 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePathways of energy transfer in LHCII revealed by room-temperature 2D electronic spectroscopy
Kym L.
Wells
a,
Petar H.
Lambrev
b,
Zhengyang
Zhang
a,
Gyözö
Garab
b and
Howe-Siang
Tan
*a
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: howesiang@ntu.edu.sg
bInstitute of Plant Biology, Biological Research Centre, Hungarian Academy of Sciences, PO Box 521, H-6726, Szeged, Hungary
First published on 29th April 2014
Abstract
We present here the first room-temperature 2D electronic spectroscopy study of energy transfer in the plant light-harvesting complex II, LHCII. Two-dimensional electronic spectroscopy has been used to study energy transfer dynamics in LHCII trimers from the chlorophyll b Qy band to the chlorophyll a Qy band. Observing cross-peak regions corresponding to couplings between different excitonic states reveals partially resolved fine structure at the exciton level that cannot be isolated by pump–probe or linear spectroscopy measurements alone. Global analysis of the data has been performed to identify the pathways and time constants of energy transfer. The measured waiting time (Tw) dependent 2D spectra are found to be composed of 2D decay-associated spectra with three timescales (0.3 ps, 2.3 ps and >20 ps). Direct and multistep cascading pathways from the high-energy chlorophyll b states to the lowest-energy chlorophyll a states have been resolved occurring on time scales of hundreds of femtoseconds to picoseconds.
Introduction
Light-harvesting antenna systems such as LHCII, the primary light-harvesting complex in plants, are essential for the photosynthetic process that eventually powers the biological world. The excitation energy transfer (EET) processes in naturally occurring light-harvesting systems are therefore of strong interest to scientists. Due to the ultrafast nature of these EET processes, femtosecond optical spectroscopy has proven to be a powerful tool to study these systems. Kwa et al.1 used both one-colour and two-colour pump–probe spectroscopy to study the EET processes in LHCII trimers with ps time resolution. They observed energy transfer from chlorophyll b (Chl b) to chlorophyll a (Chl a) occurring in <1 ps, which they were unable to temporally resolve. They also observed a 2–6 ps process attributed to EET between the luminal and stromal layers of the complex. Bittner et al.2 completed complementary studies with 250 fs time resolution which revealed a faster process of only ∼150 fs which was attributed to EET from Chl b to Chl a. As in the work of Kwa et al., a 3–7 ps process was observed, which could be interpreted as EET from a different Chl b pool, or as redistribution of energy between Chl a excited states.Separately collected transient absorption measurements of LHCII trimers by Visser et al.3 and Connelly et al.4 completed at 77 K each observed Chl b to Chl a EET processes with consistent time constants of 150–300 fs, 600 fs, and 4–9 ps. Similar measurements of LHCII monomers by Kleima et al.5 found two comparable transfer times of ∼200 fs and 3 ps. This work also involved global fitting of previously collected trimer3 data with these measurements to determine that in both cases 50% of the transfer occurred on the ∼200 fs timescale. It was concluded that the 600 fs process observed in the trimer is significantly slowed down in the monomer case to contribute to the observed ps process. The similarity between the monomer and the trimer results led the authors to conclude that Chl b to Chl a transfer is predominantly localised to individual monomers, which was later substantiated by theoretical studies that identified only one sizeable coupling between chlorophyll pigments on adjacent monomers in the trimer structure.6 Recent polarized transient absorption measurements by Marin et al. at 77 K measured inter-monomer EET occurring within 3.3–5 ps across the entirety of the Qy band.7
Significant efforts to model the spectral dynamics of LHCII have been undertaken.8–11 Novoderezhkin et al.6 and independently Linnanto et al.9 and Renger et al.12,13 using the 2.72 Å crystallographic LHCIIb structure14 determined the energies of the 14 excitonic states that comprise the Qy transitions of LHCII, encompassing both the Chl b and Chl a bands. They also modelled the heavily delocalised nature of these states using simultaneous quantitative fits of the linear absorption, linear dichroism, fluorescence spectra and transient absorption spectra. Other transient absorption experiments by van Grondelle and co-workers15 on several recombinant isoforms of LHCII separated energy transfer dynamics from “red” and “blue” Chl b states to Chl a states with a sequential global fitting routine. They reported EET from high energy (blue) Chl b to Chl a occurring with a 600 fs and 4 ps time constant, and energy transfer from low energy Chl b (red) to Chl a occurring with time constants of 150 fs and 600 fs. However, it is not always possible in transient absorption measurements to unambiguously isolate contributions to peaks in the transient spectra originating from different initially excited states. Whilst 2D spectroscopy, by virtue of an extra frequency axis, is able to resolve the pump frequency as well as the emission frequency. Recently, 2D spectroscopy has been applied to probe LHCII. Schlau-Cohen et al.16 identified cross peaks in the non-rephasing 2D spectra corresponding to Chl b to Chl a energy transfer. By observing these cross-peak amplitudes as a function of Tw, multistep dynamics occurring with a fastest, previously unrecorded <100 fs component were observed. These results suggest that multistep relaxation is occurring but no quantitative values were given.16 Calhoun et al.17 directly measured the energies of these excitonic states by observing the Fourier transform of the diagonal of the non-rephasing 2D spectra as a function of waiting time (Tw) at 77 K achieving excellent agreement with theory.
Presented here are 2D electronic spectroscopic studies of partially excitonically resolved energy transfer in LHCII at room temperature. At low temperatures the absorption spectra of LHCII are better resolved, but less representative of the physiological conditions under which light harvesting occurs. By performing the study presented in this paper at room temperature, we are moving to a regime which more closely mimics the in vivo conditions. Additionally, by tuning the spectrum of the first two interaction pulses to cover only the Chl b band, and using a white light continuum (WLC) to act as the third interaction pulse we observe the cross-peaks corresponding to Chl b to Chl a energy transfer without contribution from signals due to coherent processes which potentially complicate the interpretation of the collected 2D electronic spectra. In this study, we elucidate the energy transfer pathway from different spectral regions of the Chl b manifold to different spectral regions of the Chl a manifold.
In a similar approach to Schlau-Cohen's,16 integrals corresponding to different excitonic couplings on a purely absorptive 2D spectra are collected at a range of Tw values, to observe real-time energy transfer. To date, few room temperature multidimensional spectroscopy studies of light harvesting complexes have been completed.18–20 To develop a true understanding of the efficiency of light harvesting systems in vivo, studies must start to be tailored to more closely match physiological conditions.
Experimental
The experimental setup and theory behind the phase cycling schemes necessary for these pump–probe geometry 2D electronic spectroscopy measurements have been well described21,22 and demonstrated23 previously, as has the use of a white light continuum (WLC) probe and necessary chirp considerations associated with this.24–26 Only a brief description of the experimental set-up is provided here. By utilizing a pump–probe geometry set-up with phase cycling the absorptive 2D spectra are recovered directly. In a traditional boxcar arrangement both the 2D rephasing and 2D non-rephasing signals need to be collected, phased and summed to yield the purely absorptive 2D spectra. The phasing needs to be performed with great care to separate the real and imaginary parts of the 2D purely absorptive spectrum without causing distortion to the observed lineshape.20,27,28 In pump–probe geometry 2DES, wherein the first two interaction pulses are collinear allowing the simultaneous measurement of the rephasing and non-rephasing signals, the phasing step is unnecessary. Thus, there is no ambiguity in the peak shape arising from inadequate phasing.LHCII trimers were isolated and purified from spinach thylakoid membranes, solubilized with α-dodecyl maltoside (α-DM) by sucrose-gradient ultracentrifugation as described in the work of Caffarri et al.29 Before measurements, the LHCII suspension was diluted with nitrogen-flushed 0.06% α-DM and 10 mM HEPES buffer solution to provide an optical density of 0.27 (at the Chl a Qy maximum) in a 1 mm sapphire-windowed sample cell. All measurements were collected at room temperature (298 K). A commercially available regenerative amplifier laser system (Legend Elite, Coherent) provided pulses centred at 800 nm (0.8 W, 1 KHz, 40 fs) which pumped a home-built optical parametric amplifier (OPA). The near-infrared output of the OPA was subsequently frequency doubled to be resonant with the Qy transition of the Chl b band of LHCII at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 385 cm−1 (650 nm) with a FWHM of ∼350 cm−1 (∼14 nm), which was sufficiently broad to excite the entirety of the Chl b exciton manifold. This beam was passed through a commercially available acousto-optic programmable dispersive filter30 (Dazzler, Fastlite) to generate the first two excitation pulses, with variable time delay and relative phase. All measurements presented here were referenced to a carrier frequency of 14277 cm−1 (700 nm), to place the signal into the partially rotating frame, taken with a 1 × 2 phase cycling scheme22,23 and attenuated to 70 nJ sum energy for the two pulses. The pulse shaper further compressed the pulses to 55 fs FWHM. The combined third interaction pulse and local oscillator was a white light continuum generated by focussing a small amount of the fundamental frequency beam (800 nm) through a 2 mm sapphire window, and passing over a delay stage to enable control of the population time (Tw) after the first two excitation pulses. The shaped laser output and the WLC were overlapped on the sample cell, with the WLC subsequently dispersed with a spectrometer (Acton SP2300, Princeton Instruments) onto a CCD detector (PIXIS 100B, Princeton Instruments). The linear absorption spectra were taken before and after measurement scans to confirm that sample degradation had not occurred.
385 cm−1 (650 nm) with a FWHM of ∼350 cm−1 (∼14 nm), which was sufficiently broad to excite the entirety of the Chl b exciton manifold. This beam was passed through a commercially available acousto-optic programmable dispersive filter30 (Dazzler, Fastlite) to generate the first two excitation pulses, with variable time delay and relative phase. All measurements presented here were referenced to a carrier frequency of 14277 cm−1 (700 nm), to place the signal into the partially rotating frame, taken with a 1 × 2 phase cycling scheme22,23 and attenuated to 70 nJ sum energy for the two pulses. The pulse shaper further compressed the pulses to 55 fs FWHM. The combined third interaction pulse and local oscillator was a white light continuum generated by focussing a small amount of the fundamental frequency beam (800 nm) through a 2 mm sapphire window, and passing over a delay stage to enable control of the population time (Tw) after the first two excitation pulses. The shaped laser output and the WLC were overlapped on the sample cell, with the WLC subsequently dispersed with a spectrometer (Acton SP2300, Princeton Instruments) onto a CCD detector (PIXIS 100B, Princeton Instruments). The linear absorption spectra were taken before and after measurement scans to confirm that sample degradation had not occurred.
The first two interaction pulses were scanned over a total time range of 180 fs in 3 fs steps, whilst the Tw value remained constant. Due to the presence of a coherent artefact at short Tw, only 2D electronic spectra (2DES) at Tw larger than 150 fs were analysed. 2DES were collected at Tw values extending to 12.5 ps. In each collected 2DES presented here, the two frequency axes can be viewed as ‘pump’ (ωτ) and ‘probe’ axes (ωt) where the frequency-resolved excitation of the ‘pump’ is correlated with the frequency-resolved absorption of the ‘probe’ after a waiting time Tw. Since for these measurements a WLC is used to provide the third interaction field and local oscillator, the true Tw value for each ωt in each 2DES is different according to the chirp of the WLC. For the data analysis presented here the Tw is corrected according to the relative observation time of the coherent artefact at each ωt.
Results
2D spectra and time evolution
Fig. 1 shows the linear absorption spectrum of the Qy bands of LHCII at room temperature, with the Chl a band and Chl b band centred at ∼14800 cm−1 and ∼15375 cm−1 respectively. The spectrum of the pump pulse (the first two interaction pulses) used in these measurements is overlaid. The stick plots indicate the positions of the excitonic states comprising these bands, according to the model of Novoderezhkin et al.6
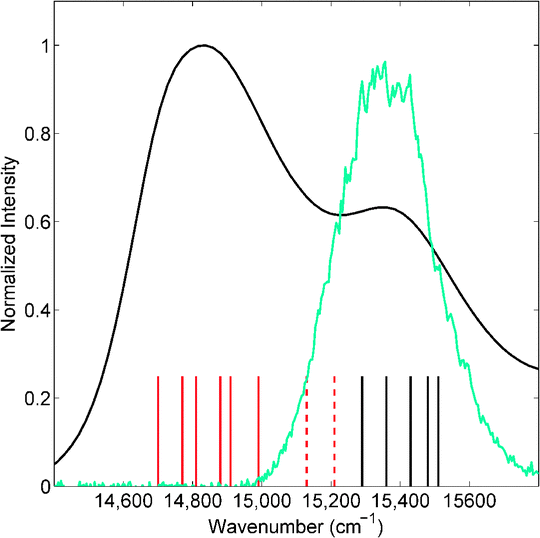 | ||
| Fig. 1 Linear absorption spectrum of LHCII trimers (black) and the spectrum of the first two interaction pulses used in the 2DES measurements (blue). Stick plots – Chl a based excitonic states (red) and predominantly Chl b based excitonic states (black).6,17 | ||
Fig. 2 shows typically collected purely absorptive 2DES at a selection of Tw values. All 2DES are plotted on the same intensity scale. The diagonal of the 2D spectrum for Tw = 0.3 ps is overlaid as a white dashed line. A qualitative description of the trends and features of the 2D spectra is provided here, followed by a quantitative study detailed below. All 2DES show two features. Firstly, a diagonal feature (ωτ = ωt) corresponds to the Chl b excitonic diagonal signals. Since the first two excitation pulses do not spectrally overlap with Chl a excitonic states, neither diagonal signals nor signals due to quantum coherences31 are observed in the Chl a spectral region. At Tw > 5 ps the Chl b diagonal signal vanishes, indicating that energy transfer out of the Chl b states is fully completed within this time.
 | ||
| Fig. 2 Purely absorptive 2DES recorded at waiting times Tw = 0.3, 0.6, 1.25, 2.5, 5 and 12.5 ps. The same colour-coding scale is used for all spectra, with red colour representing the positive signal and blue colour representing the negative signal. The axes correspond to the “pump” frequency ωτ and the “probe” frequency ωt. | ||
The second feature is an off-diagonal peak (ωτ = 15400 cm−1, ωt = 14800 cm−1), rising in amplitude over Tw < 10 ps, which corresponds to energy transfer from the initially excited Chl b excitonic states to Chl a excitonic states. The decay of the cross-peak at Tw > 10 ps is indicative of relaxation to the ground state. The broad peak is composed of a number of overlapping cross-peaks corresponding to energy transfer between different excitonic states. The principal relationship guiding the interpretation of the 2D spectra is that a cross-peak appearing at the intersection of a “pump” frequency ωτ and a “probe” frequency ωt, which we will denote as ωτ → ωt, is a result of energy transfer from an excitonic state with resonance frequency ωτ to a state with resonance frequency ωt. The intensity of the cross peak over time follows the population, or concentration, of the acceptor state resulting from the respective EET process.
Although individual exciton states cannot be resolved because of the large spectral overlap, the shape of the cross-peak reveals substantial information regarding the pathways and dynamics of EET. As can be seen from Fig. 2, at sub-ps waiting times the main cross-peak is broader and asymmetric due to the population of different excitonic states of various energies. At longer waiting times, as excitation energy is concentrated on the lowest-energy Chl a excitonic states, the peak becomes narrower, symmetric and its midpoint shifts to lower frequencies. In particular one can easily notice that at Tw = 0.3 ps there is a relatively high intensity at the intersection 15300 → 15000 cm−1. It can be inferred that an energy level corresponding to 15000 cm−1 is populated from an exciton state at ca. 15300 cm−1 over a time scale of 0.3 ps. At the same time, the cross-peak amplitude at 15500 → 15000 cm−1 is significantly lower, indicating that these two frequencies are not strongly coupled. Energy from the 15500 cm−1 level flows to exciton(s) positioned at 14800 cm−1 instead. The feature at the intersection 15300 → 15000 cm−1 disappears gradually with increasing Tw. Another conclusion that can be drawn directly from the shape of the 2D spectra is that the 15000 cm−1 state is long-lived – visible even at Tw = 12.5 ps.
The intensity of the 2D spectra at several frequency intersections is plotted as a function of waiting time in Fig. 3. The first panel (Fig. 3a) traces the intensity along the diagonal (ωτ = ωt), showing the decay of Chl b excited states. It is immediately apparent that higher-energy states decay faster than lower-energy states. The next three panels group time traces representing EET from different Chl b frequencies to a single Chl a frequency. Rather than decay, these traces show the rise in Chl a excited-state population. In Fig. 3b, high-energy Chl a states (15000 cm−1) are rapidly populated and then slowly decay over several ps. In Fig. 3d, slow, picoseconds rise kinetics is observed for the lowest-energy states (14600 cm−1). Comparing the time traces plotted in the same panel, i.e. at one ωt, some notable differences are revealed. The 15300 → 15000 cm−1 trace rises to a maximum in 1 ps, affirming the strong coupling between these two frequencies, and then slowly decays following the depopulation of the intermediate state at 15000 cm−1. In contrast, the 15500 → 15000 cm−1 curve has lower amplitude, slower rise and no significant decay. In comparison, the 15500 → 14800 cm−1 trace rises quickly to a maximum and then decays, whereas the 15300 → 14800 cm−1 trace shows a biphasic rise over the chosen time range. Finally, the traces representing EET to the lowest-energy Chl a do not significantly depend on the pump frequency. These slowly populated states are the final acceptors of excitation energy as indicated by the slow continuous rise at Tw > 5 ps (in contrast, the signal at higher frequencies decays on this time scale).
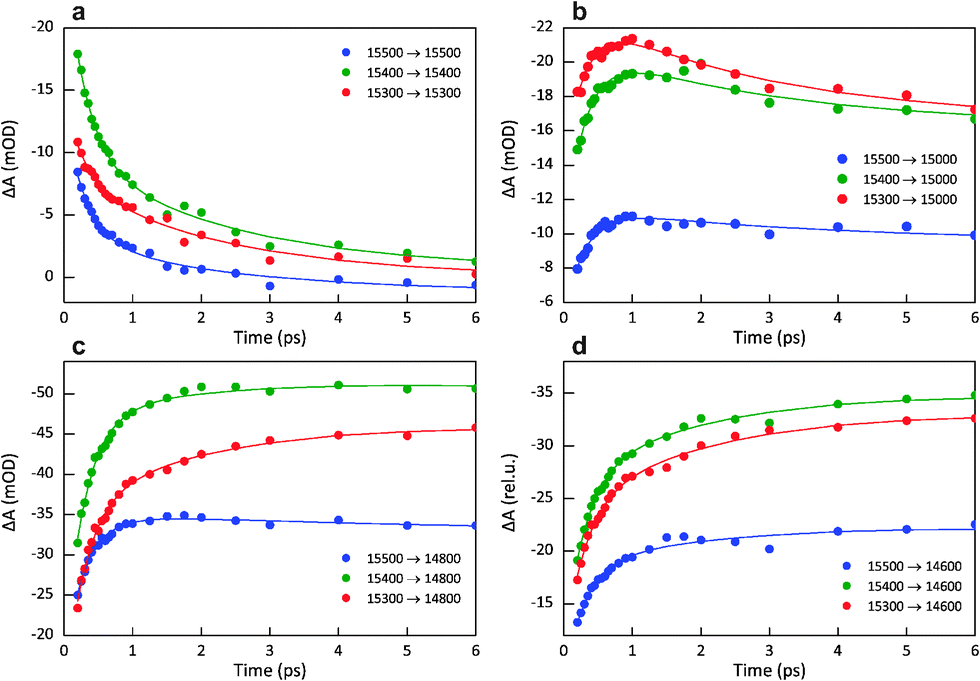 | ||
| Fig. 3 Time traces of the absorptive 2D signal at selected frequency intersections, indicated in the plots as ωτ → ωt. The top-left panel represents diagonal signals (ωτ = ωt) and the rest – cross-peak signals. Each point in the time traces is the integrated 2D signal from an area of ca. 50 cm−1 × 50 cm−1. Symbols represent the experimentally measured data points and solid lines show the best fit using a global three-exponential analysis with lifetimes of 0.3, 2.3 and 175 ps across all ωτ/ωτ. | ||
We must reiterate that due to spectral overlap the time traces for different ωτ → ωt obviously sum up the EET among multiple excitonic states. While it is tempting to relate the cross-peak frequencies to calculated excitonic levels, e.g. those calculated by Calhoun et al.,17 there are complications such as inhomogeneous broadening, spectral diffusion32 and Stokes' shifts of the excited-state emission contributing to the non-linear spectra.
Global analysis of the 2D spectral evolution
To obtain a quantitative measure of the lifetimes of the system and qualitative information about the processes occurring on each lifetime, we present the results of a global analysis of the 2D spectra to recover 2D decay-associated spectra (2D DAS). The global analysis procedure (written in MATLAB) relies on the assumption that the diagonal peaks as well as the cross-peaks in the 2D spectra follow the kinetics of the excited state population in the system, which can be described as a sum of exponential components.33,34 The time-dependent 2D absorptive signal at any given frequency pair ωτ, ωt is then described by the equation:where t = Tw, Γi are the lifetimes of the system, n is the total number of lifetime components, and Di(ωτ,ωt) are 2D DAS for the respective lifetimes. Note that the lifetimes are independent of frequency, i.e. the kinetics for every frequency pair is described with the same set of lifetimes. The absorptive 2D spectra taken at a range of population times Tw were simultaneously fitted according to such a model, whereby the lifetimes were determined by a global iterative non-linear least squares minimization algorithm. The method of variable projection was applied33 to separate the nonlinear parameters (lifetimes) from the linear parameters (pre-exponential amplitudes Di(ωτ,ωt) comprising the 2D DAS). The latter were calculated by linear regression at every iteration of the minimization. Prior to the analysis, the 2D data were corrected for dispersion (chirp) of the probe light, determined from a conventional pump–probe measurement performed with the same experimental setup. In this present study, the instrument response function (IRF) is not taken into consideration. However in future studies the IRF may need to be included in the analysis, especially for timescales comparable to the laser pulse widths.

A minimum of three lifetimes was necessary to achieve a good fit of the 2D spectra – 0.30 ps, 2.3 ps, and ≫10 ps. The number of time points and the signal-to-noise level limit the number of lifetimes that can be resolved. Since the fit was performed only in the time region of 0.2–8 ps, the longest lifetime is not accurately determined. Time trace fits produced by the analysis are displayed in Fig. 3 as solid lines.
The 2D DAS resulting from the global analysis are shown in Fig. 4. In general the DAS associated with a given lifetime is a linear combination of the spectra of all absorbing species (exciton states) whose concentration changes with this lifetime.33 In the 2D DAS, negative peaks (in blue) indicate decay, whereas positive peaks (in red) indicate rise of the bleaching signal. On a 2D DAS, the occurrence of a negative peak and a positive peak along the same ωτ frequency strongly suggests EET from frequency ωt of the negative peak to the frequency ωt of the positive peak. Thus, the 2D DAS corresponding to the first two lifetimes, 0.3 and 2.3 ps (Fig. 4a and b, respectively), indicates EET, and the 2D DAS with the longest lifetime depicted in Fig. 4c indicates decay to the ground state.
 | ||
| Fig. 4 2D decay-associated spectra resulting from a global fit of the 2D spectra with three exponential terms. The respective lifetimes are indicated in the plots (lower-right corner). Blue colours represent negative pre-exponential amplitudes, i.e. decay of the bleaching signal, and red colours represent positive amplitudes, i.e. rise of the bleaching signal. The first two 2D DAS show rise of the cross-peak, centered at ωt ∼ 14800 cm−1, and the final DAS shows decay of the cross-peak. | ||
The first 2D DAS (Fig. 4a) reveals that on a time scale of 300 fs Chl b excited states with energies around 15400 cm−1 transfer energy to Chl a excited states between 14600 and 15000 cm−1. It is rather clear from this DAS that there is ultrafast, femtosecond-scale EET to a broad range of Chl a frequencies, including the lowest ones (<14700 cm−1). The positive peak can also be seen to be non-circular and aligned in an anti-diagonal fashion. This indicates that the lower energy acceptor excitons are correlated with the higher energy donor excitons. To further aid in understanding the 2D DAS, slices of the 0.3 ps lifetime 2D DAS at selected ωτ frequencies are plotted in Fig. 5a. The positive part of the curves can be regarded as the spectrum of Chl a states that are populated by EET from the selected ωτ frequency. Note that the negative peak position, representing the decay of the donor states, approximately matches the selected pump frequency. On a sub-ps time scale, the sliced DAS show EET from a frequency of 15300 cm−1 to the high-energy intermediate (14900–15000 cm−1) and from 15500 cm−1 to the main Chl a band (14800 cm−1). This is consistent with the 15300 → 15000 cm−1 cross-peak found in the 2D spectrum at Tw = 0.3 ps (Fig. 2).
 | ||
| Fig. 5 Horizontal slices of the 2D DAS for τ = 0.3 ps and τ = 2.3 ps at selected pump frequencies ωτ, showing EET originating from excitonic states with the respective transition resonance frequency ωτ. | ||
The second 2D DAS (Fig. 4b) represents slow EET processes, occurring on a time scale of 2.3 ps. From the shape of the 2D DAS, three main conclusions can be drawn. First, EET from the higher-energy Chl b states to Chl a occurs on this slower time scale as well. Hence, there is a long-lived population of Chl b states not connected by fast EET to either Chl b or Chl a. Comparing the amplitude of the negative Chl b peak in the two DAS, it can be found out that 33% of the Chl b states decay with the slower lifetime. Second, a distinguishing feature of the 2.3 ps 2D DAS is the negative cross-peak 15300 → 15100 cm−1. The cross-peak indicates the presence of a long-lived intermediate Chl a state at ∼15100 cm−1, populated by rapid EET from a Chl b state at 15300 cm−1 and then decaying via slow (picoseconds) EET to the lower-energy Chl a. In Fig. 5b, horizontal slices at ωτ = 15200 cm−1 and 15450 cm−1 of the 2D DAS of Fig. 4b are taken. The plots reveal EET from 15500 cm−1 to the lowest-energy Chl a (14600 cm−1) as well as downstream energy flow from the intermediate level of 15000–15200 cm−1. These DAS slices corroborate the finding that high-energy Chl b states (15450 cm−1) are coupled to low-energy Chl a (14670 cm−1), with EET between them occurring on two time scales – hundreds of femtoseconds and a few picoseconds. Lastly, the slower 2.3 ps 2D DAS reveals equilibration in the lower energy Chl a manifold, where mid-energy Chl a states (14800–14900 cm−1) decay to the lowest-energy states, causing the comparatively narrow and skewed Chl a cross-peak.
The third and final 2D DAS, associated with the longest lifetime (≫10 ps), represents the decay of Chl a excited states.
The DAS do not explicitly reveal whether the EET is a direct or a multistep process involving intermediate excitonic states. In fact from the DAS slices it seems that the 2.3 ps EET from 15450 cm−1 to 14670 cm−1 is a multistep process via states at ∼14900 cm−1. For a definitive answer to the question of multistep EET, 2DES data of substantially higher dynamic range and time resolution are needed (in order to resolve more lifetimes) or, alternatively, a 3D experimental setup with an additional frequency axis.
Discussion
The pump–probe geometry 2DES results presented here partially resolve the EET pathways between Chl b and Chl a excitonic states in LHCII at room temperature. To our knowledge, this is the most detailed experimentally obtained EET kinetics scheme so far. Whilst the identified cross-peak regions studied here are not fully resolved, differences between couplings of different initially excited excitonic states are clearly evident, which pump–probe measurements alone cannot observe.In a short summary, our qualitative and quantitative global data analysis suggests: (1) fast (≤300 fs) EET from high Chl b levels (15400–15500 cm−1) to middle Chl a levels (14800 cm−1); (2) fast EET from lower Chl b levels (15200–15300 cm−1) to the intermediate level (15000–15100 cm−1); (3) on a much slower, picoseconds time scale, another population (33%) of Chl b states decays via EET to Chl a; (4) slow relaxation from the high-energy Chl a intermediate to the lowest Chl a levels; (5) equilibration in the low-energy Chl a manifold also occurs on this slower time scale.
The Chl b manifold can be divided into two groups, with one located near the stromal side and the other near the luminal side. The two groups are physically separated from each other, which can be the reason why we can observe two distinct lifetimes of EET from Chl b. According to the theoretical models and numerical simulations based on transient absorption, and other steady state spectroscopies, developed in the work of van Grondelle and Novoderezhkin,8,11 there is no fast energy relaxation from the Chl b manifold to the lowest-energy Chl a states. However, our 2DES experiments unambiguously demonstrate such EET. The model of van Grondelle and Novoderezhkin predicts fast energy relaxation from the luminal Chl b group (Chls 606 and 607) to the high-energy Chl a 604. This fast EET is probably the origin of the cross peak at 15300 → 15100 cm−1 in Fig. 2. Also in agreement with the model, and with previous low-temperature 2DES experiments,16 the high-energy Chl a state is weakly coupled to the low-energy Chls and is thus a bottleneck state that decays on a time scale of several ps. Our experiments clearly distinguish the bottleneck state with a resonant frequency of about 15000 cm−1 and further show the fast coupling of this state to the low-energy Chl b (15300 cm−1).
A parallel can also be drawn between our work and ultrafast transient absorption experiments done on CP29, another member of the LHCII family, which has a highly similar pigment arrangement, except that a few Chl b pigments found in the major LHCII are replaced by Chl a in the CP29 structure.35 Croce et al.36 have observed three lifetimes of EET from Chl b (15400 cm−1) to Chl a in CP29 – 150 fs, 1.2 ps and 5–6 ps. The present analysis of the 2D cannot resolve all lifetimes in the system but the general finding of a fast-decaying (300 fs) and a slow-decaying (2 ps) population of Chl b is in agreement with these earlier experiments. Probably the longest lifetime in CP29 involves high-energy Chl a intermediates as well. Finally, our experimental data point to a slower, picoseconds equilibration kinetics within the Chl a domain, in line with the structure-based theoretical predictions of Renger et al.13
Not all excitonic states can be unambiguously resolved in our experiments and therefore it is not possible to determine all states and pathways based on these experimental data alone. Furthermore, we have not included the contributions from direct excitation of the first vibronic replica of the Chl a Qy bands by the first two interaction pulses. However since absorptivity of these bands is much smaller than the main Chl b Qy bands, their contributions are minor, although this warrants further studies and simulations.
Whilst polarization controlled measurements have shown great promise in isolating specific spectral features in 2DES,37–39 it is unlikely that these methods will provide unambiguous assignment of the multistep mechanisms involved here. It is thus suggested that higher dimensional spectroscopy measurements, such as those already demonstrated on solvated Chl a,40,41 are needed to further clarify the intermediate excitonic states involved in these step-wise energy transfer processes, as higher dimensional spectroscopy will reveal higher order couplings. These multi-step pathways are most likely due to spatial overlap of excitonic states of mixed Chl a and Chl b composition with both the high-energy Chl b excitonic states and the low-energy Chl a excitonic states. To truly understand the efficiency of naturally occurring light-harvesting complexes, and indeed energy transfer processes in larger pigment–protein complexes, a much refined understanding of the energy transfer mechanisms at physiological temperatures is required.
Acknowledgements
This work is supported by a joint grant from the Hungarian National Innovation Office and the Singapore Agency for Science, Technology and Research, A*STAR (A*STAR SERC Grant No. 102-149-0153; NIH-A*STAR TET_10-1-2011-027) and a grant from the Singapore National Research Foundation (NRF-CRP5-2009-04). Z.Z. thanks the Nanyang President's Graduate Scholarship for support. P.L. acknowledges support from the Hungarian Scientific Research Fund (OTKA-PD 104530). The authors would like to acknowledge Jeremy Rouxel for fruitful discussions.Notes and references
- S. L. S. Kwa, H. van Amerongen, S. Lin, J. P. Dekker, R. van Grondelle and W. S. Struve, Biochim. Biophys. Acta, 1992, 1102, 202–212 CrossRef CAS.
- T. Bittner, K. D. Irrgang, G. Renger and M. R. Wasielewski, J. Phys. Chem., 1994, 98, 11821–11826 CrossRef CAS.
- H. M. Visser, F. J. Kleima, I. H. M. van Stokkum, R. van Grondelle and H. van Amerongen, Chem. Phys., 1996, 210, 297–312 CrossRef CAS.
- J. P. Connelly, M. G. Muller, M. Hucke, G. Gatzen, C. W. Mullineaux, A. V. Ruban, P. Horton and A. R. Holzwarth, J. Phys. Chem. B, 1997, 101, 1902–1909 CrossRef CAS.
- F. J. Kleima, C. C. Gradinaru, F. Calkoen, I. H. M. van Stokkum, R. van Grondelle and H. van Amerongen, Biochemistry, 1997, 36, 15262–15268 CrossRef CAS PubMed.
- V. I. Novoderezhkin, M. A. Palacios, H. van Amerongen and R. van Grondelle, J. Phys. Chem. B, 2005, 109, 10493–10504 CrossRef CAS PubMed.
- A. Marin, I. N. M. van Stokkum, V. I. Novoderezhkin and R. van Grondelle, J. Photochem. Photobiol., A, 2012, 234, 91–99 CrossRef CAS PubMed.
- R. van Grondelle and V. I. Novoderezhkin, Phys. Chem. Chem. Phys., 2006, 8, 793–807 RSC.
- J. Linnanto, J. Martiskainen, V. Lehtovuori, J. Ihalainen, R. Kananavicius, R. Barbato and J. Korppi-Tommola, Photosynth. Res., 2006, 87, 267–279 CrossRef CAS PubMed.
- V. I. Novoderezhkin and R. van Grondelle, Phys. Chem. Chem. Phys., 2010, 12, 7352–7365 RSC.
- V. Novoderezhkin, A. Marin and R. van Grondelle, Phys. Chem. Chem. Phys., 2011, 13, 17093–17103 RSC.
- F. Müh, M. Madjet and T. Renger, J. Phys. Chem. B, 2010, 114, 13517–13535 CrossRef PubMed.
- T. Renger, M. E. Madjet, A. Knorr and F. Müh, J. Plant Physiol., 2011, 168, 1497–1509 CrossRef CAS PubMed.
- Z. F. Liu, H. C. Yan, K. B. Wang, T. Y. Kuang, J. P. Zhang, L. L. Gui, X. M. An and W. R. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed.
- M. A. Palacios, J. Standfuss, M. Vengris, B. F. van Oort, I. H. M. van Stokkum, W. Kuhlbrandt, H. van Amerongen and R. van Grondelle, Photosynth. Res., 2006, 88, 269–285 CrossRef CAS PubMed.
- G. S. Schlau-Cohen, T. R. Calhoun, N. S. Ginsberg, E. L. Read, M. Ballottari, R. Bassi, R. van Grondelle and G. R. Fleming, J. Phys. Chem. B, 2009, 113, 15352–15363 CrossRef CAS PubMed.
- T. R. Calhoun, N. S. Ginsberg, G. S. Schlau-Cohen, Y. C. Cheng, M. Ballottari, R. Bassi and G. R. Fleming, J. Phys. Chem. B, 2009, 113, 16291–16295 CrossRef CAS PubMed.
- G. Panitchayangkoon, D. Hayes, K. A. Fransted, J. R. Caram, E. Harel, J. Z. Wen, R. E. Blankenship and G. S. Engel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12766–12770 CrossRef CAS PubMed.
- E. Collini, C. Y. Wong, K. E. Wilk, P. M. G. Curmi, P. Brumer and G. D. Scholes, Nature, 2010, 463, 644–U669 CrossRef CAS PubMed.
- J. M. Anna, E. E. Ostroumov, K. Maghlaoui, J. Barber and G. D. Scholes, J. Phys. Chem. Lett., 2012, 3, 3677–3684 CrossRef CAS.
- H.-S. Tan, J. Chem. Phys., 2008, 129, 124501 CrossRef PubMed.
- S. Yan and H.-S. Tan, Chem. Phys., 2009, 360, 110–115 CrossRef CAS PubMed.
- Z. Zhang, K. L. Wells, E. W. J. Hyland and H.-S. Tan, Chem. Phys. Lett., 2012, 550, 156–161 CrossRef CAS PubMed.
- P. E. Tekavec, J. A. Myers, K. L. M. Lewis and J. P. Ogilvie, Opt. Lett., 2009, 34, 1390–1392 CrossRef.
- P. F. Tekavec, J. A. Myers, K. L. M. Lewis, F. D. Fuller and J. P. Ogilvie, Opt. Express, 2010, 18, 11015–11024 CrossRef CAS PubMed.
- P. A. Tekavec, K. L. M. Lewis, F. D. Fuller, J. A. Myers and J. P. Ogilvie, IEEE J. Sel. Top. Quantum Electron., 2012, 18, 210–217 CrossRef CAS.
- J. D. Hybl, A. W. Albrecht, S. M. G. Faeder and D. M. Jonas, Chem. Phys. Lett., 1998, 297, 307–313 CrossRef CAS.
- D. B. Turner, K. E. Wilk, P. M. G. Curmi and G. D. Scholes, J. Phys. Chem. Lett., 2011, 2, 1904–1911 CrossRef CAS.
- S. Caffarri, R. Croce, J. Breton and R. Bassi, J. Biol. Chem., 2001, 276, 35924–35933 CrossRef CAS PubMed.
- A. Monmayrant, A. Arbouet, B. Girard, B. Chatel, A. Barman, B. J. Whitaker and D. Kaplan, Appl. Phys. B: Lasers Opt., 2005, 81, 177–180 CrossRef CAS PubMed.
- A. Ishizaki, T. R. Calhoun, G. S. Schlau-Cohen and G. R. Fleming, Phys. Chem. Chem. Phys., 2010, 12, 7319–7337 RSC.
- K. L. Wells, Z. Y. Zhang, J. R. Rouxel and H. S. Tan, J. Phys. Chem. B, 2013, 117, 2294–2299 CrossRef CAS PubMed.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, 2004, 1657, 82–104 CrossRef CAS PubMed.
- A. R. Holzwarth, in Biophysical Techniques in Photosynthesis, ed. J. Amesz and A. J. Hoff, Kluwer Academic Publishers, Dordrecht, 1996, pp. 75–92 Search PubMed.
- X. W. Pan, M. Li, T. Wan, L. F. Wang, C. J. Jia, Z. Q. Hou, X. L. Zhao, J. P. Zhang and W. R. Chang, Nat. Struct. Mol. Biol., 2011, 18, 309–315 CAS.
- R. Croce, M. G. Müller, R. Bassi and A. R. Holzwarth, Biophys. J., 2003, 84, 2508–2516 CrossRef CAS.
- G. S. Schlau-Cohen, T. R. Calhoun, N. S. Ginsberg, M. Ballottari, R. Bassi and G. R. Fleming, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13276–13281 CrossRef CAS PubMed.
- N. S. Ginsberg, J. A. Davis, M. Ballottari, Y. C. Cheng, R. Bassi and G. R. Fleming, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3848–3853 CrossRef CAS PubMed.
- G. S. Schlau-Cohen, E. De Re, R. J. Cogdell and G. R. Fleming, J. Phys. Chem. Lett., 2012, 3, 2487–2492 CrossRef CAS.
- Z. Zhang, K. L. Wells and H.-S. Tan, Opt. Lett., 2012, 37, 5058–5060 CrossRef CAS PubMed.
- Z. Zhang, K. L. Wells, M. T. Seidel and H.-S. Tan, J. Phys. Chem. B, 2013, 117, 15369–15385 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2014 |