 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DFT study of the structures, stabilities and redox behaviour of the major surfaces of magnetite Fe3O4†
David
Santos-Carballal
*a,
Alberto
Roldan
a,
Ricardo
Grau-Crespo
b and
Nora H.
de Leeuw
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, WC1H 0AJ, London, UK. E-mail: david.carballal.10@ucl.ac.uk; n.h.deleeuw@ucl.ac.uk; Fax: +44 (0)20 7679 7463; Tel: +44 (0)20 7679 1015
bDepartment of Chemistry, University of Reading, Whiteknights, RG6 6AD, Reading, UK
First published on 12th May 2014
Abstract
The renewed interest in magnetite (Fe3O4) as a major phase in different types of catalysts has led us to study the oxidation–reduction behaviour of its most prominent surfaces. We have employed computer modelling techniques based on the density functional theory to calculate the geometries and surface free energies of a number of surfaces at different compositions, including the stoichiometric plane, and those with a deficiency or excess of oxygen atoms. The most stable surfaces are the (001) and (111), leading to a cubic Fe3O4 crystal morphology with truncated corners under equilibrium conditions. The scanning tunnelling microscopy images of the different terminations of the (001) and (111) stoichiometric surfaces were calculated and compared with previous reports. Under reducing conditions, the creation of oxygen vacancies in the surface leads to the formation of reduced Fe species in the surface in the vicinity of the vacant oxygen. The (001) surface is slightly more prone to reduction than the (111), due to the higher stabilisation upon relaxation of the atoms around the oxygen vacancy, but molecular oxygen adsorbs preferentially at the (111) surface. In both oxidized surfaces, the oxygen atoms are located on bridge positions between two surface iron atoms, from which they attract electron density. The oxidised state is thermodynamically favourable with respect to the stoichiometric surfaces under ambient conditions, although not under the conditions when bulk Fe3O4 is thermodynamically stable with respect to Fe2O3. This finding is important in the interpretation of the catalytic properties of Fe3O4 due to the presence of oxidised species under experimental conditions.
1 Introduction
Magnetite, Fe3O4, is of significant importance as the main component of industrial catalysts in, for example, the dehydrogenation of ethylbenzene1 which is used as the primary feedstock for the production of 85% of commercial styrene.2,3 Fe3O4 is also used as a catalyst for the water gas shift (WGS) reaction, where molecular hydrogen is formed from carbon monoxide and water,4–6 the Fischer–Tropsch synthesis of hydrocarbons7 and the Haber–Bosch process for the production of ammonia.8–11 The high stability and catalytic activity as well as its low cost make Fe3O4 the catalyst of choice for these heterogeneous processes.12 Furthermore, Fe3O4 is important in other applications, such as groundwater remediation,13 and potentially in spintronic devices due to the conducting properties of only one channel of spins.14,15Above 120 K, Fe3O4 crystallizes in the spinel structure16 with space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (cubic),17 but when cooled below that temperature, it undergoes a phase transition known as the Verwey transition, where the space group changes to P2/m (monoclinic).17 Thus, at room temperature, Fe3O4 has the spinel face-centred cubic unit cell, on which we will focus in this paper. In this structure, the oxygen ions are regularly close packed along the [111] direction, separating layers of Fe ions, which appear in two different alternate arrangements. One is composed of Fe ions occupying two types of positions (octahedral (FeB) and tetrahedral (FeA)) and the other one has only FeB, shown in the scheme in Fig. 1. The experimental lattice constant for Fe3O4 is a = 8.390 Å17 and each unit cell is composed of eight formula units (four rhombohedral primitive cells). Unlike the rest of the iron oxides, Fe3O4 has Fe ions in mixed valence states, with the chemical formula often written as FeA3+[Fe2+Fe3+]BO4, where A and B represent the tetrahedral and octahedral sites, respectively. The distribution where the 3+ cations occupy the A sites, while the B sites contain a mixture of 2+ and 3+ cations, is called inverse (in contrast with the normal spinel where the 2+ cations are all located in the A site).
m (cubic),17 but when cooled below that temperature, it undergoes a phase transition known as the Verwey transition, where the space group changes to P2/m (monoclinic).17 Thus, at room temperature, Fe3O4 has the spinel face-centred cubic unit cell, on which we will focus in this paper. In this structure, the oxygen ions are regularly close packed along the [111] direction, separating layers of Fe ions, which appear in two different alternate arrangements. One is composed of Fe ions occupying two types of positions (octahedral (FeB) and tetrahedral (FeA)) and the other one has only FeB, shown in the scheme in Fig. 1. The experimental lattice constant for Fe3O4 is a = 8.390 Å17 and each unit cell is composed of eight formula units (four rhombohedral primitive cells). Unlike the rest of the iron oxides, Fe3O4 has Fe ions in mixed valence states, with the chemical formula often written as FeA3+[Fe2+Fe3+]BO4, where A and B represent the tetrahedral and octahedral sites, respectively. The distribution where the 3+ cations occupy the A sites, while the B sites contain a mixture of 2+ and 3+ cations, is called inverse (in contrast with the normal spinel where the 2+ cations are all located in the A site).
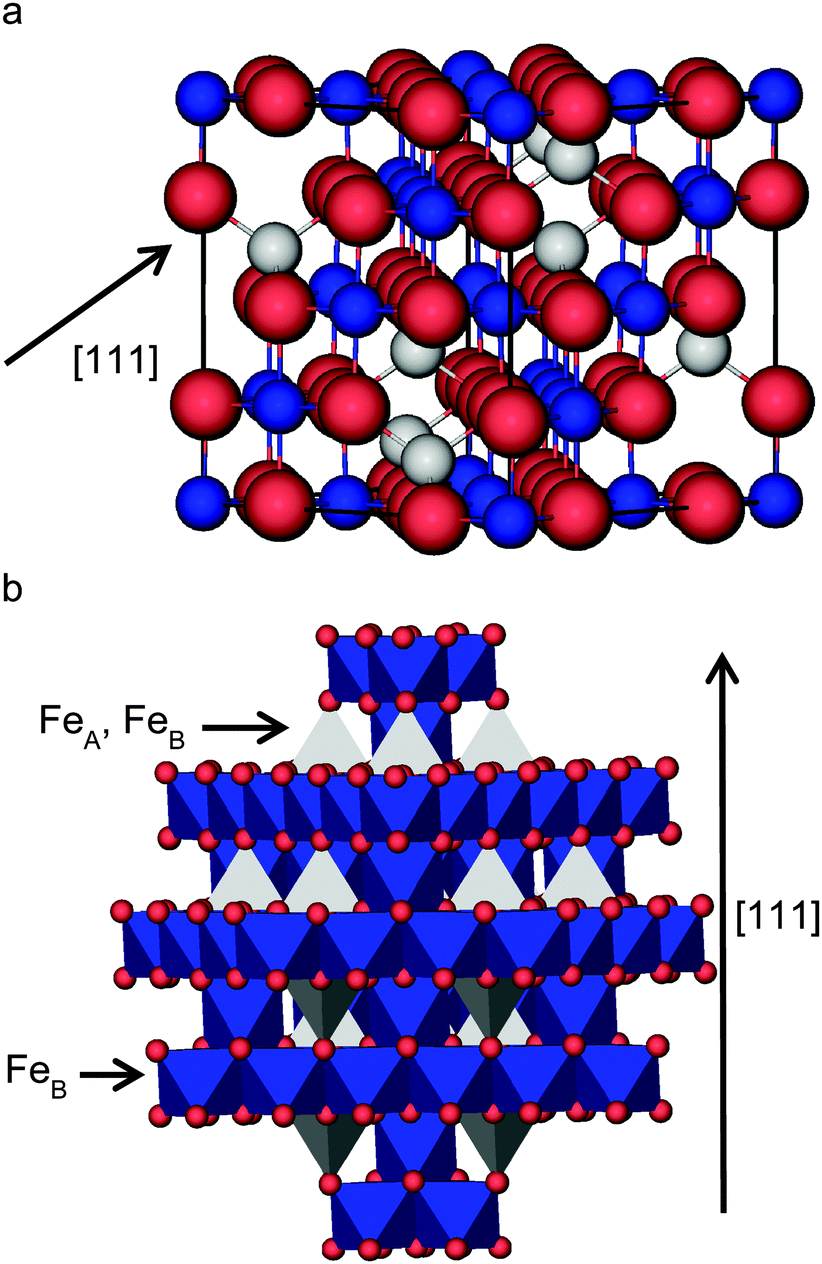 | ||
| Fig. 1 View of the bulk structure of Fe3O4: (a) ball and stick model of the cubic unit cell and (b) polyhedral model showing the alternating layers of FeB and FeA–FeB ions separated by O ions. FeA ions are in grey, FeB ions are in blue and O ions are in red. | ||
Biological,18,19 extra-terrestrial20,21 and synthetic22,23 Fe3O4 crystals have been described by several authors. Among all the crystal habits in which this mineral has been found, the three most common are the octahedral morphology enclosed by (111) surfaces; a truncated octahedron by adding the (001) plane and as twinning on the (111) surface.12,22 Zhao et al. synthesized Fe3O4 under a systematic range of conditions using a polyol process, where the crystals obtained ranged from cubic, truncated octahedral to octahedral shapes, depending on pH.22
The stacking sequence of the atomic planes perpendicular to the [001] direction can be represented as FeA–(O–FeB) (atoms inside brackets are within the same layer), leading in principle, to two different bulk-like terminations for the (001) surface, which are all polar. There are also two possible non-dipolar reconstructions of this surface, i.e. when the slab is terminated by either 0.5 mono layers (ML) FeA or 0.5 ML O–FeB in both the top and bottom surface. Experimentally, this surface has been found to have a 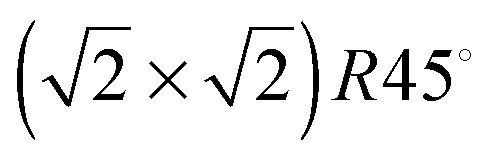 reconstruction for which different rationalizations have been given. Studies combining low-energy electron diffraction (LEED), X-ray photoelectron spectroscopy (XPS), X-ray photoelectron diffraction (XPD) and scanning tunnelling microscopy (STM),24 as well as another work combining LEED with low-energy ion scattering (LEIS),25 have suggested a (001) surface terminated by the reconstructed non-dipolar 0.5 ML of FeA. On the other hand, a different study, combining STM, LEED, LEIS and XPS, has suggested a surface terminated by the reconstructed charge-compensated O–FeB with one oxygen vacancy per unit cell.26 Meanwhile, Voogt et al.27 were unable to differentiate them based on reflection high-energy electron diffraction (RHEED) and LEED, suggesting as possible terminations: the reconstructed non-dipolar 0.5 ML FeA layer or the reconstructed charge-compensated O–FeB layer with oxygen vacancies or hydroxyl groups.27 More recently, Parkinson et al.28 have identified experimentally using STM and LEED images that the (001) surface terminations are temperature dependent. The 0.5 ML O–FeB termination or one with wavelike structure and small defects, such as hydroxyl groups, is thermodynamically more stable at 923 K, while at lower temperatures (623 K) the surface terminated by 0.5 ML of FeA is observed, although some point-defects may stabilise other terminations.28
reconstruction for which different rationalizations have been given. Studies combining low-energy electron diffraction (LEED), X-ray photoelectron spectroscopy (XPS), X-ray photoelectron diffraction (XPD) and scanning tunnelling microscopy (STM),24 as well as another work combining LEED with low-energy ion scattering (LEIS),25 have suggested a (001) surface terminated by the reconstructed non-dipolar 0.5 ML of FeA. On the other hand, a different study, combining STM, LEED, LEIS and XPS, has suggested a surface terminated by the reconstructed charge-compensated O–FeB with one oxygen vacancy per unit cell.26 Meanwhile, Voogt et al.27 were unable to differentiate them based on reflection high-energy electron diffraction (RHEED) and LEED, suggesting as possible terminations: the reconstructed non-dipolar 0.5 ML FeA layer or the reconstructed charge-compensated O–FeB layer with oxygen vacancies or hydroxyl groups.27 More recently, Parkinson et al.28 have identified experimentally using STM and LEED images that the (001) surface terminations are temperature dependent. The 0.5 ML O–FeB termination or one with wavelike structure and small defects, such as hydroxyl groups, is thermodynamically more stable at 923 K, while at lower temperatures (623 K) the surface terminated by 0.5 ML of FeA is observed, although some point-defects may stabilise other terminations.28
To date, most computational efforts have been concentrated on explaining the stability of the bulk-like dipolar O–FeB termination, leaving largely ignored the reconstructed non-dipolar 0.5 ML FeA termination. Pentcheva et al.29 have studied the stability under varying redox conditions of one ideal and reconstructed stoichiometric (0.5 ML FeA) and several non-stoichiometric terminations using spin-polarised density functional theory (DFT) calculations. These authors found that the modified polar bulk-like O–FeB termination was the most stable for the whole range of chemical potential,29 which was validated by experimental X-rays diffraction (XRD)29 and by the wavelike pattern along the [011] direction observed on experimental STM images.30 Further studies by Parkinson et al.31 of this surface termination using spin-polarized DFT + U calculations supported the Jahn–Teller distortion of this surface based on simulation of STM images of antiphase domain boundaries (APDBs).31
In the [011] direction, Fe3O4 is composed of alternating layers of (FeA–FeB–O) and (FeB–O). After reconstruction, two non-dipolar terminations are possible: one terminated by the (FeB–O) layer with 0.25 ML FeA on top and another terminated by the (FeA–FeB–O) layer with 0.25 ML of FeA vacancies. Single crystal studies carried out on this surface, involving the use of STM, LEED, scanning tunnelling spectroscopy (STS) and Auger electron spectroscopy (AES), have found a one-dimensional reconstruction along the [01![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] direction, which was concluded not to have a simple bulk iron-oxide termination.32,33 Subsequent studies supported by atomically resolved STM34–36 suggested a model based on a surface terminated by a polar (FeA–FeB–O) bulk-like layer, in order to explain the atomic rows observed on the tops of ridges along the [01] direction. However, the authors also left open the possibility of alternative models, including surface reconstruction, to interpret the STM images.34
] direction, which was concluded not to have a simple bulk iron-oxide termination.32,33 Subsequent studies supported by atomically resolved STM34–36 suggested a model based on a surface terminated by a polar (FeA–FeB–O) bulk-like layer, in order to explain the atomic rows observed on the tops of ridges along the [01] direction. However, the authors also left open the possibility of alternative models, including surface reconstruction, to interpret the STM images.34
The (111) surface is the dominant cleavage plane of Fe3O4, and the stacking of the atomic layers perpendicular to this surface is FeA1–FeB1–FeA2–O1–FeB2–O2. All of the six possible different bulk-like surface terminations are dipolar. Only two reconstructions lead to non-dipolar terminations, i.e. 0.5 ML FeB1 or 0.5 ML FeB2. Several possible terminations have been described from LEED and STM results: one dipolar plane showing close packed features (due to 0.75 ML of FeB2 and 0.25 ML of O2 over a close packed O1 layer);37 a reconstructed non-dipolar honeycomb plane (due to 0.5 ML of FeB1), which was the most stable one;37 a reconstructed dipolar 0.25 ML FeA1 plane;38 as well as a regular bulk-like dipolar FeA1 termination and an intermediate case between the former two, which were obtained as a function of the annealing temperature.39 Again, most of the computational studies have been directed towards the dipolar bulk-like terminations of the (111) surface. Martin et al.40 used spin-polarized DFT calculations to study the dipolar non-stoichiometric bulk-like FeB1 and FeA1 terminations of the (111) surface and they found FeB1 to be the more stable of the two, which they validated through comparison with experimental STM images.40 Berdunov et al.41 used DFT to study the dipolar non-stoichiometric bulk-like O2 termination of the (111) surface which was also validated via comparison with experimental STM images.41 Kiejna et al.42 studied the non-stoichiometric bulk-like dipolar terminations of the (111) surface using DFT + U, and although they did not calculate the stoichiometric slab, they predicted the FeA1 termination as the most stable for the whole range of chemical potential they considered.42 Reduced surfaces of the Fe3O4(111) surface show superstructures with Fe1−xO(111) islands,43 which makes the surface even richer in possible terminations.
Following the seminal work by Tasker44 on the surface properties of ionic solids, in the present work we have used DFT + U to investigate the non-dipolar stoichiometric terminations of the low Miller index surfaces of Fe3O4, in order to complement previous experimental and computational studies. We report the equilibrium morphology of the crystals enclosed by stoichiometric non-dipolar surfaces and the factors that govern the redox properties of the most common surfaces, (001) and (111), which are also the most prominent surfaces of Fe3O4 moieties.45,46 We have also calculated the STM images of the different stoichiometric non-dipolar terminations of these surfaces to determine the most likely to appear in nanocrystals through comparisons with available experimental STM data. We have studied the redox processes by the systematic formation of single O vacancies and the adsorption of single O on the surface, as opposed to previous computational studies which have focused on bulk-like terminations and their modifications. This approach allows us to explore how these redox processes determine the surface properties by finely tuning the conditions of temperature and oxygen partial pressure on the stoichiometric non-dipolar surfaces.
2 Computational methods
2.1 Calculation details
We have used the Vienna Ab initio Simulation Package (VASP)47–50 to carry out quantum mechanical calculations within the usual Kohn–Sham (KS) implementation of DFT. The Perdew–Burke–Ernzerhof (PBE)51,52 version of the generalized gradient approximation (GGA) was employed as the exchange–correlation potential, together with the semiempirical method of Grimme53 to model the long-range dispersion interactions. The core electrons, up to and including the 3p levels of Fe and the 1s of O, were frozen and their interaction with the valence electrons was described by the projector augmented wave (PAW) method.54,55 KS valence states were expanded in a plane-wave basis set with a cutoff of 400 eV for the kinetic energy. An energy threshold-defining self-consistency of the electron density was set to 10−5 eV and the optimization of the structures was conducted via a conjugate gradients technique, which stops when the Hellmann–Feynman forces on all atoms are less than 0.01 eV Å−1.All calculations were spin-polarised, but spin–orbit coupling was not considered. Within the VASP code, it is possible to assign an initial spin population and orientation to each atom of the system, to converge to a particular spin configuration. Thus, the initial magnetic moments were set following a high-spin ferrimagnetic structure, i.e. with opposite spins in the tetrahedral and octahedral sites, in agreement with experiment.56,57 In order to describe the electronic and magnetic behaviour properly, an accurate treatment of the electron correlation in the localized d-Fe orbitals is crucial. Hence, we have used the Dudarev et al.58 approach within the DFT + U59 for improving the description of these localized states. This is a correction typically used where standard LDA and GGA functionals fail to describe the electronic structure properly.60 The value for the on-site Coulomb interaction term in this study was Ueff = 3.7 eV, which was obtained following the procedure described in ref. 15 but adjusted to a different DFT functional. The limitation of this approximation is the difficulty in choosing an adequate value for the Ueff parameter, which is usually property dependent.61–63 An alternative computational approach is the use of hybrid functionals, although in that case the calculated properties also depend on the fraction of the exact Hartree–Fock exchange that is added to the DFT exchange term,60,64–67 and the calculations are too computationally expensive to be applied to the large surface models employed in this study.
Bulk calculations were performed using the rhombohedral primitive unit cell containing 14 atoms (Fe6O8). Integrations in the reciprocal space were performed using a Monkhorst–Pack grid of 7 × 7 × 7 Γ-centred k-points,68–70 which ensured electronic and ionic convergence. Test calculations with a higher number of k-points led to an energy difference smaller than 1 meV per cell. k-Point grids for the surface calculations were chosen in a such a way that a similar spacing of points in the reciprocal space was maintained.
Within this setup, we calculated a lattice constant for the bulk Fe3O4 unit cell of a = 8.398 Å, in excellent agreement with the experimental value of 8.390 Å,17 and an equilibrium volume of ![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) = 74.043 Å3 per formula unit. The calculated total spin magnetization per formula unit, MS = 4.00 μB lies very close to the 4.05 μB, measured experimentally at 4.2 K,71 and the atomic spin densities, ms(FeA) = 4.032 μB, ms(FeB) = 3.906 μB and ms(O) = 0.055 μB have the ferrimagnetic character observed before,15,57 following very closely the Néel model,56 where the electronic configurations are
= 74.043 Å3 per formula unit. The calculated total spin magnetization per formula unit, MS = 4.00 μB lies very close to the 4.05 μB, measured experimentally at 4.2 K,71 and the atomic spin densities, ms(FeA) = 4.032 μB, ms(FeB) = 3.906 μB and ms(O) = 0.055 μB have the ferrimagnetic character observed before,15,57 following very closely the Néel model,56 where the electronic configurations are 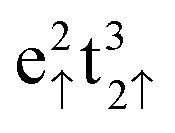 for FeA and
for FeA and 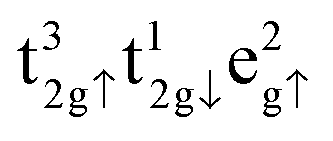 as well as
as well as 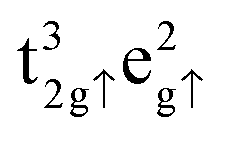 for FeB. Calculated charges for FeA, FeB and O atoms are 1.589, 1.521 and −1.158 e− respectively. For further information, the density of states of the Fe3O4 bulk phase are provided in Fig. S1 of the ESI.†
for FeB. Calculated charges for FeA, FeB and O atoms are 1.589, 1.521 and −1.158 e− respectively. For further information, the density of states of the Fe3O4 bulk phase are provided in Fig. S1 of the ESI.†
2.2 Surface models
In order to build slab models of the Fe3O4 surfaces, two models are used in the literature to explain the reconstructions found in polar surface terminations: the electron-counting72 and the dipole method.44 Both models are based on the condition that the net surface charge or dipole perpendicular to the surface, respectively, must be zero.A surface structure satisfies the model of electron-counting (i.e., it is charge- or auto-compensated) if all the partially filled dangling bonds in the cations are empty and the partially filled dangling bonds in the anions are full. It assumes that the atomic orbitals are in the conduction or valence band respectively. To achieve this, the model postulates that a stable surface structure will be the one that (after reconstruction) is able to accommodate exactly all the electrons of the partially filled orbitals of the cations (in the conduction band) into the partially filled orbitals of the anions (valence band). However, the disadvantage of this approach is that this condition directly links to the conducting properties of the material under investigation. If the surface satisfies this model, it will be a semi-conductor; otherwise the remnant electrons will lead to a metallic surface.
The dipole method for reconstructing dipolar surfaces is a more robust option, at least with half-metallic materials like Fe3O4,15 because it is not connected to the conducting properties of the structure. This method, pioneered by Tasker,44 considers the crystal as a stack of planes, where three possibilities can arise.44 In type 1, each plane has overall zero charge because it is composed of anions and cations in stoichiometric ratio which makes it non-dipolar, whereas in type 2 the stacking of three symmetrically charged planes cancels out the dipole moment perpendicular to the surface. In type 1 and 2, no reconstruction of the surface is needed because the repeat unit is non-dipolar perpendicular to the surface. However, in type 3 surfaces, alternating charged planes stack in a sequence that produces a dipole moment perpendicular to the surface, but the surface can be reconstructed through moving half of the ions with the same charge from the top to the bottom of the slab. This method also guarantees that the surface does not generate an electrical field within the crystal and therefore the potential felt at each ion site reaches the constant bulk value, a condition that is not necessarily satisfied by the electron-counting model.
All the surfaces in this study were created by cutting the geometry optimised bulk using the dipole method implemented in METADISE.73 The resulting slabs are represented by keeping fixed the bottom atoms at their ab initio relaxed bulk positions to simulate the bulk phase of Fe3O4 and by relaxing the rest of the slab during the optimization, giving a single relaxed surface. The slabs comprise 56 atoms (8 formula units), with 24 Fe and 32 O atoms. The Fe3O4(001), (011) and (111) surfaces were modelled with slabs having surface areas of 70.5, 99.7 and 61.1 Å2, respectively, and they were constructed of 9, 5 and 13 atomic layers, respectively. Fig. 2(c) provides a representation of their stacking sequence in each direction. For the (001) and (111) surfaces, the simulation slabs were symmetrical along the z axis. Top species in the (001) surface were (0.5 ML) FeA atom and 2 FeB and 4 O atoms (equivalent to 0.5 ML for each of the ions) for terminations A and B respectively, see Fig. 2. For the (111) surface, terminations A and B were terminated by half of the (FeB)6 and (FeB)2 bulk layers respectively. However, the simulation slabs for the (011) surface were asymmetrical along the z axis, with complementary top and bottom layers. Top layer of termination A was a single (0.25 ML) FeA atom above a bulk-like O–FeB layer, while its bottom layer was a bulk-like FeA–FeB–O layer with one (0.25 ML) FeA vacancy. For termination B, top and bottom layers were the other way round.
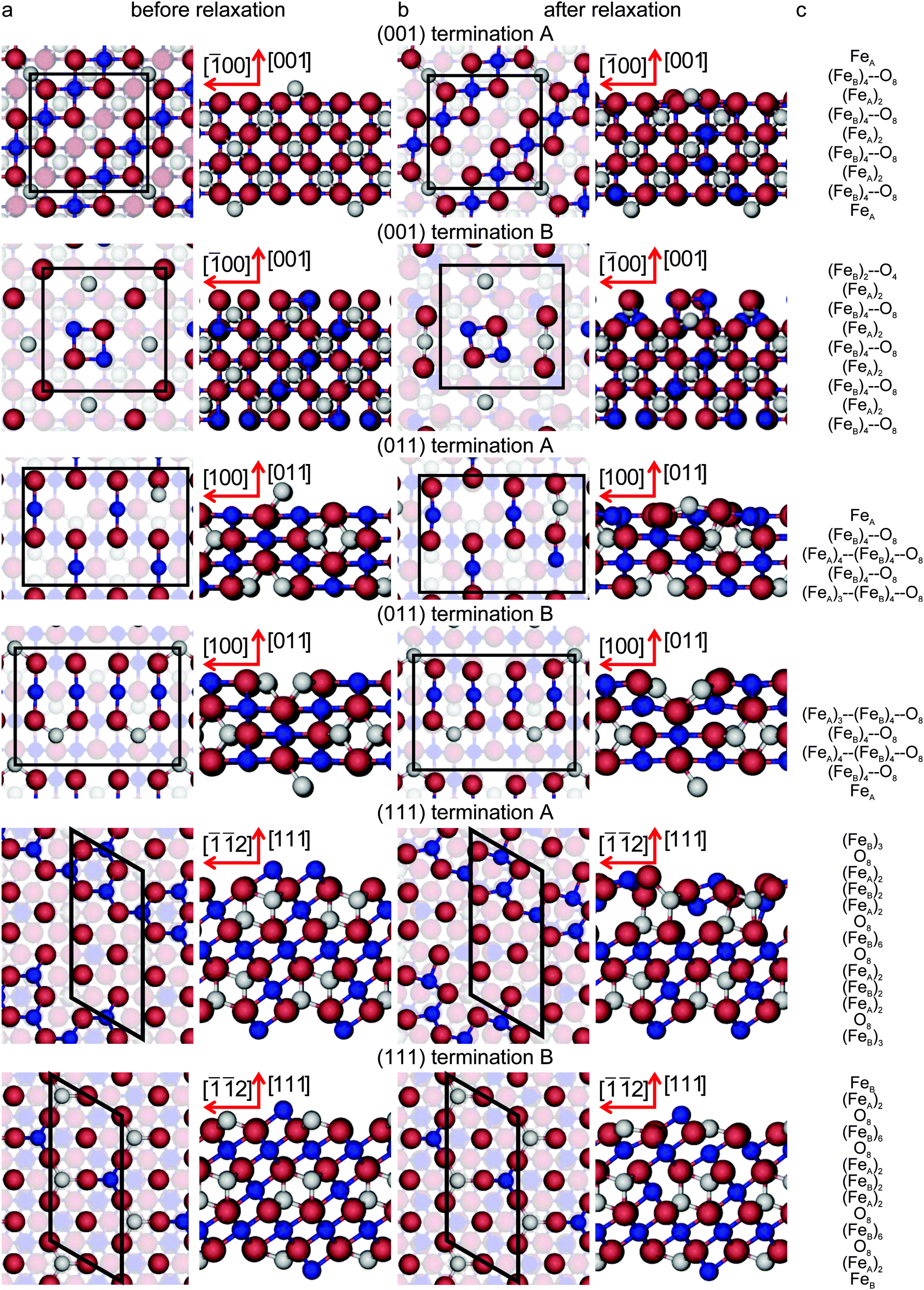 | ||
| Fig. 2 Top and side view of the simulation slabs for the low-index surfaces of Fe3O4. The surfaces are shown (column a) before, (column b) after relaxation and (column c) their stacking sequence. For the colour code see Fig. 1. Layers with atoms with dangling bonds are highlighted. Crystallographic directions for the top view of (001) surface terminations is [100] for the abscissae towards the right; for the (011) surface terminations it is [01] for the abscissae towards the right; and for the (111) surface terminations it is [01] for the longest axis towards the top. | ||
In every simulation cell, a vacuum region of 12 Å was added perpendicular to the surface to avoid interaction between the periodic slabs. Different slab and vacuum thicknesses as well as numbers of relaxed layers were tested until convergence within 1 meV per cell was achieved. Since we are going to remove and add O atoms to the surfaces at one side of the slab only, we applied dipole corrections perpendicular to all surfaces in the calculations to enhance the electronic convergence.74,75 We have used Bader analysis76 in the implementation of Henkelman and co-workers77–79 to analyse the charge transfer around the defects introduced in the stoichiometric surfaces. We have chosen this methodology for partitioning atomic charges, as it is based upon the charge density, which is insensitive to the metal oxidation state and the basis set used, unlike wavefunction-based population schemes.80–82 The DOS of the optimised surfaces is presented in Fig. S1 of the ESI.†
2.3 Calculation of surface energies
We have carried out energy minimisations of the (001), (011) and (111) slabs to obtain their surface energies. We derived the surface energy of the unrelaxed surface (γu) from a single point calculation of the pristine symmetric stoichiometric slab before relaxation, via equation: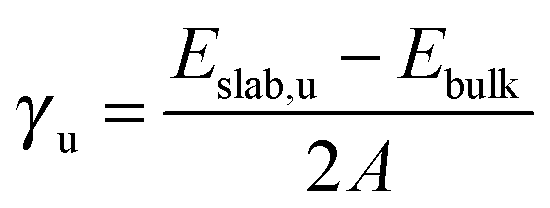 | (1) |
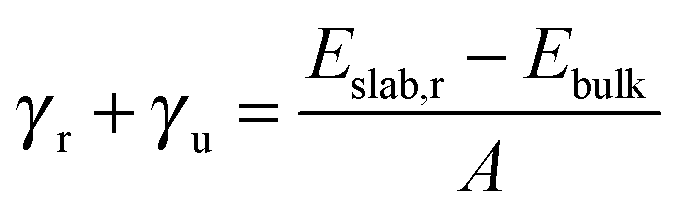 | (2) |
We have also calculated the degree of relaxation of each surface as a percentage (for (011) γr ∼ Eclev/2):
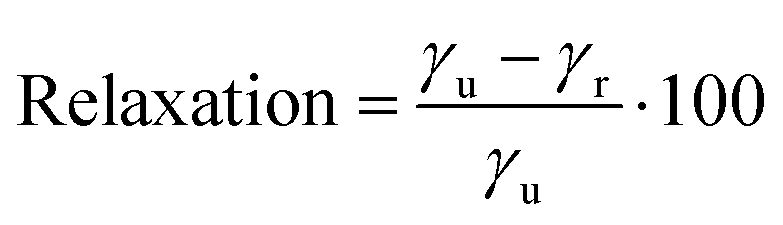 | (3) |
The equilibrium morphology of a Fe3O4 crystal is determined by the surface free energies and the related growth rates of the various surfaces, which provides a measure of the relative stabilities of the surfaces. The morphology is constructed according to Wulff's theorem,83 where the distance from the centre of the particle to the surface is proportional to the surface energy. It is based on the Gibbs approach,84 who proposed that under thermodynamic control the equilibrium form of a crystal should possess minimal total surface free energy for a given volume. Previous studies have shown85,86 that using surface energies to calculate crystal morphologies provides good agreement with experiment as the difference in entropy between bulk and surface is small.
2.4 Redox processes of the (001) and (111) surfaces
We have also examined the redox properties of the most common Fe3O4 surfaces, the (001) and (111), by removing or adding O atoms to form non-stoichiometric compositions of the top atomic layer. We have based the discussion of the stabilities of the non-stoichiometric terminations on the ab initio thermodynamics formalism87 where the surface free energy (σ) is calculated according to the equation: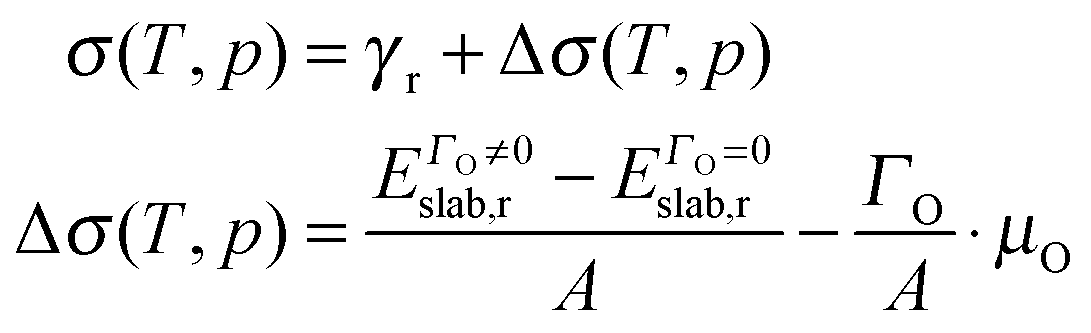 | (4) |
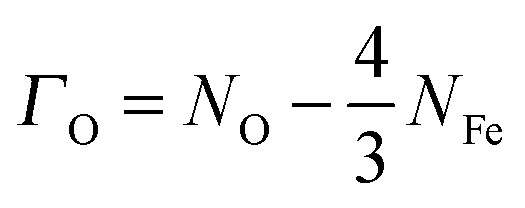 | (5) |
It is possible to express the chemical potential of molecular O2 (μO) in the gas phase as:
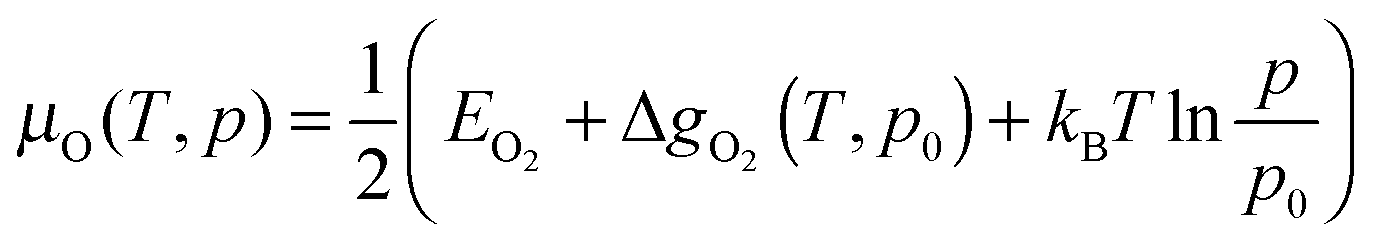 | (6) |
Here the first term within the bracket is the DFT energy of the O2 molecule. The second term is the difference in the Gibbs free energy per O2 molecule at p0 = 1 bar between 0 K and T, which in this study has been extracted from thermodynamic tables88 to avoid its calculation in the gas phase.89,90 The last term represents the change in free energy of the O2 gas (assuming ideal gas behaviour) at constant temperature (T) when its partial pressure changes from p0 to p.
We express μO with respect to half the energy of the O2 molecule. The above convention makes μO a function of only experimental quantities. For consistency in the evaluation of the slab energies, we must subtract half of the energy of the O2 molecule for each O atom in the slab. Expressing μO as described, it is possible to plot the surface free energies given by eqn (4) for different surface compositions as a function of μO, and discuss the redox behaviour of the surface.89,90
Finally, for the calculation of the energy required to create an O atom vacancy or to add the atom on the surfaces, we need the energy of the O2 molecule. However, it is known that GGA calculations fail in the description of the binding energy for this particular molecule, as is shown in the (over)binding of the O2 molecule.52
According to our calculations, the O2 triplet ground state has an equilibrium bond length of 1.23 Å and a binding energy of −6.08 eV (with respect to triplet oxygen atoms), comparing well with previous computational studies.62,91,92 However, this value lies 0.91 eV below the experimental binding energy (−5.17 eV).93 Therefore, we have considered that half of the over-binding of the O2 molecule, 0.46 eV, will be added to correct the adsorption or vacancy formation energies with respect to one O atom. The redox processes in the following sections are all reported with respect to the corrected value. A comparative analysis with uncorrected energy values is provided in the ESI.†
2.5 Calculation of scanning tunnelling microscopy (STM) images
The STM images were simulated according to the basic formulation of the Tersoff–Hamann approach94 where the STM tip was approximated to an infinitely small point source. The tunnelling current between the surface and the tip in the STM experiments is proportional to the local density of the states (LDOS) integrated between the Fermi energy and the sample bias. We have used the program HIVE95 for the production of our STM topographic images, where the DFT-based partial charge density was integrated from −2.5 eV to the Fermi energy. In the constant current mode, the tip of the STM is moved across the surface where its height varies to keep the charge density at a constant value, which is given by a constant LDOS. We map the simulated STM images by means of the heights as a function of the position of the tip over the surface. More details about the method can be found elsewhere.963 Results and discussion
3.1 Stoichiometric surfaces
We have modelled different terminations of the three lowest Miller index surfaces of Fe3O4, shown in Fig. 2, whereas Table 1 summarises their surface energies before and after energy minimization. Before relaxation, the order of increasing surface energies, and therefore decreasing stability, is (001) < (111) < (011), which remains the same after relaxation. Note that this order was established by taking into account only the most stable termination (with lowest γ) per surface, as these terminations would be the most likely to appear for each plane.| Surface | γ u (J m−2) | γ r (J m−2) | Relaxation (%) |
|---|---|---|---|
| a Note that for the (011) surface it is only possible to report the average surface energy, as termination A and B are complementary. | |||
| (001) Termination A | 1.45 | 0.96 | 34.2 |
| (001) Termination B | 3.28 | 2.17 | 33.9 |
| (011) Terminations A and Ba | 2.13 | 1.37 | 35.5 |
| (111) Termination A | 2.75 | 1.09 | 60.3 |
| (111) Termination B | 1.58 | 1.10 | 30.4 |
Before geometry optimisation, termination A of the (001) slab was terminated by 0.5 ML of 2-coordinated FeA ions occupying a bridge site (above two O ions) with a 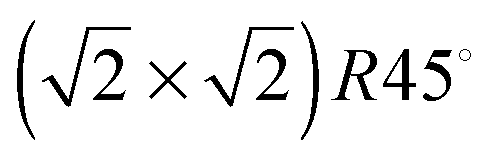 symmetry according to Wood's notation,97 which is a vectorial description of the surface structure. Beneath the surface, the slab shows a bulk structure consisting of single rows in the [110] direction of 5-coordinated FeB ions alternating every two single rows of O ions with cubic packing, see Fig. 2. During energy minimization, the protruding FeA ions move 0.53 Å towards the bulk, i.e. they experienced ∼50% inward relaxation based on ∼1.05 Å as the layer interspacing, thereby becoming closer to the nearest two O (0.25 ML of the 2nd layer), which displace 0.35 Å to the surface to accommodate this relaxation, see Table 2. The relaxation pattern of the top atomic layer of the surface slab agrees semi-quantitatively with the ∼40% inward relaxation reported for the topmost layer of this termination based on LEIS analysis,25 which is generally regarded to fit better than the more complex relaxation pattern reported before for this surface termination by Chambers et al.24 Previous studies, purely theoretical98 or combined with experiments,99 have concluded that the Fe3O4(001) surface terminates with Fe ion dimers with
symmetry according to Wood's notation,97 which is a vectorial description of the surface structure. Beneath the surface, the slab shows a bulk structure consisting of single rows in the [110] direction of 5-coordinated FeB ions alternating every two single rows of O ions with cubic packing, see Fig. 2. During energy minimization, the protruding FeA ions move 0.53 Å towards the bulk, i.e. they experienced ∼50% inward relaxation based on ∼1.05 Å as the layer interspacing, thereby becoming closer to the nearest two O (0.25 ML of the 2nd layer), which displace 0.35 Å to the surface to accommodate this relaxation, see Table 2. The relaxation pattern of the top atomic layer of the surface slab agrees semi-quantitatively with the ∼40% inward relaxation reported for the topmost layer of this termination based on LEIS analysis,25 which is generally regarded to fit better than the more complex relaxation pattern reported before for this surface termination by Chambers et al.24 Previous studies, purely theoretical98 or combined with experiments,99 have concluded that the Fe3O4(001) surface terminates with Fe ion dimers with 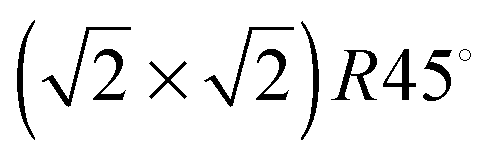 symmetry. The second Fe may migrate from a sub-surface layer98 or from a dipolar bulk-like FeA terminated (001) surface.99 However, we have not included dimers here as this lies outside the scope of the present study. The surface energy of termination B of the (001) surface is also reported in Table 1, but we do not consider this plane for further analysis because of its high surface energy, which makes it very unlikely to appear in the Fe3O4 crystal morphology.
symmetry. The second Fe may migrate from a sub-surface layer98 or from a dipolar bulk-like FeA terminated (001) surface.99 However, we have not included dimers here as this lies outside the scope of the present study. The surface energy of termination B of the (001) surface is also reported in Table 1, but we do not consider this plane for further analysis because of its high surface energy, which makes it very unlikely to appear in the Fe3O4 crystal morphology.
| (001) Termination A | (011) Termination A | (011) Termination B | (111) Termination A | (111) Termination B | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Layer | Species | Δdz | Layer | Species | Δdz | Layer | Species | Δdz | Layer | Species | Δdz | Layer | Species | Δdz | ||
| 1st | FeA | −0.53 | 1st | FeA | −0.98 | 1st | FeA | −0.27 | 1st | FeB | −0.59 | 1st | FeB | −0.09 | ||
| 2nd | O | 0.50 ML | 0.02 | 2nd | O | −0.02 | FeB | −0.11 | 2nd | O | 0.75 ML | −0.10 | 2nd | FeA | −0.31 | |
| FeB | −0.05 | FeB | −0.11 | O | 0.08 | 0.25 ML | 0.62 | 3rd | O | −0.03 | ||||||
| O | 0.25 ML | 0.35 | O | −0.03 | 2nd | O | −0.06 | 3rd | FeA | 0.09 | 4th | FeB | 0.06 | |||
| 0.25 ML | −0.08 | 3rd | FeA | −0.04 | FeB | 0.23 | 4th | FeB | 0.41 | 5th | O | 0.03 | ||||
| 3rd | FeA | 0.11 | FeB | 0.06 | O | −0.04 | 5th | FeA | −0.21 | 6th | FeA | 0.02 | ||||
| O | −0.03 | O | 0.01 | 3rd | FeA | 0.02 | 6th | O | −0.08 | 7th | FeB | 0.03 | ||||
| 4th | FeB | 0.03 | Bulk | FeB | 0.00 | 7th | FeB | 0.00 | Bulk | |||||||
| O | 0.05 | O | 0.00 | Bulk | ||||||||||||
| 5th | FeA | −0.01 | Bulk | |||||||||||||
| Bulk | ||||||||||||||||
The stacking sequence of the Fe3O4(011) surface is shown in Fig. 2 and the vertical shifts of the ions towards the vacuum after energy minimization are listed in Table 2. One of the two lowest energy surface terminations, termination A, terminates with 0.25 ML of mono-coordinated FeA at the surface followed by a bulk-like structure consisting of single rows of 4-coordinated FeB ions shifted 25% in the [01] direction and alternating with single rows of O ions with cubic packing. During energy minimisation, the protruding FeA ions move 0.98 Å towards the bulk, thereby compressing the surface layer atoms underneath which move horizontally to accommodate this relaxation. Termination B has essentially the same relaxed surface energy as termination A but it differs in its structure. It is terminated with a bulk-like structure consisting of a single row of 4-coordinated FeB ions between two rows of O ions. The latter O atoms are in cubic packing and alternate with double rows of 3-coordinated FeA ions in rhombohedral packing along the [01] direction. The double row of FeA ions is partially vacant by 0.25 ML with p(1 × 2) symmetry. During energy minimization, the top FeA and FeB ions shift towards the bulk by 0.27 Å and 0.11 Å respectively which generates a 0.23 Å movement towards the surface of the FeB ions in the sub-surface layer. Based on the similarity between the relaxed structure of termination B, differing only by 0.25 ML FeA vacancy from the bulk-like FeA–FeB–O termination proposed in ref. 34, we can still compare some structural characteristics between them. The calculated FeB–FeB or O–O distance of the atoms lying in the same row along the [01] direction is 2.77 Å in termination B, which agrees well with their reported 3.0 ± 0.3 Å.34 Moreover, along the [001] direction, the calculated FeB–O distance of 1.92 Å also compares well with 2.1 ± 0.3 Å from STM experiments.34
Finally, the bottom two panels of Fig. 2 represent the stacking sequence of the Fe3O4(111) surface terminations, while the vertical displacement of the ions in the surface regions during the optimisation are listed in Table 2. One of the two lowest energy terminations, termination A, contains 0.5 ML of 3-coordinated FeB ions with c(2 × 4) symmetry, occupying hexagonal close packed (hcp) hollow positions in the top layer. The next layer has a bulk-like structure consisting of rows of O ions along the [01] direction with rhombohedral packing. The percentage relaxation experienced by this surface termination is the largest of this study. During its geometry optimisation, the top FeB ions move towards the bulk by 0.59 Å, causing 0.25 ML of the O in the layer underneath to move towards the surface by 0.62 Å. As we can see in Table 2, the fourth and fifth atomic layers are also affected by the surface relaxation. Termination B terminates with 0.5 ML of 3-coordinated FeB with p(1 × 2) symmetry, where these ions occupy hcp hollow sites, followed by a bulk-like structure consisting of rows of FeA alternating along the [01] direction with two rows of O ions with rhombohedral packing. During energy minimization, the top FeB and FeA ions move towards the bulk by 0.09 Å and 0.31 Å respectively. The mean FeA–FeB distance in the surface layer of the relaxed structure is 3.55 Å (as opposed to the calculated bulk value of 3.48 Å), which is in excellent agreement with 3.6 ± 0.4 Å, the experimental distance reported between the two features (FeA and FeB) from an STM image.37
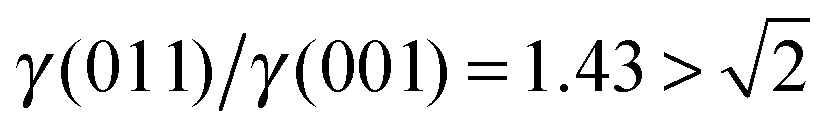 , see Fig. 3(b), and, as shown in Fig. 3(d), the (011) surface would only become present in the crystal morphology if
, see Fig. 3(b), and, as shown in Fig. 3(d), the (011) surface would only become present in the crystal morphology if 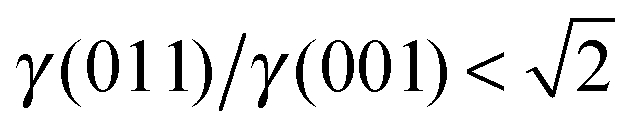 .
.
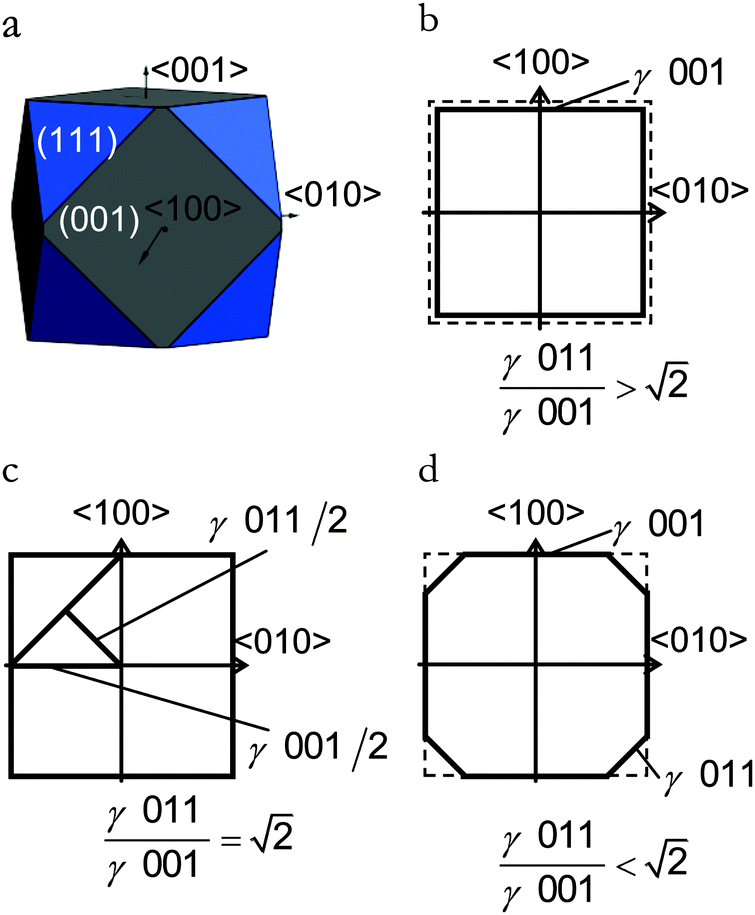 | ||
| Fig. 3 (a) Equilibrium morphology for a Fe3O4 crystal derived from a Wulff construction. (b–d) Schemes of the crystal cross-sectional planes along the 〈100〉 and 〈010〉 axes for different ratios of stabilities of the lateral surfaces, which illustrate why the (011) surface is absent in the equilibrium morphology. | ||
There are many ways to modify the shape of nanoparticles, such as solvent, media, capping agents, temperature or viscosity, but the Wulff morphology shown in Fig. 3(a) expresses a particle produced in conditions of perfect thermodynamic equilibrium, vacuum and at 0 K. Nevertheless, our results compare well with the morphologies of crystals synthesised by Zhao et al.,22 who described the formation of Fe3O4 under different pH conditions. They increased the OH− concentration and the resulting crystal shapes changed from cubic (or spherical – depending on other conditions) at low pH via truncated octahedral to octahedral at high pH values. All their crystals showed mainly the (001) and (111) surfaces but, in some cases, a little (011) surface was expressed due to certain conditions which may modify the surfaces' relative energies. The occasional appearance of the (011) surface is rationalised in terms of kinetically-controlled anisotropic growth of the Fe3O4 nanoparticles. Zhao et al.,22 suggested that a high concentration of KOH in the solution can lead to selective adsorption of the hydroxyl anions to certain planes of the crystal, which slows down considerably their growth process. Therefore, the presence of these ions can affect the relative stabilities of the different crystal surfaces. The inversion of the nature of the inequality 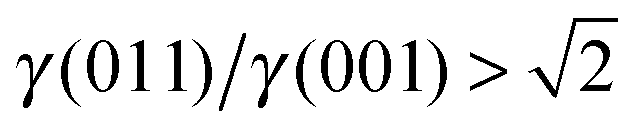 , which already lies close to
, which already lies close to 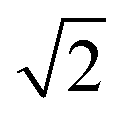 , will cause the (011) plane to show up in the morphology.
, will cause the (011) plane to show up in the morphology.
The STM images in Fig. 4 are calculated on pristine Fe3O4(001) and (111) surfaces. Fig. 4(a) shows the STM image of the Fe3O4(001) surface, termination A, acquired at a distance (d) of 1.90 Å to the tip and at a density (ϱ) of 0.0059 e Å−3. This image resolves the protruding 2-coordinted FeA as the brightest spots with 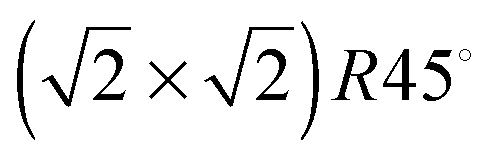 symmetry. The O ions from the layer below are also clearly well-defined circles forming rows along the [110] direction and with cubic packing. The STM image of termination A of the (001) surface does not show the atomic positions of the FeB placed in the same layer as the O ions due to their low partial charges at this bias. We observed the reproduction of the FeA ions in the same symmetry in the STM image obtained from annealed Fe3O4 at 623 K.28
symmetry. The O ions from the layer below are also clearly well-defined circles forming rows along the [110] direction and with cubic packing. The STM image of termination A of the (001) surface does not show the atomic positions of the FeB placed in the same layer as the O ions due to their low partial charges at this bias. We observed the reproduction of the FeA ions in the same symmetry in the STM image obtained from annealed Fe3O4 at 623 K.28
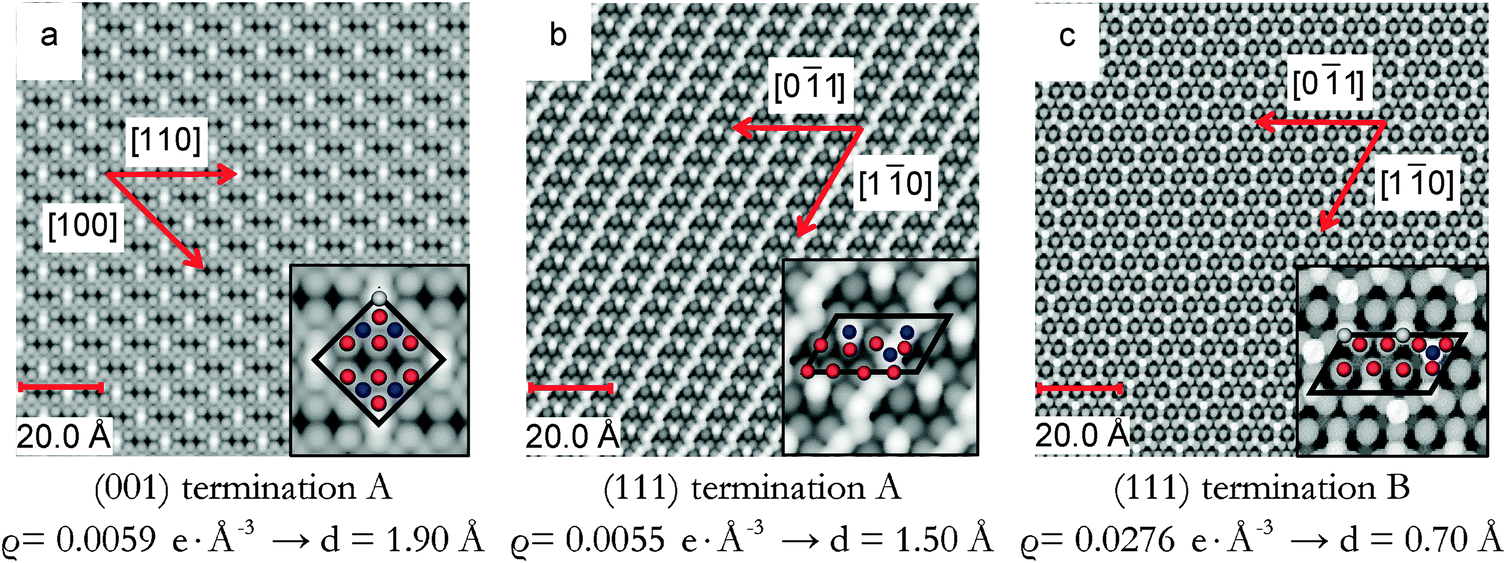 | ||
| Fig. 4 Simulated STM images of (a) termination A of (001), (b) termination A of (111) and (c) termination B of (111) surfaces obtained using a bias of −2.5 eV. Density (ϱ) and tip distance (d) are also indicated. Insets show enlargements of the STM images. In the inset, FeA ions are in grey, FeB ions are in blue and O ions are in red. | ||
The STM image of the Fe3O4(111) surface termination A is shown in Fig. 4(b) acquired at a density of 0.0055 e Å−3 and a distance of 1.50 Å to the tip. The image resolves the protruding FeB as the brightest spots along rows in the [10] direction and as dots in between these rows. The undulation of the rows is due to the 0.25 ML of O atoms in the 2nd layer that have moved towards the surface after minimization. From the modelled STM, we can even observe the rhombohedral packing of the sub-layer O ions.
The last STM image in Fig. 4(c) corresponds to termination B of the Fe3O4(111) surface obtained with a density of 0.0276 e Å−3 and the tip at 0.70 Å from the highest atom. The image acquired resolves the protruding FeB as the brightest spots in the STM image with a p(1 × 2) symmetry and the FeA ions from the layer below which are always bonded to three O atoms immediately underneath. This atomic arrangement forms a pattern of incomplete hexagons (with Fe atom vacancies in one vertex of the imaginary hexagon) which can be seen as a thermally equilibrated structure with vacancies evenly distributed. Details of the layers further below are also visible in our STM image. Experimental studies of the Fe3O4(111) surface37 have shown that among the two different terminations considered there, the one with 0.50 ML of Fe atoms is more stable than the one with 0.75 ML of Fe atoms and 0.25 ML of O atoms, agreeing well with our model of termination B of the (111) surface, whose simulated STM is shown in Fig. 4(c). The calculated vertical distance between the FeA in the vertex of the hexagon and the O ion in its centre is 0.50 Å, which also agrees well with the value reported experimentally, 0.5 Å.37 This experimental termination shows regions with full hexagons and others with incomplete hexagons (due to Fe vacancies). This atomic rearrangement may be a consequence of the high temperatures to which the surface was exposed.
3.2 Redox behaviour
We have studied the redox properties of the most stable terminations, A and B, of the Fe3O4(001) and (111) surfaces, respectively, by comparing the surface free energies corresponding to different O to Fe ratios at the surface. We maintained the number of Fe atoms in the slab as in the stoichiometric surface, but we modified the number of O atoms in the top layer by Γ (given by eqn (5)), as we were interested in studying the effect of different temperature and oxygen pressure on the stoichiometric non-dipolar surfaces. Because of the size of our supercells, and assuming that O atoms occupy bulk-like positions around the surface Fe atoms, 17 values of Γ are possible if we constrain the calculations to a maximum of one ML of adatoms or vacancies. However, due to the complexity of the (001) and (111) surfaces and in order to reduce the number of Γ to small values that reflect realistic μO, we have used only five values of Γ: Γ = 0 is the stoichiometric surface, Γ = +1, +2 are the partially oxidized surfaces, and Γ = −1, −2 are the partially reduced surfaces. We have represented all of them schematically in Fig. 5. | ||
| Fig. 5 Top view of the schematic representation of the Fe3O4 before and after relaxation; (column a) contains (001) and (column b) contains (111) surfaces, both with different Γ. Stoichiometric (Γ = 0); partially reduced (Γ = −1, −2) and partially oxidized (Γ = +1, +2). FeA ions are in grey, FeB ions are in blue and O ions are in red, removed O ions are in pale red and added O atoms are in dark red. Only the closest defects are highlighted indicating their relative position, while all of them are shown. The arrows in (a) and (b) indicate the [110] and [01] directions respectively. Black lines indicate the surface unit cell. | ||
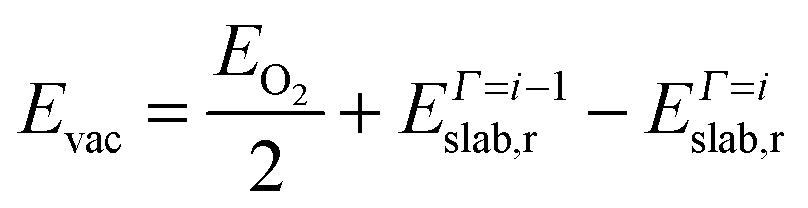 | (7) |
We proceed with the second reduction of Fe3O4(001) leading to Γ = −2. We removed an O located in the pristine row along the [110] direction, see Fig. 5(a) for Γ = −2, which is at intermediate distance to FeA. This second vacancy is 3.23 eV less favourable than the previous state but it is just more likely than removing a more distant O ion from the row where the vacancy is now being created, 3.31 eV. This indicates that although the first vacancy is created preferentially in a position far away from the 2-coordinated FeA, the second reduction might lead to a vacancy in the following O row at an intermediate distance from FeA. As the energies to create the second vacancy in the two positions already described are within the DFT error, it might also be possible to find vacancies in the O positions further away from the row where the vacancy is now being created.
We have characterised the Γ = −1 surface by means of a Bader analysis and compared the atomic charges with those on the pristine surface. The positive charge of the protruding FeA ion was increased by a negligible amount (<0.05 e−), where this small variation can be accounted for by the defect that was created at the farthest O location. The surface FeB ions, however, are reduced, especially the ones closest to the vacancy with a variation in charge of 0.25–0.37 e−. This can be interpreted in terms of the number of O ions directly coordinated to the FeB ions, see Fig. 5(a) for Γ = −1, where just over 80% of the electron density is transferred to the FeB ions after removing the O atoms.
Creating a second vacancy among the atoms coordinated to the 3-coordinated FeA, Fig. 5(b) for Γ = −2 costs 3.45 eV, which is less costly by 0.19 eV than removing the left O within the hexagon. These energies provide information about the consecutive reduction mechanism, where the first O vacancy is created in the centre of the incomplete Fe-hexagons and the next in one of the atom positions coordinated to the 3-coordinated FeA.
The Bader analysis indicates that upon vacancy formation on the Γ = −1 surface, charge transfer on the FeA is negligible and the protruding 3-coordinated FeB is only slightly reduced. However, the FeB ions whose charge is affected more are those in the 4th atomic layer (see Fig. 2) below the removed O atom to which they were previously directly coordinated. Altogether, the charge on those three FeB is reduced by ∼0.89 e−, i.e. they have accepted 78.5% of the electron density previously held by the removed O.
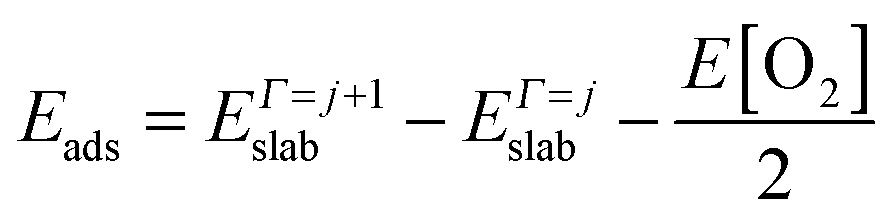 | (8) |
Adding a second O atom (Γ = +2) is also an exothermic process, releasing 0.96 eV per adatom. The second O atom preferentially coordinates the protruding FeA and a 5-coordinated FeB, forming another O bridging structure collinear with the [10] direction, Fig. 5(a) for Γ = +2. As for Γ = +1, the top atomic contraction leads to short Fe–O distances, 1.85 Å. Another conformation for the second O adsorption is coordinating equivalent atoms but forming a V-shaped structure, leading to a weaker adsorption (Eads = −0.80 eV).
At this point, it is worth mentioning that although we started from the ideal terminations similar to the bulk when we added the first and second oxygen atom, this did not prevent them to relax to a different position. In fact, we can see in Fig. 5(a) for Γ = +1 (and +2), that after surface relaxation, the added oxygen has moved from its bulk site to another position, closer to the protruding FeA. This finding agrees with the work of Reuter and Scheffler,90 who found for RuO2(110) that terminations at positions different from the bulk can be important in non-stoichiometric compositions.
The Bader analysis on the density of the (Γ = +1) oxidised (001) surface shows the oxidation of the top layer FeB by 0.60 e− while the protruding 2-coordinated FeA ion only donates 0.04 e− to the newly added O atom. Hence, the O adatom gains 1.00 e− mainly from the surface metals whereas the charge of the surface anions (all of them more negative than the adatom) change by about 0.02 e−.
The addition of a second O-adatom coordinating the protruding FeB and one of the other two closest unoccupied 3-coordinated FeA releases 2.30 eV; see the schematic representation in Fig. 5(b) for Γ = +2. During the optimisation, this second oxygen pushes the protruding 3-coordinated FeB out of its equilibrium position in the stoichiometric surface, thereby forming a FeA–O–FeB–O–FeA row of atoms along the [01] direction. The equilibrium bond lengths, FeA–O and FeB–O are 1.86 Å and 1.80 Å respectively, which compares well with values reported before (between 1.80–1.85 Å) for the Fe–O distance at the Fe3O4(111) surface.40 In the next most favourable conformation the second O is located atop one surface O coordinating only the FeA, but this process is endothermic by 0.43 eV. The calculations thus show that both the first and second adsorbed O preferentially coordinate the protruding 3-coordinated FeB and two of its FeA neighbours with a resulting bridging structure in the [01] direction.
Unlike the (001) surface, where the added oxygen atoms moved during the energy minimization, in the (111) we observed instead that the protruding FeB ion moved from its bulk position after the addition of two defects (Γ = +2), see Fig. 5(b). This validates in our methodology the possibility of exploring non-bulk-like relaxed positions for any atom in the surface of our slab, as long as all non-equivalent bulk-like positions for the defects are carefully investigated.
The Bader analysis indicates that in the preferred structure for Γ = +1, the adatom gains 1.04 e−, where the charge of the other surface O atoms decreased as little as in the (001) surface. Amongst the two Fe ions coordinated to the added O atom, FeB increases its charge by 0.08 e−, but FeA by 0.39 e−. The charge on other surface FeA and FeB ions upon addition of the O atom changed by an average of 0.03 e− and −0.01 e− respectively.
3.3 Temperature and pressure effects
In this section, we discuss the thermodynamics of the redox processes at the (001) and (111) surfaces as a function of temperature and O2 partial pressure in the gas phase. We express these macroscopic parameters by μO.In Fig. 6(a), we have plotted μO in terms of temperature and the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) p, along abscissas for easy comparison with the plots in Fig. 6(b) and (c). All the information used for the construction of Fig. 6(a) comes from experiment88 and is independent from the calculations (see Computational methods section). Variations in T and p are necessary to modify the value of μO as required and to reflect different reducing or oxidising conditions. For example, the oxygen chemical potential is −0.3 eV (which is a typical oxidising value) at ambient conditions, i.e. at the intercept of T = 298.15 K and p = 0.21 bar, while more reducing conditions (lower values of μO) can be achieved by increasing T while keeping the pressure constant (i.e. horizontal solid line in Fig. 6(a)).
p, along abscissas for easy comparison with the plots in Fig. 6(b) and (c). All the information used for the construction of Fig. 6(a) comes from experiment88 and is independent from the calculations (see Computational methods section). Variations in T and p are necessary to modify the value of μO as required and to reflect different reducing or oxidising conditions. For example, the oxygen chemical potential is −0.3 eV (which is a typical oxidising value) at ambient conditions, i.e. at the intercept of T = 298.15 K and p = 0.21 bar, while more reducing conditions (lower values of μO) can be achieved by increasing T while keeping the pressure constant (i.e. horizontal solid line in Fig. 6(a)).
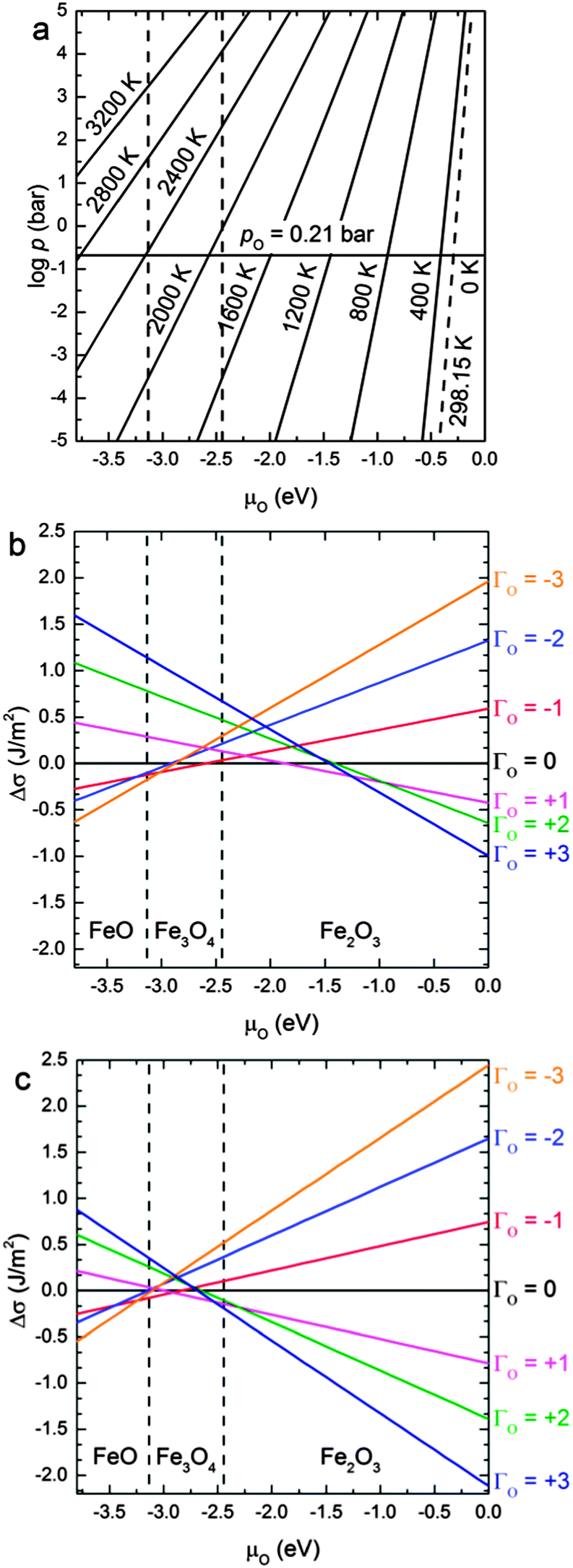 | ||
| Fig. 6 (a) μO in the gas phase as a function of the temperature and the logarithm of the oxygen partial pressure and relative surface free energies (Δσ) for the Fe3O4 (b) (001) and (c) (111) surfaces as a function of the oxygen chemical potential (μO). The areas corresponding to μO smaller than −3.13 eV, bigger than −2.44 eV and between these two values represent the approximate conditions under which bulk FeO, Fe2O3 and Fe3O4 respectively are the stable oxides. | ||
The area between the two vertical dashed lines (μO from −3.13 to −2.44 eV) in Fig. 6 corresponds to the conditions where the Fe3O4 bulk material is thermodynamically stable with respect to both FeO and Fe2O3 bulk. We have derived these conditions (μO) from the experimental formation enthalpy of the three oxides93 and their increasing oxidation from FeO to Fe2O3, see eqn (9). Under normal conditions, Fe2O3 is the thermodynamically stable bulk phase, while the synthesis of Fe3O4 requires high temperatures or a low pressure of O2 (which ultimately can lead to FeO).
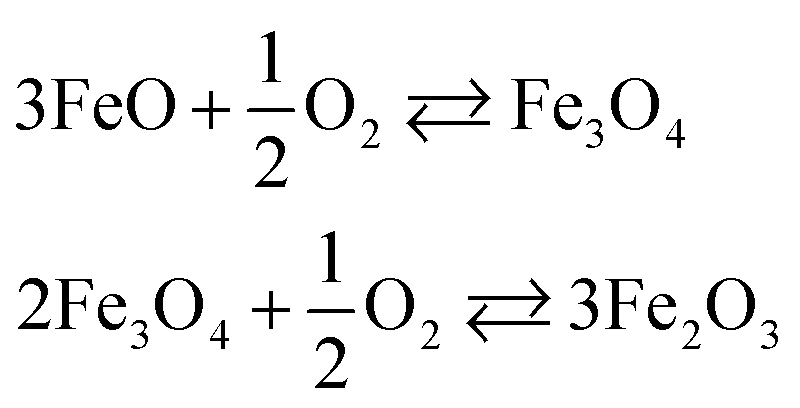 | (9) |
Fig. 6(b) and (c) show the variation of the surface free energies (Δσ) of each surface compositions versus μO. Note that we have only used the most stable configuration for Γ = −2, −1, 0, +1, +2. Further degrees of reduction/oxidation (Γ = ±3) could also be investigated but instead of exploring many different positions where to remove or add the O atoms, we have linearly fitted the intercept of the linear regressions in Fig. 6 as a function of Γ for inferring the intercepts of further oxidation/reduction of these surfaces. We have limited this treatment to three defects, as a higher number can lead to the formation of molecular oxygen in the case of oxidation or a new oxide phase in the case of reduction.
At μO = −0.3 eV (ambient conditions), the (Γ = +3) oxidation of the Fe3O4(001) surface will take place, see Fig. 6(b). For the conditions where bulk Fe3O4 is the most stable oxide, the (Γ = +3) oxidized (001) remains the most stable surface up to μO = −1.25 eV, from where the surface experiences a progressive reduction. In the early stages of this reduction, the unit cell loses two O atoms and remains so until μO = −1.85 eV. Beyond that chemical potential and until μO = −2.60 eV, which is just beyond the conditions in which the phase transition from Fe2O3 to Fe3O4 takes place, the most stable surface is the stoichiometric one. At lower values of μO = −3.00 eV (but still within the conditions in which Fe3O4 is the most stable phase), the (Γ = −3) reduced surface is the favoured system, until reaching the conditions where the reduced bulk phase of FeO is the most stable oxide. A recent publication by Nie et al.100 reports that the Fe3O4(001) surface is oxidised under exposure to 4 × 10−9 bar of oxygen at 923 K. They used low-energy electron microscopy (LEEM) and Raman spectroscopy to prove that Fe3O4 grows at the expenses of Fe ions migrating from the bulk towards the surface. The Fe ion vacancies in the bulk, in turn, transform it into α-Fe2O3 (hematite), which is the equilibrium iron oxide phase at the temperature and pressure of the experiment. Our results hence agree well with these experimental findings, although they are close to the limit in which the stoichiometric surface is the most stable one. The experimental conditions described above correspond to μO = −1.75 eV according to eqn (6), a value at which Fe3O4(001) surface is prone to suffer oxidation (see Fig. 6(b)), by adding one O atom per surface unit cell around the protruding FeA. Our results, however, agree partially with those reported in a DFT study by Pentcheva et al.29 as those authors found that the modified non-stoichiometric polar bulk-like Fe3O4(001) surface (FeB–O layer) is the most stable under any chemical potential. However, the surface proposed by Pentcheva et al. is a generic oxidised (001) surface, created from a bulk-like termination, whereas our surface is gradually oxidised or reduced. However, regardless of terminations and reconstructions, we also predict our non-dipolar surface to be (Γ = +3 and +1) oxidized up to μO = −1.85 eV, but from this value of chemical potential onwards, our results predict a gradual reduction, which no longer agrees with the work by Pentcheva et al. as they predict the same oxidized surface for any value of μO.
The redox behaviour of the Fe3O4(111) surface is shown in Fig. 6(c). It indicates that the redox properties of the (111) surface are similar to the (001) surface, although the oxidized character extends to lower chemical potentials. The surface tends to be (Γ = +3) oxidised under the condition where Fe2O3 is the most stable phase and up to μO = −2.45 eV, which is within the region where Fe3O4 is the thermodynamically most stable iron oxide. From here, the surface loses two O atoms for a very short range of chemical potential, until μO = −2.95 eV, from where the surface loses a further two more O atoms, becoming now reduced up to μO = −3.25 eV. From this final value of the chemical potential, the surface becomes reduced (Γ = −3). In a previous DFT + U study,42 Kiejna et al. studied the redox properties of the Fe3O4(111) surface. They only studied the non-stoichiometric dipolar bulk-like terminations and found that the FeA1 surface, which corresponds with a generic oxidized one, is the most stable one up to μO = −2.6 eV, from which point their surfaces started to reduce gradually. Although we cannot make a direct comparison of our results due to the different terminations considered in both works, owing to our gradual redox processes, our results show the same trend.
Comparing both (Γ = +3) oxidised surfaces, the (111) is lower in energy than the (001) for the whole μO scale considered. Therefore the (111) surface will remain oxidised even at μO where Fe3O4(001) is not. We suggest then, from Fig. 6, that under the conditions in which the bulk Fe3O4 is the most stable oxide, the phase transformation of the reduction of Fe3O4 towards FeO will start from the (001) surface. On the other hand, Fe3O4 oxidation to Fe2O3 would take place initially on the (111).
4 Conclusions
In this paper, we have modelled three different surface orientations of Fe3O4 crystals by using DFT methods within the GGA + U approximation. We have investigated the stabilities of their multiple (reconstructed) non-dipolar stoichiometric surface terminations and studied the redox properties of the most prominent surfaces. We have modified the redox conditions by creating O vacancies or adding O atoms to the most stable non-dipolar stoichiometric surface termination, under a wide range of chemical potentials, including ambient conditions and those conditions where bulk Fe3O4 is the thermodynamically most stable oxide. In the initial stages of oxidation, the excess O atoms form bridging structures with the Fe ions at the surface, and in particular the Fe ions protruding from the surface. We found that some oxidised (non-stoichiometric) structures relaxed in such a way that it broke the bulk-like termination.We conclude that the Fe-terminated (001) and (111) planes are the most stable Fe3O4 surfaces, in agreement with previous experiments as shown by STM images. The equilibrium morphology of Fe3O4 was found to be cubic with truncated corners, which means that (001) and (111) are the main surfaces exposed in the crystals. Although both (001) and (111) surfaces will be oxidized under ambient conditions, both surfaces suffer a gradual reduction, that starts at lower chemical potentials for the (001) surface including the stoichiometric plane.
The reduction of the (001) and (111) surfaces is thermodynamically favourable at the low end of the μO values in the region where Fe3O4 is the most stable oxide. We found that, in both cases, the O vacancies are likely to migrate towards the bulk, thereby changing the phase structure.
Acknowledgements
We would like to thank Thomas A. Mellan for useful discussions. We acknowledge the Engineering & Physical Sciences Research Council (grant EP/G036675) for funding. Via our membership of the UK's HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work made use of the facilities of HECToR, the UK's national high-performance computing service, which is provided by UoE HPCx Ltd at the University of Edinburgh, Cray Inc and NAG Ltd, and funded by the Office of Science and Technology through EPSRC's High End Computing Programme. The authors also acknowledge the use of the UCL Legion High Performance Computing Facility (Legion@UCL), and associated support services, in the completion of this work. DS-C thanks UCL for a Graduate Global Excellence Award and an Overseas Research Scholarship from the UCL Industrial Doctorate Centre in Molecular Modelling and Materials Science. A Roldan is grateful to the Ramsay Memorial Trust and University College London for the provision of a Ramsay Fellowship.References
- W. Weiss and W. Ranke, Prog. Surf. Sci., 2002, 70, 1–151 CrossRef CAS.
- S.-S. Chen and Updated by Staff, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2006 Search PubMed.
- D. H. James and W. M. Castor, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 529–544 Search PubMed.
- J. S. Campbell, P. Craven and P. W. Young, Catalyst Handbook, Wolfe Scientific Books, 1970, pp. 97–126 Search PubMed.
- D. G. Rethwisch, J. Phillips, Y. Chen, T. F. Hayden and J. A. Dumesic, J. Catal., 1985, 91, 167–180 CrossRef CAS.
- J. C. Gonzalez, M. G. Gonzalez, M. A. Laborde and N. Moreno, Appl. Catal., 1986, 20, 3–13 CrossRef CAS.
- S. Li, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2001, 105, 5743–5750 CrossRef CAS.
- G. C. Bond, Heterogeneous Catalysis; Principles and Applications, Clarendon Press, Oxford, 1974 Search PubMed.
- G. A. Somorjai and M. Salmeron, in Homogeneous and Heterogeneous Photocatalysis, ed. E. Pelizzetti and N. Serpone, D. Reidel Publ. Co., Doordrecht, The Netherlands, NATO ASI Series C, 1986, vol. 174, pp. 445–478 Search PubMed.
- S. Topham, in Catalysis: Science and Technology, ed. J. R. Anderson, Springer, Berlin, 1985, vol. 7, pp. 1–50 Search PubMed.
- G. W. Bridger and C. B. Snowden, Catalyst Handbook, Wolfe Scientific Books, 1970, pp. 126–147 Search PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2nd edn, 2003 Search PubMed.
- F. dos Santos Coelho, J. D. Ardisson, F. C. C. Moura, R. M. Lago, E. Murad and J. D. Fabris, Chemosphere, 2008, 71, 90–96 CrossRef CAS PubMed.
- Z. Zhang and S. Satpathy, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 13319–13331 CrossRef CAS.
- A. Roldan, D. Santos-Carballal and N. H. de Leeuw, J. Chem. Phys., 2013, 138, 204712 CrossRef CAS PubMed.
- R. J. Hill, J. R. Craig and G. V. Gibbs, Phys. Chem. Miner., 1979, 4, 317–339 CrossRef CAS.
- J. P. Wright, J. P. Attfield and P. G. Radaelli, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 214422 CrossRef.
- U. Lins, C. N. Keim, F. F. Evans, M. Farina and P. R. Buseck, Geomicrobiol. J., 2007, 24, 43–50 CrossRef CAS.
- D. Faivre and D. Schüler, Chem. Rev., 2008, 108, 4875–4898 CrossRef CAS PubMed.
- J. P. Bradley, R. P. Harvey and H. Y. McSween Jr., Geochim. Cosmochim. Acta, 1996, 60, 5149–5155 CrossRef CAS.
- D. S. McKay, E. K. Gibson Jr., K. L. Thomas-Keprta, H. Vali, C. S. Romanek, S. J. Clemett, X. D. F. Chillier, C. R. Maechling and R. N. Zare, Science, 1996, 273, 924–930 CrossRef CAS.
- L. Zhao, H. Zhang, Y. Xing, S. Song, S. Yu, W. Shi, X. Guo, J. Yang, Y. Lei and F. Cao, Chem. Mater., 2008, 20, 198–204 CrossRef CAS.
- D. Faivre, N. Menguy, F. Guyot, O. Lopez and P. Zuddas, Am. Mineral., 2005, 90, 1793–1800 CrossRef CAS.
- S. A. Chambers, S. Thevuthasan and S. A. Joyce, Surf. Sci., 2000, 450, L273–L279 CrossRef CAS.
- A. V. Mijiritskii and D. O. Boerma, Surf. Sci., 2001, 486, 73–81 CrossRef CAS.
- B. Stanka, W. Hebenstreit, U. Diebold and S. A. Chambers, Surf. Sci., 2000, 448, 49–63 CrossRef CAS.
- F. C. Voogt, T. Fujii, P. J. M. Smulders, L. Niesen, M. A. James and T. Hibma, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 11193–11206 CrossRef CAS.
- G. S. Parkinson, Z. Novotný, P. Jacobson, M. Schmid and U. Diebold, Surf. Sci., 2011, 605, L42–L45 CrossRef CAS PubMed.
- R. Pentcheva, F. Wendler, H. L. Meyerheim, W. Moritz, N. Jedrecy and M. Scheffler, Phys. Rev. Lett., 2005, 94, 126101 CrossRef CAS.
- M. Fonin, R. Pentcheva, Y. S. Dedkov, M. Sperlich, D. V. Vyalikh, M. Scheffler, U. Rüdiger and G. Güntherodt, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 104436 CrossRef.
- G. S. Parkinson, T. A. Manz, Z. Novotný, P. T. Sprunger, R. L. Kurtz, M. Schmid, D. S. Sholl and U. Diebold, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 195450 CrossRef.
- R. Jansen, V. A. M. Brabers and H. van Kempen, Surf. Sci., 1995, 328, 237–247 CrossRef CAS.
- R. Jansen, H. van Kempen and R. M. Wolf, J. Vac. Sci. Technol., B, 1996, 14, 1173 CAS.
- G. Maris, O. Shklyarevskii, L. Jdira, J. G. H. Hermsen and S. Speller, Surf. Sci., 2006, 600, 5084–5091 CrossRef CAS PubMed.
- G. Maris, L. Jdira, J. G. H. Hermsen, S. Murphy, G. Manai, I. V. Shvets and S. Speller, Jpn. J. Appl. Phys., 2006, 45, 2225–2229 CrossRef CAS.
- G. Maris, L. Jdira, J. G. H. Hermsen, S. Murphy, G. Manai, I. V. Shvets and S. Speller, IEEE Trans. Magn., 2006, 42, 2927–2929 CrossRef CAS.
- A. R. Lennie, N. G. Condon, F. M. Leibsle, P. W. Murray, G. Thornton and D. J. Vaughan, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 10244–10253 CrossRef CAS.
- M. Ritter and W. Weiss, Surf. Sci., 1999, 432, 81–94 CrossRef CAS.
- N. Berdunov, S. Murphy, G. Mariotto and I. V. Shvets, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 085404 CrossRef.
- G. J. Martin, R. S. Cutting, D. J. Vaughan and M. C. Warren, Am. Mineral., 2009, 94, 1341–1350 CrossRef CAS.
- N. Berdunov, S. Murphy, G. Mariotto and I. V. Shvets, Phys. Rev. Lett., 2004, 93, 057201 CrossRef CAS.
- A. Kiejna, T. Ossowski and T. Pabisiak, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 125414 CrossRef.
- N. G. Condon, F. M. Leibsle, T. Parker, A. R. Lennie, D. J. Vaughan and G. Thornton, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 15885–15894 CrossRef CAS.
- P. W. Tasker, J. Phys. C: Solid State Phys., 1979, 12, 4977–4984 CrossRef CAS.
- I. Kostov, Mineralogy, Oliver Boyd, Edinburgh, London, 1968 Search PubMed.
- R. V. Gaines, H. C. W. Skinner, E. E. Foord, B. Mason and A. Rosenzweig, Dana's New Mineralogy: The System of Mineralogy of James Dwight and Edward Salisbury, Wiley-Blackwell, 1997 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- L. Néel, Ann. Phys., 1948, 3, 137–198 Search PubMed.
- C. G. Shull, E. O. Wollan and W. C. Koehler, Phys. Rev., 1951, 84, 912–921 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- V. I. Anisimov, M. A. Korotin, J. Zaanen and O. K. Andersen, Phys. Rev. Lett., 1992, 68, 345–348 CrossRef CAS.
- I. de P. R. Moreira, F. Illas and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 155102 CrossRef.
- C. Loschen, J. Carrasco, K. M. Neyman and F. Illas, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035115 CrossRef.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- R. Grau-Crespo, F. Corà, A. A. Sokol, N. H. de Leeuw and C. R. A. Catlow, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 035116 CrossRef.
- D. Muñoz, N. M. Harrison and F. Illas, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 085115 CrossRef.
- I. Ciofini, F. Illas and C. Adamo, J. Chem. Phys., 2004, 120, 3811–3816 CrossRef CAS PubMed.
- F. Illas and R. L. Martin, J. Chem. Phys., 1998, 108, 2519–2527 CrossRef CAS PubMed.
- F. Corà, Mol. Phys., 2005, 103, 2483–2496 CrossRef.
- A. Baldereschi, Phys. Rev. B: Condens. Matter Mater. Phys., 1973, 7, 5212–5215 CrossRef CAS.
- D. J. Chadi and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 1973, 8, 5747–5753 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- Z. K
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) kol and J. M. Honig, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 9090–9097 CrossRef.
kol and J. M. Honig, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 9090–9097 CrossRef. - M. D. Pashley, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 10481–10487 CrossRef CAS.
- G. W. Watson, E. T. Kelsey, N. H. de Leeuw, D. J. Harris and S. C. Parker, J. Chem. Soc., Faraday Trans., 1996, 92, 433–438 RSC.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014–4022 CrossRef CAS.
- J. Neugebauer and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 16067–16080 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford (UK), 1990 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- K. B. Wiberg and P. R. Rablen, J. Comput. Chem., 1993, 14, 1504–1518 CrossRef CAS.
- J. G. Ángyán, G. Jansen, M. Loss, C. Hättig and B. A. Heß, Chem. Phys. Lett., 1994, 219, 267–273 CrossRef.
- F. De Proft, C. Van Alsenoy, A. Peeters, W. Langenaeker and P. Geerlings, J. Comput. Chem., 2002, 23, 1198–1209 CrossRef CAS PubMed.
- G. Wulff, Z. Kristallogr. Mineral., 1901, 34, 449–530 CAS.
- J. W. Gibbs, Collected Works, Longman, New York, 1928 Search PubMed.
- T. G. Cooper and N. H. de Leeuw, J. Cryst. Growth, 2006, 294, 137–149 CrossRef CAS PubMed.
- N. H. de Leeuw and T. G. Cooper, Geochim. Cosmochim. Acta, 2007, 71, 1655–1673 CrossRef CAS PubMed.
- X.-G. Wang, W. Weiss, S. K. Shaikhutdinov, M. Ritter, M. Petersen, F. Wagner, R. Schlögl and M. Scheffler, Phys. Rev. Lett., 1998, 81, 1038–1041 CrossRef CAS.
- M. W. J. Chase, NIST JANAF Termochemical Tables, American Chemical Society and American Institute of Physics for the National Institute of Standards and Technology, Washington DC, 1998 Search PubMed.
- R. Grau-Crespo, C. R. A. Catlow and N. H. de Leeuw, J. Catal., 2007, 248, 77–88 CrossRef CAS PubMed.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 035406 CrossRef.
- R. Grau-Crespo, I. de P. R. Moreira, F. Illas, N. H. de Leeuw and C. R. A. Catlow, J. Mater. Chem., 2006, 16, 1943–1949 RSC.
- T. A. Mellan and R. Grau-Crespo, J. Chem. Phys., 2012, 137, 154706 CrossRef PubMed.
- W. M. Haynes, in CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC; London: Taylor & Francis [distributor], Boca Raton, Fla., 93rd edn, 2012 Search PubMed.
- J. Tersoff and D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 805–813 CrossRef CAS.
- D. E. P. Vanpoucke and G. Brocks, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 241308 CrossRef.
- S. Irrera, A. Roldan, G. Portalone and N. H. de Leeuw, J. Phys. Chem. C, 2013, 117, 3949–3957 CAS.
- E. A. Wood, J. Appl. Phys., 1964, 35, 1306–1312 CrossRef CAS PubMed.
- J. R. Rustad, E. Wasserman and A. R. Felmy, Surf. Sci., 1999, 432, L583–L588 CrossRef CAS.
- N. Spiridis, J. Barbasz, Z. Łodziana and J. Korecki, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 155423 CrossRef.
- S. Nie, E. Starodub, M. Monti, D. A. Siegel, L. Vergara, F. El Gabaly, N. C. Bartelt, J. de la Figuera and K. F. McCarty, J. Am. Chem. Soc., 2013, 135, 10091–10098 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp00529e |
| This journal is © the Owner Societies 2014 |