nitrogen were used as driver gas; diaphragms were made from 80 or 100 μm thick aluminium foil. The experimental conditions behind the incident and reflected shock waves were calculated from pre-shock conditions and the shock wave velocity, which was measured by four fast piezo-electric sensors (PCB Piezotronics M113A21). A frozen-chemistry code was applied taking into account real gas effects and the measured shock wave damping, which was on the order of 1% per meter. Storage gas mixtures of 500–750 ppm NCN3 and 1000 ppm C2H5I in argon were prepared using the partial pressure method and were further diluted with argon using calibrated mass flow controllers (Aera, FC-7700CU). The low pressure section of the shock tube was flushed for about 5 min at p ≈ 30 mbar to reduce possible adsorption effects on the shock tube wall.NCN precursor
The thermal decomposition of cyanogen azide (NCN3)| NCN3 + M → 1NCN + N2 + M | (3) |
| 1NCN + M → 3NCN + M | (4) |
The CIISC efficiency is strongly dependent on the nature of the collider, reveals a non-linear pressure dependence due to a pressure saturation effect, and increases with increasing temperature.18,28 In order to accurately model the initial formation rate of 3NCN (denoted NCN in the following), the CIISC rate constant has been allowed to vary within the error limit reported by Dammeier et al.28
The highly toxic and explosive precursor molecule NCN3 has been directly synthesized using a procedure described previously.29,30 Briefly, a small amount of gaseous cyanogen bromide (BrCN, ∼20 mbar) was passed into an evacuated 1 L glass flask containing a huge excess of solid sodium azide (NaN3). After a 8–10 h reaction time, the gaseous products were analyzed by FTIR spectroscopy. Almost no water and carbon dioxide (∼0.01%), which serves as an indicator for a potential gas leak, were present in the reaction samples and the impurities of remaining cyanogen bromide were well below 4%, in some cases <0.1%. A slow decomposition of about 10% NCN3 per day took place in the storage flask, therefore mixtures were used up within 3 days. Accurate initial NCN3 mole fraction in the actual reaction mixtures were determined by modeling the maximum NCN yield in the experiments and were found to be consistent with the expected NCN3 content in the storage gas mixtures in all cases.
H precursor
Hydrogen atoms were generated by the thermal unimolecular decomposition of ethyl iodide (C2H5I). Under typical experimental conditions behind shock waves, the reaction can be assumed to take place close to the high pressure limit39 and exhibits two main reaction channels:| C2H5I → C2H5 + I | (5a) |
| C2H5I → C2H4 + HI | (5b) |
H atom formation proceeds through the fast subsequent decomposition of the ethyl radical,
| C2H5 + M → C2H4 + H + M. | (6) |
Although ethyl iodide has been widely used as an H atom precursor, until recently the assumed absolute values and temperature dependences of the H atom yield were uncertain and often represented the most significant source of error in such studies. Selected values of reported branching ratios ϕ = k5a/(k5a + k5b) are collected in Fig. 2. The most frequently used value of ϕ = 0.87 ± 0.11 is based on direct H and I atom resonance absorption spectroscopic (ARAS) measurements performed by Kumaran et al.31 at temperatures of 946 K < T < 1303 K. Also based on ARAS experiments, Herzler and Roth34 reported a coinciding value, Yamamori et al.32 used lower values of ϕ < 0.78 at overall higher temperatures, Miyoshi et al.33 determined a higher value of ϕ = (0.92 ± 0.06), and Giri et al.35 assumed ϕ = 1 by considering the hydrogen forming pathway only. Recently, Weber et al.,38 Yang and Tranter,37 and Bentz et al.36 revisited the ethyl iodide pyrolysis. Whereas Yang and Tranter found excellent agreement with their laser-schlieren densitometry shock tube measurements by assuming ϕ = 0.87, Weber et al. report a significantly lower value of ϕ = (0.7 ± 0.1) from a mass spectrometric investigation of the flash pyrolysis of ethyl iodide. However, Bentz et al. could show by a combination of H- and I-ARAS measurements and statistical rate calculations that the abstraction reaction H + C2H5I → C2H5 + HI, which had been neglected in former studies, needs to be taken into account for an accurate analysis of the branching ratio. Their experimental data, together with other available literature data, have been very recently reanalyzed by Varga et al.39 in a follow-up publication using a new global optimization method developed by Turányi.40 The simultaneous optimization of the rate constant expressions of all relevant reactions yielded a considerably temperature dependent branching ratio decreasing from ϕ(T = 962 K) = 0.96 to ϕ(T = 2450 K) = 0.71 over the temperature range of this study. A very low 3σ uncertainty level of ±0.035 has been specified by Varga et al. at a temperature of T = 1200 K. As can be seen in Fig. 2 the recommended branching ratios are consistent with most of the previous literature data. We consider these results to be most reliable and therefore adopted the ethyl iodide decomposition mechanism as reported by Varga et al. It is included in the list of reactions in Table 1.
 | ||
| Fig. 2 Selected literature values for the branching ratio ϕ of the thermal decomposition of ethyl iodide yielding C2H5 + I (channel (5a)) and C2H4 + HI (channel (5b)), respectively. H and/or I atom resonance absorption measurements: Kumaran et al.,31 Yamamori et al.,32 Miyoshi et al.,33 Herzler and Roth,34 Giri et al.,35 and Bentz et al.;36 laser schlieren technique: Yang and Tranter;37 mass spectrometry: Weber et al.;38 global optimization: Varga et al.39 The gray area represents the assumed uncertainty range of ±7%. | ||
| No. | Reaction | A | n | E a | Ref. |
|---|---|---|---|---|---|
| a Rate expression of Teng et al.4 scaled by a factor of 1.6. b Assuming ΔfHo298K(NCN) = 450 kJ mol−1; this corresponds to k1a(CH + N2 → NCN + H) = 2.3 × 1010 × T0.53 × exp(−71.2 kJ mol−1/RT). c ρ = 3.5 × 106 mol cm−3; for other densities see ref. 28. | |||||
| 3 | NCN3 ⇌ 1NCN + N2 | 4.9 × 109 | 71 | 28 | |
| 4 | 1NCN → NCN | 2.0 × 106 | 31 | 28 | |
| 2 | NCN + H → products | See text | |||
| 2a | NCN + H ⇌ CH + N2 | 4.2 × 1015 | −0.69 | 2.0 | This workb |
| 2b | NCN + H ⇌ HCN + N | 7.9 × 1012 | 0.41 | 22.8 | This worka |
| 7 | NCN + M ⇌ C + N2 + M | 8.9 × 1014 | 260 | 17 | |
| 8 | NCN + NCN ⇌ CN + CN + N2 | 3.7 × 1012 | 28 | ||
| 9 | NCN + C ⇌ CN + CN | 1.0 × 1014 | 28 | ||
| 10 | NCN + N ⇌ N2 + CN | 1.0 × 1013 | 2 | ||
| 11 | NCN + CN ⇌ C2N2 + N | 1.3 × 1014 | 33.5 | 2 | |
| 11 | NCN + CH ⇌ HCN + CN | 3.2 × 1013 | −3.6 | 2 | |
| 13 | NCN + CH2 ⇌ H2CN + CN | 8.0 × 1013 | 26.9 | 2 | |
| 5a | C2H5I ⇌ C2H5 + I | 3.4 × 1013 | 203 | 39 | |
| 5b | C2H5I ⇌ C2H4 + HI | 4.7 × 1013 | 226 | 39 | |
| 6 | C2H5 + M ⇌ C2H4 + H + M | 1.0 × 1018 | 140 | 39 | |
| 14 | C2H5I + H ⇌ C2H5 + HI | 1.0 × 1015 | 21.6 | 39 | |
| 15 | C2H5I + I ⇌ C2H5 + I2 | 4.0 × 1013 | 69.9 | 43 | |
| 16a | C2H5 + H ⇌ CH3 + CH3 | 4.2 × 1013 | 44 | ||
| 16b | C2H5 + H ⇌ C2H4 + H2 | 1.2 × 1012 | 45 | ||
| 17 | H + HI ⇌ H2 + I | 6.6 × 1013 | 4.1 | 39 | |
We have recommended the use of a different expression with opposite temperature dependence of ϕ in a previous paper dating back to the year 2002.41 That recommendation was based on a theoretical treatment of the unimolecular decomposition of ethyl iodide with barrier heights taken from the paper of Kumaran et al. Then, the energy barrier E0 of the I atom forming C–I bond fission channel (5a) was assumed to be about 15 kJ mol−1 higher than of the HI elimination channel (5b). However, the recent accurate CCSD(T) ab initio data of Bentz et al.36 showed that both barriers are more or less energetically equal, which is more consistent with the reported decrease of ϕ with increasing temperature. For a more detailed treatment of the unimolecular decomposition reactions of alkyl iodides in the framework of statistical rate theories we refer to the work of Kumaran et al., Miyoshi et al., and Bentz et al.31,33,36
NCN detection
Triplet NCN radicals were detected by time-resolved difference amplification laser absorption spectroscopy at![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 30383.11 cm−1 (λ = 329.1302 nm). The absorption feature at this wavelength mainly stems from a superposition of the 3Π1 sub band of the Ã3Πu(000) −
= 30383.11 cm−1 (λ = 329.1302 nm). The absorption feature at this wavelength mainly stems from a superposition of the 3Π1 sub band of the Ã3Πu(000) − ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 3Σg(000) and the Q1 band head of the 3Σ+(010) − 3Π(010) transition.21 About 1 mW UV laser radiation was generated by intra-cavity frequency doubling of a continuous-wave ring-dye laser (Coherent, 899) with DCM-Special as dye pumped with a solid state Nd:YVO4 laser using 8 W at λ = 532 nm (Coherent Verdi V10). The wavelength of the laser fundamental was measured interferometrically by a wavemeter (MetroLux) with an accuracy of Δ
3Σg(000) and the Q1 band head of the 3Σ+(010) − 3Π(010) transition.21 About 1 mW UV laser radiation was generated by intra-cavity frequency doubling of a continuous-wave ring-dye laser (Coherent, 899) with DCM-Special as dye pumped with a solid state Nd:YVO4 laser using 8 W at λ = 532 nm (Coherent Verdi V10). The wavelength of the laser fundamental was measured interferometrically by a wavemeter (MetroLux) with an accuracy of Δ![[small upsilon, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e131.gif) ≈ ±0.015 cm−1. The UV laser beam was split into a detection and a reference beam by a (50
≈ ±0.015 cm−1. The UV laser beam was split into a detection and a reference beam by a (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) beam splitter plate. The detection beam was passed through the shock tube and coupled into an optical fiber (Thorlabs BF H22-550) connected to a balanced photodetector and amplifier (Thorlabs PDB 150A-EC). The intensity of the reference laser beam was fine-tuned by a variable neutral density filter to match the intensity of the detection beam. The resulting difference signals were low-pass-filtered (1.4 MHz), amplified (Ortec Fast Preamp 9305, 18 dB), and stored by an analog input board (Measurement Computing, PCI-DAS4020/12, 20 MHz, 12 bit). The NCN concentration-time profiles were calculated from the detected absorption profiles based on NCN absorption cross sections log(σ(base e)/(cm2 mol−1)) = 8.9–8.3 × 10−4 × T/K previously measured with an accuracy of ±25% using the same apparatus at similar temperatures and pressures.16
:50) beam splitter plate. The detection beam was passed through the shock tube and coupled into an optical fiber (Thorlabs BF H22-550) connected to a balanced photodetector and amplifier (Thorlabs PDB 150A-EC). The intensity of the reference laser beam was fine-tuned by a variable neutral density filter to match the intensity of the detection beam. The resulting difference signals were low-pass-filtered (1.4 MHz), amplified (Ortec Fast Preamp 9305, 18 dB), and stored by an analog input board (Measurement Computing, PCI-DAS4020/12, 20 MHz, 12 bit). The NCN concentration-time profiles were calculated from the detected absorption profiles based on NCN absorption cross sections log(σ(base e)/(cm2 mol−1)) = 8.9–8.3 × 10−4 × T/K previously measured with an accuracy of ±25% using the same apparatus at similar temperatures and pressures.16
3 Results
The reaction of NCN radicals with hydrogen atoms has been investigated behind incident and reflected shock waves in the temperature and pressure ranges 962 K < T < 2425 K and 290 mbar < p < 2130 mbar, respectively, at three different total densities of ϱ ≈ 3.5 × 10−6, 7.4 × 10−6, and 1.5 × 10−5 mol cm−3. Reaction gas mixtures contained 72–363 ppm ethyl iodide and 3–31 ppm NCN3 in argon. In most cases, a large excess of ethyl iodide with [C2H5I]0/[NCN3]0 ratios up to 60 was applied, hence the hydrogen atom was used as the excess species. Experimental conditions are listed in Table 2.| T/K | ϱ × 106/mol cm−3 | x(NCN3) ppm | x(C2H5I) ppm | k 2 × 10−13/cm3 mol−1 s−1 | T/K | ϱ × 106/mol cm−3 | x(NCN3) ppm | x(C2H5I) ppm | k 2 × 10−13/cm3 mol−1 s−1 |
|---|---|---|---|---|---|---|---|---|---|
| 1186 | 3.78 | 7.4 | 75 | 3.3 | 962 | 3.52 | 3.8 | 185 | 4.5 |
| 1714 | 3.43 | 19.0 | 72 | 6.1 | 1000 | 3.57 | 4.0 | 185 | 4.4 |
| 1720 | 3.43 | 21.0 | 72 | 5.5 | 1192 | 3.77 | 4.6 | 185 | 3.5 |
| 1813 | 3.46 | 21.8 | 72 | 5.8 | 1230 | 3.80 | 5.2 | 185 | 3.8 |
| 1870 | 3.46 | 22.8 | 72 | 6.5 | 1552 | 4.03 | 9.5 | 185 | 5.0 |
| 1936 | 4.20 | 9.0 | 76 | 7.2 | 1747 | 3.44 | 27.5 | 184 | 6.0 |
| 1991 | 3.51 | 27.8 | 72 | 6.8 | |||||
| 2070 | 3.54 | 23.5 | 72 | 7.2 | 1482 | 3.99 | 5.0 | 299 | 4.6 |
| 2227 | 3.57 | 25.0 | 72 | 7.2 | 1509 | 4.01 | 3.0 | 104 | 4.4 |
| 2242 | 3.55 | 26.0 | 72 | 7.9 | 1578 | 3.37 | 24.5 | 712 | 5.3 |
| 2425 | 2.89 | 28.0 | 72 | 8.5 | 1714 | 3.41 | 23.7 | 40 | 6.2 |
| 1774 | 3.31 | 31.0 | 363 | 6.2 | |||||
| 996 | 3.56 | 7.0 | 138 | 4.5 | 1988 | 2.11 | 11.9 | 155 | 6.4 |
| 1013 | 3.59 | 4.6 | 137 | 3.8 | |||||
| 1023 | 3.60 | 6.9 | 138 | 4.0 | 983 | 7.10 | 7.0 | 138 | 4.6 |
| 1101 | 3.69 | 3.2 | 137 | 3.8 | 993 | 7.13 | 6.5 | 138 | 4.5 |
| 1113 | 3.70 | 5.3 | 138 | 4.2 | 1059 | 7.25 | 5.8 | 138 | 4.0 |
| 1151 | 3.73 | 8.5 | 136 | 3.6 | 1119 | 7.41 | 6.4 | 138 | 3.9 |
| 1217 | 3.80 | 12.5 | 138 | 4.1 | 1153 | 7.48 | 6.5 | 138 | 4.8 |
| 1314 | 3.88 | 4.0 | 137 | 3.9 | 1646 | 7.19 | 4.0 | 185 | 5.5 |
| 1413 | 3.95 | 6.3 | 136 | 4.0 | 1760 | 7.43 | 7.4 | 138 | 5.7 |
| 1416 | 3.94 | 4.6 | 137 | 4.7 | 1805 | 7.50 | 3.9 | 137 | 5.9 |
| 1576 | 4.05 | 6.3 | 136 | 4.7 | |||||
| 1688 | 3.41 | 16.4 | 131 | 5.2 | 1713 | 14.7 | 5.0 | 138 | 5.4 |
| 1735 | 14.8 | 5.0 | 138 | 5.5 |
Fig. 3a illustrates a typical experiment behind the incident shock wave at a temperature T = 1720 K and a total density ϱ = 3.43 × 10−6 mol cm−3 with initial mole fractions of x(NCN3) = 21 ppm and x(C2H5I) = 72 ppm. After the arrival of the incident shock wave, the NCN signal increases within 20 μs. Obviously, both the thermal decomposition of NCN3 and the singlet-triplet relaxation of NCN are fast. The subsequent NCN decay is well resolved and is more or less complete at the end of the experimental time window set by the Schlieren signal of the reflected shock wave at t = 460 μs.
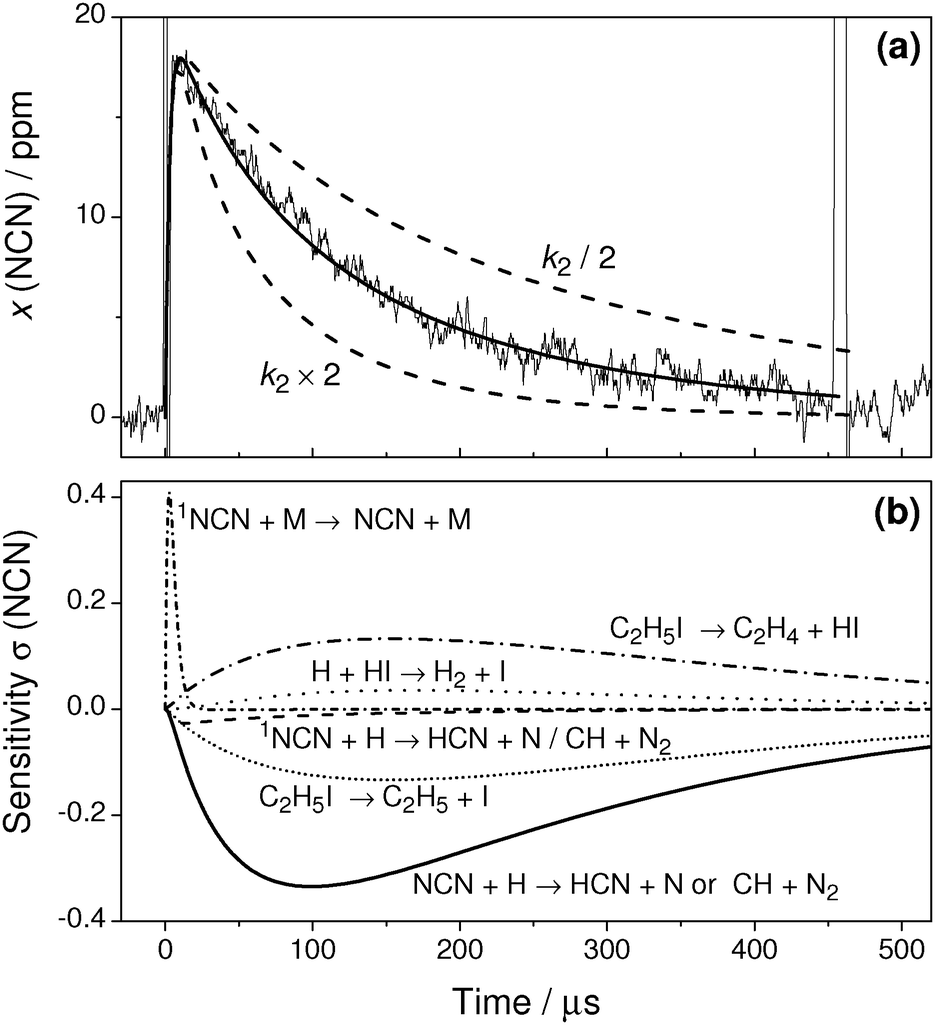 | ||
| Fig. 3 (a) Experimental NCN profile in comparison with numerical simulations. T = 1720 K, ϱ = 3.43 × 10−6 mol cm−3, p = 490 mbar, x(NCN3) = 21 ppm, x(C2H5I) = 72 ppm, k2 = 5.5 × 1013 cm3 mol−1 s−1. (b) Sensitivity analysis. | ||
In order to extract rate constants for reaction (2), NCN + H → products, the NCN profiles were numerically simulated using the CHEMKIN-II package.46 The GRI-Mech 3.0 was used as a base mechanism42 supplemented by an iodine submechanism adopted from our previous work41 and the reactions outlined in Table 1. The mechanism for NCN secondary chemistry was assembled from literature data, in particular from our previous measurements that have been validated to reproduce NCN concentration-time profiles of pure NCN3–argon mixtures over a wide range of experimental conditions. The ethyl iodide decomposition has been modeled by including the recently optimized submechanism reported by Varga et al.39 (vide supra). In order to identify potential contributions of 1NCN secondary chemistry, all triplet NCN reactions have been duplicated for singlet NCN. Although this treatment neglects the presumably different 1NCN reactivity, it can be safely assumed that 1NCN secondary chemistry is dominated by its relaxation reaction forming triplet NCN within the first few μs of the experiments. Thermodynamic data were taken from Burcat's thermodynamic database47 with updated NASA polynomial parameters for NCN from Goos et al.22 Note that the assumed value for the enthalpy of formation of NCN, although of utmost importance for the discussion of the branching ratio of reaction (2) (vide infra), is not important for the determination of the total rate constant from the experimental profiles.
The solid curve in Fig. 3a reflects the best fit of the data using k2 as an adjustable parameter. Two additional simulations using k2 varied by a factor of two (dashed curves) are shown as well. They deviate strongly from the experimental profile demonstrating the high sensitivity of reaction (2). Assuming either the products of reaction channel (2a), CH + N2, or reaction channel (2b), HCN + N, did not change the extracted k2 value within error limits. The high sensitivity of reaction (2) is further outlined in the sensitivity analysis shown in Fig. 3b. Here, the sensitivity coefficient σ(i,t) for reaction i at time t was normalized with respect to the maximum concentration [NCN]max over the time history, σ(i,t) = 1/[NCN]max × (∂[NCN]/∂lnki). For the analysis, a branching ratio of ϕ = k2b/k2 = 0.5 has been assumed. Following the initial increase of the signal, which is determined by the NCN relaxation reaction (4), reaction (2) dominates the NCN decay. The relatively high sensitivity coefficients for reactions (5a) and (5b) directly reflect the influence of the branching ratio of reaction (5) and hence the assumed H atom yield from ethyl iodide pyrolysis. Presuming that this branching ratio is accurate, the sensitivity analysis reveals that the rate constant of reaction (2) could be directly measured under nearly pseudo first-order conditions.
Whereas the highest feasible experimental temperature was limited by the increasingly fast thermal decomposition of NCN, towards lower temperatures non-NCN secondary chemistry becomes significant as well. This is illustrated by the T = 1150 K experiment and sensitivity analysis shown in Fig. 4. Both reaction (14), H + C2H5I, and the assumed products of reaction (2) become important. Assuming the products of channel (2a), HCN + N, at longer reaction times the reaction N + NCN significantly consumes NCN. Similarly, assuming the products of channel (2b), CH + N2, the reaction C + NCN gains importance. Here, C atoms are efficiently generated by the reaction CH + H → C + H2. Consistent with the expected diminishing role of the activation controlled channel (2b), somewhat better agreement with the experiment is obtained by assuming channel (2a). However, as the remaining differences between simulation and experiment could not be clearly attributed to a specific secondary reaction, no attempt was made to further improve the simulation at longer reaction times. Instead, the rate of reaction (2) was extracted from the NCN decay by fitting the transient at short reaction times where secondary chemistry did not yet exert a significant influence.
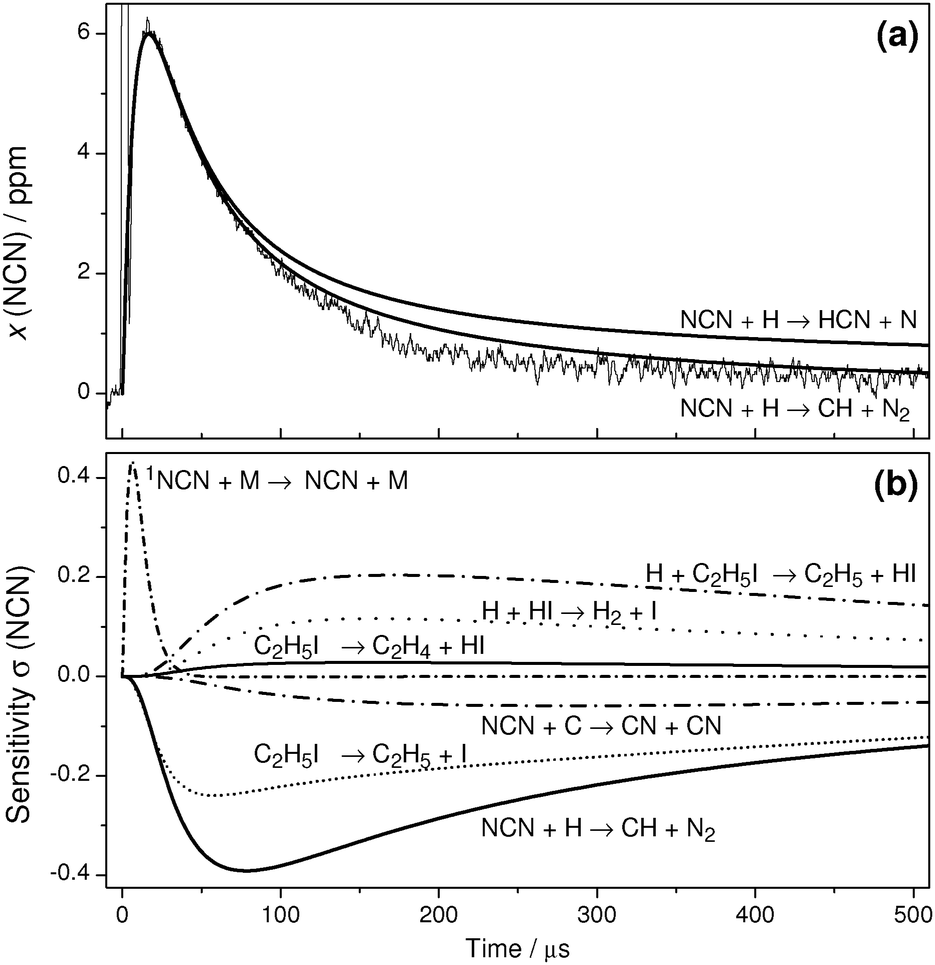 | ||
| Fig. 4 (a) Experimental NCN profile in comparison with numerical simulations. T = 1150 K, ϱ = 7.48 × 10−6 mol cm−3, p = 720 mbar, x(NCN3) = 6.5 ppm, x(C2H5I) = 138 ppm, k2 = 4.8 × 1013 cm3 mol−1 s−1. (b) Sensitivity analysis. | ||
All measured rate constants k2 are listed in Table 2 and are shown in Arrhenius form in Fig. 5. Overall, the rate constants follow the same trend independent of total density (varied by a factor of 4) and mixture composition (varied within 1.7 < [C2H5I]0/[NCN3]0 < 60). The data reveal a shallow minimum at temperatures around 1050–1200 K indicating that at least two reaction channels are active, presumably channels (2a) and (2b) with (2b) becoming more important towards higher temperatures. Accordingly, in the temperature range 962 K < T < 2425 K the total rate constant can be best represented by the sum of two Arrhenius expressions,
| k2/(cm3 mol−1 s−1) = 3.49 × 1014 exp(−33.3 kJ mol−1/RT) + 1.07 × 1013 exp(+10.0 kJ mol−1/RT), | (I) |
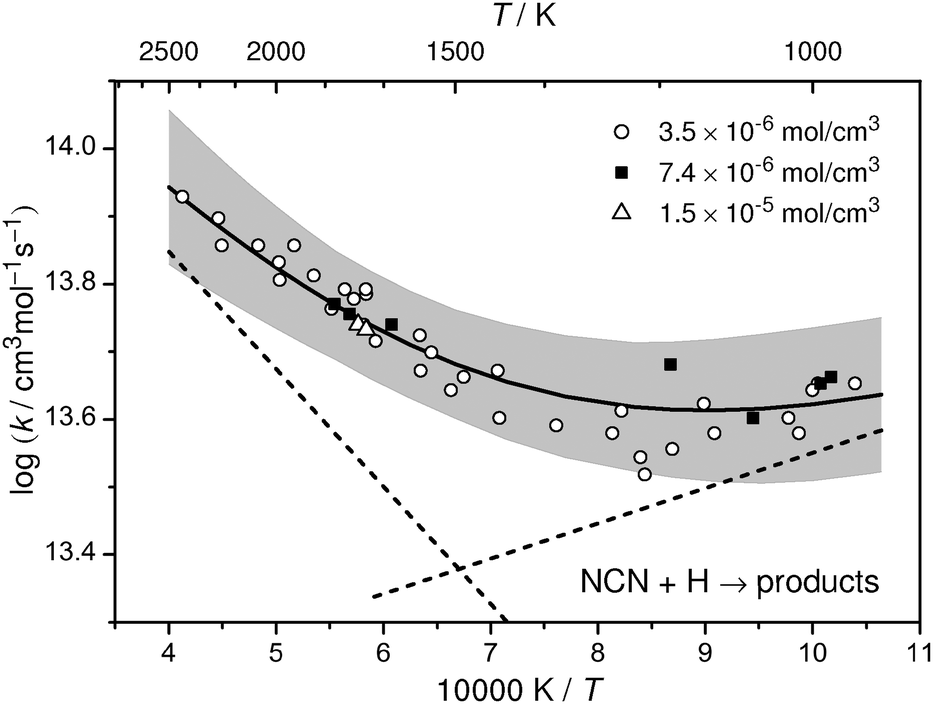 | ||
| Fig. 5 Arrhenius plot for k2 measured at three different total densities. The solid curve corresponds to a fit of the data using a sum of two Arrhenius expressions (dashed lines). The gray area represents the uncertainty range based on a comprehensive error analysis. | ||
An error analysis has to take into account uncertainties resulting from the scatter of the data (±6%), the mixture composition (in particular the initial ethyl iodide mole fraction, ±3%), the channel branching ratio of the ethyl iodide decomposition (estimated from Fig. 2 to be ±7%), the NCN absorption cross section (±25%, resulting in a 3% uncertainty in k2), and the secondary chemistry. In the middle of the investigated temperature range (T ≈ 1600 K), a direct pseudo first-order evaluation was possible and hence errors from secondary chemistry are minor. Nevertheless, we allow for a 10% error due to a possibly large uncertainty of the rate constant of the reaction (10), NCN + N, which has not been directly measured so far. Increasing its rate constant from 1 × 1013 cm3 mol−1 s−1 to 1 × 1014 cm3 mol−1 s−1 would make this reaction sensitive because N atoms are formed in reaction (2b) and hence slightly too high k2 values would have been determined by our analysis. Taking into account partial error compensation, we estimate the overall uncertainty of k2 to be ±20% at T = 1600 K, increasing to ±30% due to higher uncertainties resulting from secondary chemistry and the employed ethyl iodide branching ratio at the high and low temperature limit of the experiments. A corresponding uncertainty range is indicated by the grey shaded area in Fig. 5.
4 Discussion
The obtained total rate constant expression for k2 is compared with selected literature values and further analyzed in order to derive a consistent set of rate constants for the two main high-temperature reaction channels (2a) and (2b) as well as the NCN enthalpy of formation ΔfHo298K (ΔH in the following) in Fig. 6. As it was shown by the high level ab initio calculations of Teng et al.,4 from the four feasible reaction channels| 3NCN + 2H → 2CH + 1N2, | (2a) |
| → 1HCN + 4N, | (2b) |
| → 1HNC + 4N, | (2c) |
| → 2HNCN, | (2d) |
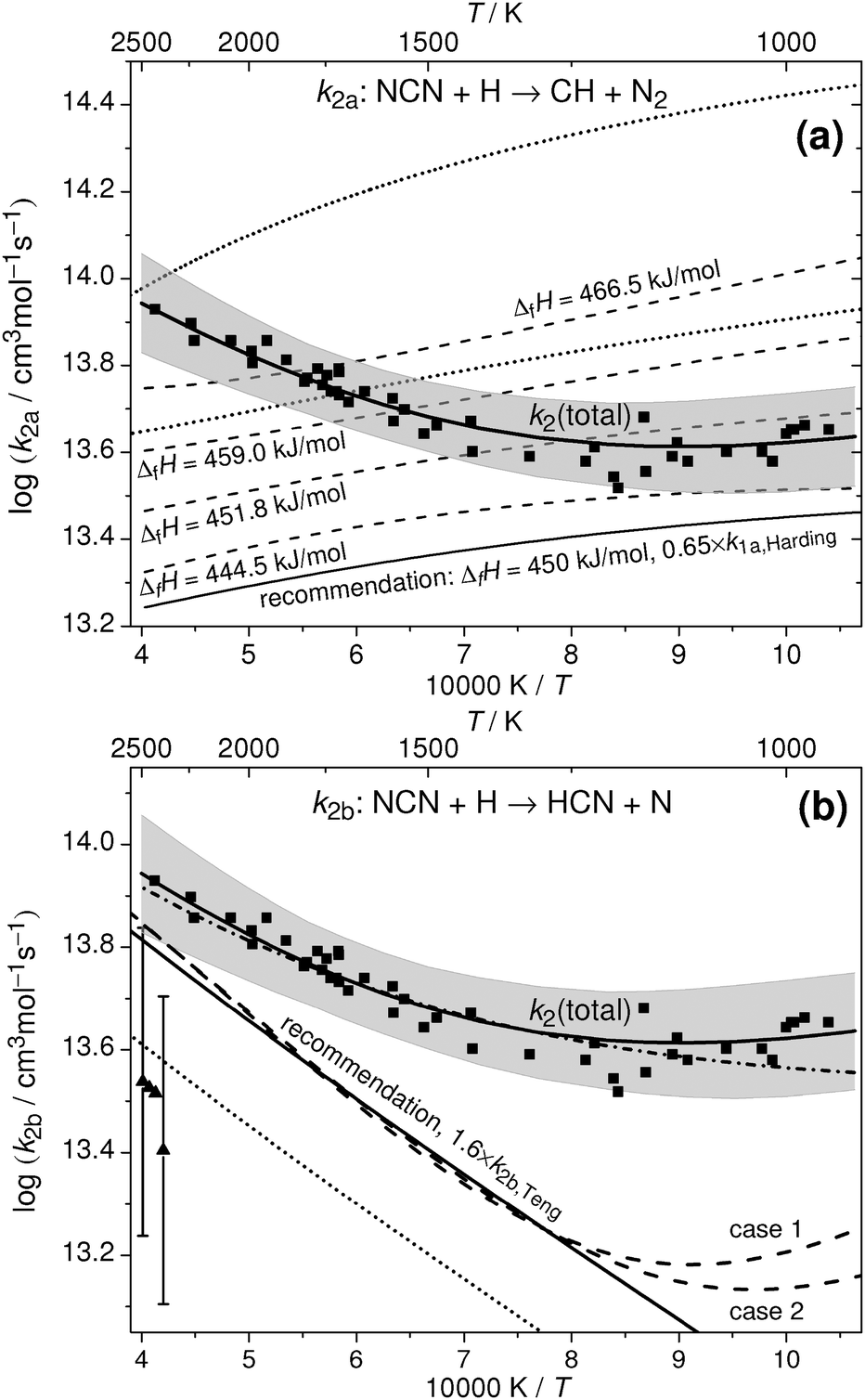 | ||
| Fig. 6 Analysis of total rate constant data in terms of ΔfHo298K(NCN) (ΔH in the following). The squares, the corresponding solid curves, and the shaded areas reflect the experimental data for k2 of this work and their uncertainty limits. Upper plot a: analysis with respect to channel (2a). The upper dotted curve depicts the original expression for k2a adopted from Teng et al.,4 the lower dotted curve a re-evaluation of their data using ΔH = 458 kJ mol−1. The dashed curves reflect k2a = k1a/K with k1a from Harding et al.3 derived for different ΔH values using eqn (II). For the lower solid curve, eqn (II) was scaled by a factor of 0.65. Lower plot b: Analysis with respect to channel (2b). Triangles with error bars and the dotted line reflect the experimental and theoretical data reported by Vasudevan et al.6 and Teng et al.,4 respectively. The dashed curves depict k2b = k2 − k2a expressions obtained from the k2 values of this work and k2a derived from Harding et al.; case 1: k2 × 1.0, k1a × 1.0, ΔH = 440 kJ mol−1; case 2: k2 × 1.0, k1a × 0.65, ΔH = 450 kJ mol−1. The lower solid curve reflects the recommended k2b expression, which is equal to the Teng et al.4 expression scaled by a factor of 1.6. The dash-dotted curve corresponds to k2 = k2a + k2b using the two recommended rate expressions. | ||
Analysis method a
In Fig. 6a, the upper dotted curve depicts the calculated rate expression for k2a from Teng et al.4 The very high rate constant values for k2a as well as the very high total rate constants k2 (dashed curve included in Fig. 1) are unreasonable. Compared to our experimentally determined total rate constant values, the expression yields up to 6 times higher rate constants for channel (2a) already. Moreover, there seems to be an inconsistency in the reported rate constants for the equilibrium CH + N2 ⇌ NCN + H in the paper of Teng et al. Using their values for the rate constant of the forward reaction (1a) and their preferred value for the enthalpy of formation, ΔH = 458 kJ mol−1, we calculate reverse rate constants k2b. Thermodynamic data for CH, N2, H, and NCN were taken from Burcat's database47 with updated heat capacity data for NCN from Goos et al.,22 where 7-term NASA polynomial parameter a6 has been scaled to adjust ΔH(NCN). Obtained k2b values are 2–3 times lower than reported by Teng et al. (lower dotted curve in Fig. 6a). Another indication that the reported rate constants may be flawed comes from the reported total rate constant values; for example, the recommended room temperature value k2 ≈ 7 × 1014 cm3 mol−1 s−1 is higher than the Lennard-Jones collision limit of kLJ ≈ (3.5–5.5) × 1014 cm3 mol−1 s−1, which can be estimated based on the Lennard Jones parameters reported in the literature (parameters for H:48–50σ = (2.00–3.26) Å and ε/kB = (2.7–145) K; parameters for NCN:48σ = 3.83 Å and ε/kB = 232 K).To the best of our knowledge, no other experimental or theoretical values for k2a have been reported explicitly in the literature yet. Therefore, we continue our analysis by calculating k2a values form the reverse reaction k1a, which has been thoroughly studied both experimentally and theoretically. For an overview of available literature data we refer to the work of Harding et al.3 who performed high-level ab initio and transition state theory calculations on the reaction CH + N2 using multi-reference electronic structure methods. Using their recommended value of the enthalpy of formation, ΔH = 459 kJ mol−1, the theoretical prediction was found to be in quantitative agreement with the most recent shock tube data of Vasudevan et al.6 over the temperature range 2100 K < T < 3350 K. At these high temperatures, the predicted rate constant is less dependent on the assumed value of ΔH. Towards lower temperatures and in the practically important temperature range of 1000–2000 K, however, an accurate enthalpy of formation is crucial. In an Arrhenius plot, Harding et al.3 present their results of temperature dependent calculations of the rate constant of reaction (1a) assuming different values for the enthalpy of formation of NCN (Fig. 13 in their paper). For example, it was shown that changing ΔH by ±8 kJ mol−1 yields a factor of 1.6 higher (−8 kJ mol−1) or 1.9 lower (+8 kJ mol−1) k1a value at T = 1000 K. In order to take this pronounced thermodynamic effect into account in our analysis and to derive rate constant estimates for k2a = k1a/K as function of the assumed NCN enthalpy of formation, we reparametrized the original data of Harding et al. and used the expression
| k1a/(cm3 mol−1 s−1) = e(274.5−0.556x) × (T/K)(−31.24+0.0706x) × e(−71.2kJmol−1/RT) | (II) |
Analysis method b
A second analysis of our data focusing on the rate constant of channel (2b) is shown in Fig. 6b. The indirect experimental data of Vasudevan et al.6 (triangles with error bars) and the most recent theoretical estimate of Teng et al.4 (dotted curve) are shown as well. In the light of the negative temperature dependence of channel (2a) it becomes clear that the experimentally determined positive temperature dependence of k2 towards higher temperatures arises from the increasingly dominant activation controlled channel (2b). Moreover, the high temperature activation energy Ea = 33 kJ mol−1 estimated from the two channel fit of our experimental data (see Fig. 5 and eqn (I)) is in very good agreement with the theoretically predicted activation energies of 35 kJ mol−1 and 28 kJ mol−1 reported by the M. C. Lin group2,4 (see Fig. 1 and 6b). Hence, we consider the activation energy of reaction channel (2b) a well-constrained quantity with a preferred value of Ea ≈ 28 kJ mol−1 adopted from the most recent ab initio study.4 Having the temperature dependence of k2b fixed, it is possible to arrive at a consistent value for the enthalpy of formation. Here, ΔH has been choosen in a way such that the rate constant expression for k2b = k2 − k2a, with k2 values taken from eqn (I) of this work and k2a values calculated via thermodynamic equilibrium from the k1a expression eqn (II), yields a temperature dependence that is consistent with 28 kJ mol−1. A matrix of appropriate enthalpy of formations with k2 and k1a varied within their uncertainty limits is given in Table 3. Here, uncertainties of k2 ± 30% as obtained in this work and k1a ± 35% as reported for the experimental shock tube results of Vasudevan et al.6 (which are in turn consistent with the k1a expression of Harding et al.) have been assumed. Table 3 reveals a large range of possible enthalpy of formations, 423 kJ mol−1 < ΔH < 456 kJ mol−1. Nevertheless, two conclusions can be drawn from this analysis. First, increasing k1a yields unfeasible enthalpy values that are even well below the results of the single-reference computations (about 444.5 kJ mol−1), which can be regarded a reasonable lower limit for ΔH. Even with k1a unchanged, the highest value of 445 kJ mol−1 would be close to this limit. Secondly, the upper limit of 456 kJ mol−1, corresponding to a scenario with k2 + 30% and k1a − 35%, is in agreement with the upper limit of 454 kJ mol−1 inferred from analysis method a. Therefore, the high experimental value of Bise et al.24 (466.5 ± 2.9 kJ mol−1), the results of the high-level basis set extrapolation and multi-reference computations (about 459 kJ mol−1),3,4 and the most recent recommendation of the Active Thermochemical Tables as cited in Goos et al.22 (457.8 ± 2) are hardly compatible with this work.| ΔfHo298K(NCN) | k 2 + 30% | k 2 | k 2 − 30% |
|---|---|---|---|
| k 1a + 35% | 436 | 430 | 423 |
| k 1a | 445 | 440 | 432 |
| k 1a − 35% | 456 | 450 | 443 |
Resulting k2b expressions are illustrated in Fig. 6b for two selected cases. Case 1, assuming that both k2 and k1a exhibit values as given by eqn (I) and (II), yields ΔH = 440 kJ mol−1. Case 2, assuming k2 from eqn (I) and k1a from eqn (II) scaled by a factor of 0.65, yields ΔH = 450 kJ mol−1. For all other cases outlined in Table 3, similar k2b curves have been obtained, of course somewhat offset for different assumed k1a values (not shown). It is obvious from Fig. 6b that the calculated k2b expressions deviate from linearity at temperatures below 1250 K. With decreasing temperatures and hence a decreasing contribution of k2b the analysis procedure gets less reliable, hence, part of this deviation may be attributed to inaccuracies of the analysis method itself. However, it may also indicate the onset of the low temperature reaction channel (2d), which has been neglected in the analysis. In this sense, the increase of k2b at low temperatures would simply arise from the neglected contributions of this channel.
Overall, relying on the direct k2 determination of this work, an enthalpy value of 450 kJ mol−1 is most consistent with both the enthalpy limits set by the single-reference computations and our analysis, 444.5 kJ mol−1 < ΔfHo298K(NCN) < 454 kJ mol−1, the experimental and theoretical values for k1a from Vasudevan et al.6 and Harding et al.,3 the activation energy of reaction channel (2b) reported by Teng et al.,4 and the indirect shock tube measurements for k2b from Vasudevan et al.6 This enthalpy value is also in very good agreement with the experimental electron affinity measurements of Clifford et al.51 (451.8 ± 16.7 kJ mol−1) that has been, for example, used in the GDFkin3.0_NCN flame modelling mechanism as well.8
Using ΔH = 450 kJ mol−1, the recommended rate constant expressions for k1a, k2a, and k2b are as follows: compatible with the lower experimental uncertainty limit of Vasudevan et al., k1a is set to 0.65 times the values of Harding et al. (eqn (II)):
| k1a/(cm3 mol−1 s−1) = 2.3 × 1010 × (T/K)0.53 × exp(−71.2 kJ mol−1/RT) |
Using the updated NASA polynomial parameters for NCN from Goos et al.22 (scaled to ΔH = 450 kJ mol−1), this corresponds to a reverse reaction rate constant k2a of
| k2a/(cm3 mol−1 s−1) = 4.2 × 1015 × (T/K)−0.69 × exp(−2.0 kJ mol−1/RT) |
Adopting the temperature dependence of Teng et al., their rate expression is recommended for k2b adjusted by a factor of 1.6 to fit the case 2 data in Fig. 6b:
| k2b/(cm3 mol−1 s−1) = 7.94 × 1012 × (T/K)0.41 × exp(−22.8 kJ mol−1/RT) |
This expression is also compatible with the upper limit of the indirect shock tube measurements of Vasudevan et al.6
Finally, the sum of k2a and k2b is shown in Fig. 6b as dash-dotted curve. It is in close agreement with the k2 rate expression given by eqn (I), except at the lowest temperatures where channel k2d presumably starts to play a role. The recommended rate expression for k2b corresponds to a branching ratio ϕ = k2b/k2 that increases from ϕ = 0.21 at T = 1000 K to ϕ = 0.74 at T = 2500 K. Hence, in the temperature range relevant for flame modelling, channel switching between channel (2a) dominating at low temperatures and channel (2b) dominating at high temperatures takes place.
5 Concluding remarks
The overall rate constant of the reaction NCN + H has been directly measured at temperatures between 962 K and 2425 K behind shock waves using the thermal decomposition of NCN3 and C2H5I as suitable precursors for NCN radicals and H atoms, respectively. A conservative error analysis revealed that comparatively narrow error limits of ±20% at T = 1600 K, increasing to ±30% at the upper and lower temperature limits of the measurements, could be achieved. A main error arises from the possibly large uncertainty of the potentially important secondary reaction (10), NCN + N, which has not been measured yet. If the theoretical estimate of Moskaleva and Lin2 turns out to be right, the influence of reaction (10) would be very small and the error estimate could be further reduced. The second most important uncertainty is related to the assumed overall H atom yield from C2H5I decomposition. However, relying on the very recently published global analysis data on the ethyl iodide composition by Varga et al.,39 this error contribution could be safely assumed to be not more than 7% (an error estimate of 3.5% at T = 1200 K has been stated in the original paper).The high reliability of the rate constant data enabled us to analyze the k2 data in terms of branching ratio and the crucial value of the enthalpy of formation of NCN. Taking into account experimental and theoretical literature data for the rate constant of reaction (1a) and the temperature dependence of reaction channel (2b), ΔfHo298K = 450 kJ mol−1 was found to be most consistent. With a robust upper limit of ΔfHo298K < 456 kJ mol−1 derived from the k2 values of this work, significantly higher literature values – about 459 kJ mol−1 from high-level ab initio calculations3,4 and 466.5 kJ mol−1 from NCN photodissociation experiments24 – are at odds with our analysis. Clearly, more work is needed to further constrain the uncertainty of the enthalpy of formation of NCN.
Modelling of NOx formation in flames critically depends on the branching ratio of the reaction NCN + H. Whereas channel (2a) constitutes the reverse reaction of the main prompt-NO formation reaction (1a), CH + N2, it is in particular reaction channel (2b) with the products HCN + N that brings the overall reaction forward on the prompt-NO pathway. The results of this study with branching fractions ϕ = k2b/k2 increasing from ϕ(T = 1000 K) = 0.21 to ϕ(T = 2500 K) = 0.74 verifies the expected strong temperature dependence of this quantity. However, again the actual value of the derived branching ratio strongly depends on the assumed value of the enthalpy of formation of NCN. In fact, accurate measurements of the branching ratio would be very useful to constrain the enthalpy of formation of NCN. Moreover, in conjunction with the already compiled theoretical and experimental rate constant data, accurate branching fractions would help to draw final conclusions on this reaction system including the contributions of the recombination channel (2d), which may play a role even at temperatures as high as 1000 K.
Acknowledgements
We would like to thank the German Science Foundation (DFG, FR1529/4) for funding, M. C. Lin for sharing data prior to publication, and Friedrich Temps for continued scientific support.References
- C. P. Fenimore, Proc. Combust. Inst., 1971, 13, 373–380 CrossRef.
- L. V. Moskaleva and M. C. Lin, Proc. Combust. Inst., 2000, 28, 2393–2401 CrossRef CAS.
- L. B. Harding, S. J. Klippenstein and J. A. Miller, J. Phys. Chem. A, 2008, 112, 522–532 CrossRef CAS PubMed.
- W.-S. Teng, L. V. Moskaleva, H.-L. Chen and M. C. Lin, J. Phys. Chem. A, 2013, 117, 5775–5784 CrossRef CAS PubMed.
- G. P. Smith, Chem. Phys. Lett., 2003, 367, 541–548 CrossRef CAS.
- V. Vasudevan, R. K. Hanson, C. T. Bowman, D. M. Golden and D. F. Davidson, J. Phys. Chem. A, 2007, 111, 11818–11830 CrossRef CAS PubMed.
- A. A. Konnov, Combust. Flame, 2009, 156, 2093–2105 CrossRef CAS PubMed.
- N. Lamoureux, P. Desgroux, A. El Bakali and J. F. Pauwels, Combust. Flame, 2010, 157, 1929–1941 CrossRef CAS PubMed.
- R. S. Zhu and M. C. Lin, J. Phys. Chem. A, 2007, 111, 6766–6771 CrossRef CAS PubMed.
- R. S. Zhu, M. T. Nguyen and M. C. Lin, J. Phys. Chem. A, 2009, 113, 298–304 CrossRef CAS PubMed.
- R. S. Zhu and M. C. Lin, Int. J. Chem. Kinet., 2005, 37, 593–598 CrossRef CAS.
- C.-L. Huang, S. Y. Tseng, T. Y. Wang, N. S. Wang, Z. F. Xu and M. C. Lin, J. Chem. Phys., 2005, 122, 184321 CrossRef PubMed.
- T.-J. Yang, N. S. Wang, L. C. Lee, Z. F. Xu and M. C. Lin, J. Phys. Chem. A, 2008, 112, 10185–10192 CrossRef CAS PubMed.
- A. Busch, PhD thesis, Karlsruher Institut für Technologie, 2010.
- A. Busch and M. Olzmann, Shock-Tube Study of the Thermal Decomposition of NCN, Proc. European Combust. Meeting Vienna, paper P810138, 2009.
- J. Dammeier and G. Friedrichs, J. Phys. Chem. A, 2010, 114, 12963–12971 CrossRef CAS PubMed.
- J. Dammeier, N. Faßheber and G. Friedrichs, Phys. Chem. Chem. Phys., 2012, 14, 1030–1037 RSC.
- J. Dammeier and G. Friedrichs, J. Phys. Chem. A, 2011, 115, 14382–14390 CrossRef CAS PubMed.
- J. A. Sutton, B. A. Williams and J. W. Fleming, Combust. Flame, 2012, 159, 562–576 CrossRef CAS PubMed.
- P. Glarborg, M. U. Alzueta, K. Dam-Johansen and J. A. Miller, Combust. Flame, 1998, 115, 1–27 CrossRef CAS.
- N. Lamoureux, C. M. Western, X. Mercier and P. Desgroux, Combust. Flame, 2013, 160, 755–765 CrossRef CAS PubMed.
- E. Goos, C. Sickfeld, F. Mauß, L. Seidel, B. Ruscic, A. Burcat and T. Zeuch, Proc. Combust. Inst., 2013, 34, 657–666 CrossRef CAS PubMed.
- S. Canneaux, A. Wallet, M. Ribaucour and F. Lousi, Comput. Theor. Chem., 2011, 967, 67–74 CrossRef CAS PubMed.
- R. T. Bise, A. A. Hoops and D. M. Neumark, J. Chem. Phys., 2001, 114, 9000–9011 CrossRef CAS PubMed.
- M. Colberg and G. Friedrichs, J. Phys. Chem. A, 2006, 110, 160–170 CrossRef CAS PubMed.
- H. Bock and R. Dammel, Angew. Chem., Int. Ed. Engl., 1987, 26, 504–526 CrossRef.
- D. J. Benard, C. Linnen, A. Harker, H. H. Michels, J. B. Addison and R. Ondercin, J. Phys. Chem. B, 1998, 102, 6010–6019 CrossRef CAS.
- J. Dammeier, B. Oden and G. Friedrichs, Z. Phys. Chem., 2012, 45, 30–40 Search PubMed.
- D. E. Milligan, M. E. Jacox and A. M. Bass, J. Chem. Phys., 1965, 43, 3149–3160 CrossRef CAS PubMed.
- J. Dammeier, PhD thesis, Universität Kiel, 2011.
- S. S. Kumaran, M.-C. Su, K. P. Lim and J. V. Michael, Proc. Combust. Inst., 1996, 26, 605–611 CrossRef.
- Y. Yamamori, K. Takahashi and T. Inomata, J. Phys. Chem. A, 1999, 103, 8803–8811 CrossRef CAS.
- A. Miyoshi, N. Yamauchi, K. Kosaka and M. Koshi, J. Phys. Chem. A, 1999, 103, 46–53 CrossRef CAS.
- J. Herzler and P. Roth, Phys. Chem. Chem. Phys., 2002, 4, 5259–5264 RSC.
- B. R. Giri, T. Bentz, H. Hippler and M. Olzmann, Z. Phys. Chem., 2009, 223, 539–549 CrossRef CAS.
- T. Bentz, M. Szõri, B. Viskolcz and M. Olzmann, Z. Phys. Chem., 2011, 225, 1117–1128 CrossRef CAS.
- X. Yang and R. S. Tranter, Int. J. Chem. Kinet., 2012, 44, 433–443 CrossRef CAS.
- K. H. Weber, J. M. Lemieux and J. S. Zhang, J. Phys. Chem. A, 2009, 113, 583–591 CrossRef CAS PubMed.
- T. Varga, G. Zsély, T. Turányi, T. Bentz and M. Olzmann, Int. J. Chem. Kinet., 2014, 46, 295–304 CrossRef CAS.
- T. Turányi, T. Nagy, G. Zsély, M. Cserháti, T. Varga, B. T. Szabó, I. Sedyó, P. T. Kiss and H. J. C. A. Zempléni, Int. J. Chem. Kinet., 2012, 44, 284–302 CrossRef.
- G. Friedrichs, D. F. Davidson and R. K. Hanson, Int. J. Chem. Kinet., 2002, 34, 374–386 CrossRef CAS.
- G. P. Smith, D. M. Golden, M. Frenklach, N. W. Moriarty, B. Eiteneer, M. Goldenberg, C. T. Bowman, R. Hanson, S. Song, W. C. Gardiner Jr., V. Lissianski and Z. Qin, GRI-Mech Version 3.0, 1999 Search PubMed.
- C. K. Westbrook, Proc. Combust. Inst., 1982, 19, 127–141 CrossRef.
- D. L. Baulch, C. T. Bowmann, C. J. Cobos, R. A. Cox, T. Just, J. A. Kerr, M. J. Pilling, D. Stocker, J. Troe, W. Tsang, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 2005, 34, 757–1397 CrossRef CAS PubMed.
- P. Camilleri, R. M. Marshall and H. Purnell, J. Chem. Soc., 1974, 70, 1434–1444 CAS.
- R. J. Kee, F. M. Ruply and J. A. Miller, Chemkin-II: A Fortran Chemical Kinetics Package for the Analysis of Gas-Phase Chemical Kinetics, Sandia National Laboratories Sandia Report SAND89-8009, 1989.
- E. Goos, A. Burcat and B. Ruscic, Extended Third Millenium Ideal Gas and Condensed Phase Thermochemical Database for Combustion with Updates from Active Thermochemical Tables, 2009, http://garfield.chem.elte.hu/Burcat/burcat.html, last access: 22 April 2014.
- R. J. Kee, F. M. Rupley, J. A. Miller, M. E. Coltrin, J. F. Grcar, E. Meeks, H. K. Moffat, A. E. Lutz, G. Dixon-Lewis, M. D. Smooke, J. Warnatz, G. H. Evans, R. S. Larson, R. E. Mitchell, L. R. Petzold, W. C. Reynolds, M. Caracotsios, W. E. Stewart, P. Glarborg, C. Wang and O. Adigun, CHEMKIN Collection (transport database), Release 3.6, Reaction Design, Inc., San Diego, CA, 2000 Search PubMed.
- M. Freindorf, Y. Shao, T. R. Furlani and J. Kong, J. Comput. Chem., 2005, 26, 1270–1278 CrossRef CAS PubMed.
- A. W. Jasper and J. A. Miller, Combust. Flame, 2014, 161, 101–110 CrossRef CAS PubMed.
- E. P. Clifford, P. G. Wenthold, W. C. Lineberger, G. A. Petersson and G. B. Ellison, J. Phys. Chem. A, 1997, 101, 4338–4345 CrossRef CAS.
| This journal is © the Owner Societies 2014 |