Two-dimensional silicene nucleation on a Ag(111) surface: structural evolution and the role of surface diffusion†
Haibo
Shu
*ab,
Dan
Cao
*c,
Pei
Liang
a,
Xiaofang
Wang
b,
Xiaoshuang
Chen
b and
Wei
Lu
b
aCollege of Optical and Electronic Technology, China Jiliang University, 310018 Hangzhou, China. E-mail: shu123hb@gmail.com
bNational Laboratory for Infrared Physics, Shanghai Institute of Technical Physics, Chinese Academy of Science, 200083 Shanghai, China
cCollege of Science, China Jiliang University, 310018 Hangzhou, China. E-mail: caodan@cjlu.edu.cn
First published on 21st October 2013
Abstract
The structural evolution of planar Si clusters and the nucleation mechanism of silicene in the initial stages of silicene epitaxial growth on a Ag(111) surface are studied by using ab initio calculations and two-dimensional nucleation theory. The ground-state SiN clusters (1 ≤ N ≤ 25) on the Ag(111) surface are found to undergo a significant structural transition from non-hexagonal plane structures to fully-hexagonal ones at N = 22, which is a crucial step for growing a high-quality silicene nanosheet. Furthermore, important parameters for controlling silicene growth, including the diffusion barriers of Si clusters, nucleation barrier, nucleus size, and nucleation rate are explored. Compared to graphene nucleation on transition-metal (TM) surfaces, the low diffusion barrier of Si atoms and the low nucleation barrier are responsible for the rapid nucleation of silicene on a Ag(111) surface. Our calculations demonstrate that silicene should be synthesized at a relatively low growth temperature (∼500 K) in order to reduce the defect density. The results can be successfully applied to explain the broad experimental observations where the growth temperature of silicene is below 550 K.
1. Introduction
Since the experimental synthesis of graphene in 2004,1 monolayer two-dimensional (2D) materials,2–8 such as graphene, MoS2, and hexagonal BN, have attracted considerable attention due to their intriguing physical and chemical properties as well as numerous potential applications. Inspired by the great achievements with 2D materials, recently there has been an increasing interest in group VI (Si and Ge) analogs of graphene.9–11 The silicon version of graphene, in which a one-atom thick Si sheet is arranged in a honeycomb lattice, is called silicene. Theoretical calculations11,12 predict that silicene exhibits many similar electronic properties to graphene, including that of its charge carriers behaving as massless Dirac fermions. Compared to graphene, the compatibility of silicene with existing Si-based technology makes the material more attractive for use with nanoelectronic devices.Motivated by the outstanding electronic properties and potential applications, extensive effects have been devoted to the synthesis of a high-quality silicene nanosheet. Mechanical exfoliation is an important method to obtain single-layer (SL) graphene.1,2 Graphene can be exfoliated from graphite due to the layered structure of graphite with weak van der Waals interactions and the high-stability sp2 bonding among carbon atoms in graphene. However, this method cannot be applied in the fabrication of silicene because the sp3 hybridization of Si atoms is more stable than the sp2 one. Therefore, the synthesis of silicene is expected to be carried out via the epitaxial growth of silicon atoms on substrate surfaces. The synthesis of monolayer silicene was first reported by Lalmi et al.13 on a Ag(111) surface. However, the reported lattice parameter of silicene was about 17% smaller than that of bulk silicon based on their experimental measurements. Thus, such a structural model remains questionable. Vogt et al.14 have provided compelling evidence for 2D silicene on a Ag(111) surface, with a superstructure of (3 × 3) silicene on a (4 × 4) Ag(111) surface. In this structure, the Ag(111) substrate was found to induce symmetry breaking in silicene, which causes the lack of massless Dirac fermions in single-layer silicene.15,16 Feng et al.17 further observed that the structure of epitaxial silicene on a Ag(111) surface depends on the coverage of the deposited Si atoms and temperature. Various different structural phases have been identified by adjusting the growth conditions, including (4 × 4),14–19 ,17–19
,17–19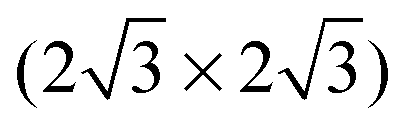 ,19,20
,19,20 17,19 (with respect to the surface lattice of Ag(111)), and
17,19 (with respect to the surface lattice of Ag(111)), and  17,21,22 (with respect to the (1 × 1) silicene). Apart from the Ag(111) substrate, recent experimental studies also demonstrated the growth of silicene on other substrates, such as (0001)-oriented ZrB223 and Ir(111)24 surfaces.
17,21,22 (with respect to the (1 × 1) silicene). Apart from the Ag(111) substrate, recent experimental studies also demonstrated the growth of silicene on other substrates, such as (0001)-oriented ZrB223 and Ir(111)24 surfaces.
Despite these preliminary achievements, the detailed growth process is not still clearly known, especially in the initial nucleation stage of silicene. The complete epitaxial growth process of silicene can be divided into three stages: (i) the Si clustering stage, (ii) the silicene nucleation stage, and (iii) the continuous growth stage of silicene islands. In the first stage, the deposited Si atoms may aggregate into Si clusters by surface diffusion. Meanwhile, the formed Si clusters can also dissociate into Si atoms or smaller Si clusters. With an increase in deposited Si atoms, the concentration of surface Si atoms and clusters increases. Therefore, the Si cluster formations have the chance to become larger. Once the size of Si clusters goes beyond a critical value (or critical nucleus), stage (ii) will start. During this stage, the probability of growth of a silicene nucleus is larger than its probability of dissociation. Stage (iii) starts once the silicene nucleus becomes a matured nucleus that does not disappear. In this stage, the silicene nuclei will gradually grow into silicene islands by the continuous attachment of Si atoms. As the size of the silicene islands increases, neighboring islands will merge and coalesce. In this process, differently orientated silicene islands may induce the formation of grain boundaries.
Among the above three stages, previous studies14–26 mainly focused on the third stage, but very little work27 has been reported on the first and second stages. Two possible difficulties are responsible for the lack of investigations about stages (i) and (ii). One is that existing experimental techniques are difficult to use to identify the structural evolution and morphology of Si clusters in the initial nucleation stage, due to their short lifetime and small size (1–2 nm). The other is that there exist many potential Si isomers with different structures. To explore the ground-state structures of different-sized Si clusters on substrate surfaces by theoretical calculations would be very time-consuming. Although few studies have been carried out, stages (i) and (ii) are very important for synthesizing high-quality silicene. First of all, the Si clustering stage is the basic step to develop the small Si clusters into large silicene islands. Secondly, the nucleation barrier and nucleation size in stage (ii) determine the incubation time of silicene. Thirdly, the nucleation barrier and the diffusion barrier of Si atoms on the substrate surface determine the density of silicene islands, which strongly affects the quality of silicene. Therefore, an understanding of the initial growth process of silicene is important for improving the silicene quality in experiments. Recently, Gao and Zhao27 reported the formation of Si clusters on a Ag(111) surface and revealed the importance of Ag–Si interactions for stabilising the planar Si clusters and 2D silicene sheet. However, there are still many unsolved fundamental problems about silicene growth on a Ag(111) substrate, such as the configuration of the ground-state Si clusters, the role of surface diffusion in the nucleation of silicene, and the relationship between the parameters controlling the nucleation of silicene (e.g. nucleation barrier, nucleus size, and nucleation rate) and the growth conditions, such as temperature and the chemical potential of silicon.
In this work, we systematically investigate the structural evolution of SiN clusters (1 ≤ N ≤ 25) and the nucleation of silicene on the most studied surface, Ag(111), by combining density-functional theory calculations and the 2D crystal nucleation theory. Our results demonstrate that the Si clusters on Ag(111) undergo a significant transition for the ground-state (GS) Si clusters from non-hexagonal planar structures to fully-hexagonal ones at N = 22. This structural transition is important for producing high-quality silicene on the Ag(111) surface. Compared to graphene nucleation on transition-metal (TM) surfaces, silicene growth on a Ag(111) surface exhibits a very high nucleation rate due to the low diffusion barriers of Si clusters and the low nucleation barrier. The high nucleation rate will result in high-density nuclei on the Ag(111) surface, which causes a large number of grain boundaries among merged silicene islands. Therefore, silicene growth should be maintained at a relatively low temperature (∼500 K), which is in good agreement with the experimental observations17–24 in which the synthesis temperature of silicene is below 550 K.
2. Computational details
All calculations in the present study are performed within the DFT frame as implemented by the Vienna ab initio Simulation Package (VASP).28,29 The ion–electron interactions are treated as projector augmented wave (PAW) potentials.30 The exchange–correlation functional is treated as the local density approximation (LDA) which is proven to give a good result for describing van der Waals interactions in a metal–graphene system31–35 and for layered materials,36 such as h-BN and graphite. The energy cutoff for the plane-wave expansion is set to 400 eV. The Monkhorst–Pack k-point mesh of 2 × 2 × 1 is found to provide sufficient accuracy in the Brillouin zone integration. All structures are optimized by a conjugate gradient method until the forces acting on each atom are less than 0.01 eV Å−1. The climbing-image nudged elastic band (cNEB) method37 is employed to search the transition states of the diffusion of Si atoms and Si24 clusters on the Ag(111) surface.The Ag(111) surface is created by using a slab geometry with three Ag atomic layers in which the bottom-layer atoms are fixed to mimic the bulk. For the geometry optimization, all atoms except for the fixed bottom-layer atoms have been fully relaxed and then SiN clusters (1 ≤ N ≤ 25) are put on the Ag(111) surface for further optimization. A (7 × 7) surface slab with dimensions of 19.95 Å × 19.95 Å is used to avoid interactions of adjacent Si clusters on the Ag(111) surface due to the periodic boundary conditions. To explore the stable configuration of Si clusters on the Ag(111) surface, three different types of SiN isomers are investigated, including chains, planar network structures, and polyhedral Si clusters. However, our calculations show that the polyhedral Si clusters on the Ag(111) surface are still energetically unfavorable in all considered size ranges, which is in agreement with the recent theoretical report.27 The relatively high stability of the Si chains and planar Si clusters originates from their strong Ag–Si interactions. Therefore, we will only consider Si clusters in the configurations of chains and 2D planar network structures in the following discussion. For each cluster of N ≥ 10, at least six different configurations are explored and the most stable structure is taken as the ground state. Taking Si10 as an example, we present eight possible stable configurations of Si10/Ag(111) in Fig. S1† and the corresponding formation energies have been listed. The stability of Si clusters on the Ag(111) surface is evaluated by calculating their formation energy, Ef, which is defined as follows,
| Ef = ET − Esub − NSiESi | (1) |
3. Results and discussion
3.1 Geometries and stability of SiN clusters (1 ≤ N ≤ 25)
We first address the geometries and structural stability of various Si clusters on the Ag(111) surface. It is useful for us to understand the structural evolution and nucleation of Si clusters on the Ag(111) surface. From a recent theoretical investigation,27 small Si clusters (e.g. Si6) with the planar triangle-based (PTB) structure on Ag(111) show a high stability, while the PTB structure becomes unstable for larger Si clusters (e.g. Si10). Similar results have been also observed in our calculations, but some new important results need to be clarified: (i) in the size range of 3 ≤ N ≤ 6 (see Fig. S2†), Si chains are still energetically unfavorable relative to PTB structures. Moreover, the energy difference ΔEf between a Si chain and its equivalent PTB structure becomes larger and larger. This result is very different from the case of graphene nucleation on TM surfaces in which the small carbon clusters are dominated by C chains.38–40 (ii) The PTB structure of Si clusters becomes unstable when the size of the Si cluster is larger than 6. Instead, the most stable configuration of SiN clusters (N > 6) shows an sp2-like planar structure (see Fig. S3†).When N is beyond 10, the size of the planar Si clusters gradually becomes larger, thus the number of possible isomers increases rapidly. Here only some important isomers of SiN (10 ≤ N ≤ 24) are presented in Fig. 1 and Fig. S4.† An interesting finding is that the ground-state structures of Si10–Si19 contain at least one pentagon (labelled as SiN-P) and the fully hexagonal network structures (labelled as SiN-H) are just metastable states, but the energy difference ΔEf between SiN-P and SiN-H becomes smaller and smaller as the cluster size increases. For instance, the ΔEf is 0.57 eV for Si10 and only 0.02 eV for Si19, as shown in Fig. 1. A structural transition of the GS from SiN-P to SiN-H occurs when the cluster size reaches N = 22. Moreover, the stability of SiN-H is enhanced as the cluster size increases (see Fig. S5†). In other words, SiN-P is the most stable structure for the smaller Si clusters (10 ≤ N ≤ 19) and SiN-H is energetically more favourable when the cluster size N is larger than 22. The structural transition of the ground state from SiN-P to SiN-H can be understood for the following reasons.
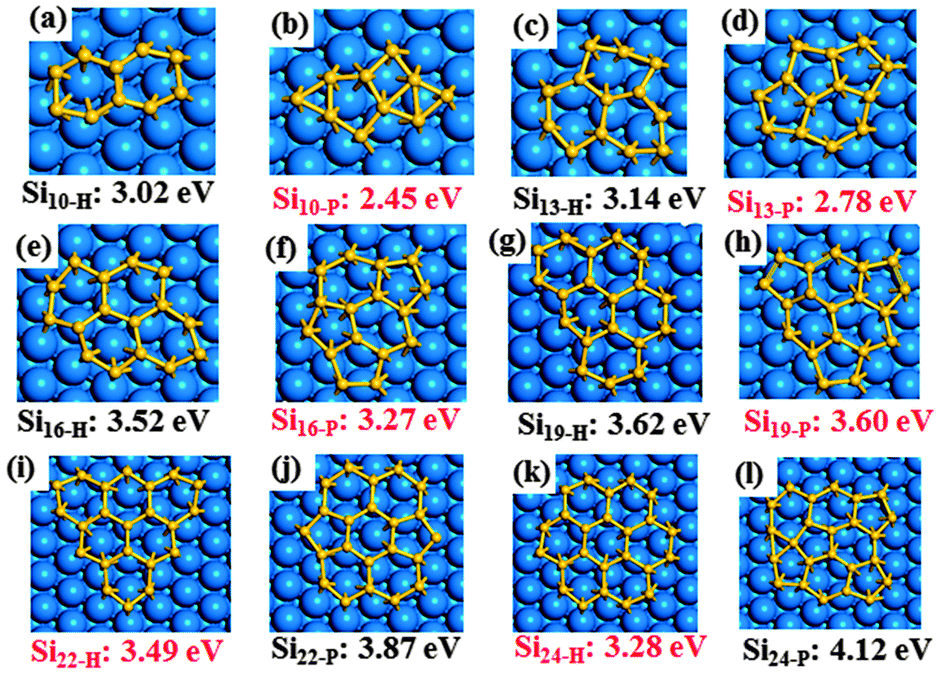 | ||
| Fig. 1 Geometries and formation energies of planar SiN clusters on the Ag(111) surface (10 ≤ N ≤ 24). For each sized cluster, the ground-state structure is highlighted by a red label. | ||
For the smaller and medium sized planar Si clusters (3 ≤ N ≤ 19), the edge atoms in the clusters are a relatively high proportion of the total. Because the inner atoms of Si clusters are very similar to the Si atoms in silicene, the formation energy of a planar Si cluster is mainly from the contribution of its edge atoms. A previous theoretical study27 has found that both the inner and edge atoms of Si clusters interact with the Ag(111) substrate. The Ag–Si interactions ensure that the energy of inner atoms in the Si clusters are very similar to that of Si atoms in bulk (see discussion below). The edge atoms of Si clusters are active relative to the inner atoms of Si clusters. So the strength of the Ag–Si interactions from the edge atoms of a Si cluster is larger than that from its inner atoms. This result can be proved by the binding energies of Si clusters on Ag(111). For instance, the calculated binding energies are 2.10, 1.53, 1.44 eV per atom for Si3, Si13, and Si24 clusters on the Ag(111) surface. The decreasing binding energies with the increasing cluster size originate from the increasing proportion of inner atoms in the Si clusters. The highly active edge atoms in a Si cluster have a high energy state. Thus, the reduction of edge atoms in Si clusters is energetically preferred. Incorporating pentagon rings into the planar Si network structures changes their shape to bowl-like ones, this results in a reduction of the number of edge atoms in Si clusters. For example, the numbers of edge atoms are 8, 9, 10, and 11 for the fully-hexagonal Si10, Si13, Si16, and Si19, respectively. In contrast, the numbers of edge atoms are 4, 7, 8, and 9 for the corresponding clusters with pentagons. Hence, the SiN-P structure is energetically preferred in the smaller Si clusters.
When the size of Si clusters is larger than 22 (N ≥ 22), the proportion of edge atoms becomes small. Although the incorporation of pentagon rings reduces the number of edge atoms in clusters, simultaneously it also increases the curvature of the Si clusters which results in an increase in strain energy. Moreover, increased curvature in the Si clusters reduces the interaction between inner atoms of the Si cluster and Ag(111), which also causes an increase in the formation energy of the Si cluster. Therefore, the stability of a planar Si cluster on Ag(111) can be attributed to a competition mechanism between the strain energy and the edge formation energy. For the large SiN-P clusters, the decreasing edge formation energy cannot compensate for the increasing strain energy. In contrast, the fully-hexagonal SiN-H structures are energetically preferred in the large Si clusters. A recent theoretical study41 has found that the core–shell structured C21 with three pentagons is a very stable magic cluster during the chemical vapor deposition (CVD) of graphene on Rh(111) and Ru(0001) surfaces. However, the Si21 version of the structure is found to be unstable on the Ag(111) surface (Fig. S4g†) and the ground-state structure of Si21 includes only one pentagon (Fig. S4h†). The low-curvature characteristic of Si21 clusters implies that the large Si clusters prefer to form fully-hexagonal structures. Such a structural evolution of Si clusters has a significant impact on the nucleation and growth of silicene on Ag(111). For instance, the coalescence of the fully-hexagonal SiN-H clusters can reduce the formation of defects (e.g. 5|7 defect pairs), which favors the production of high-quality silicene.
3.2 Role of surface diffusion of Si clusters in the nucleation of silicene
Surface diffusion is known to be crucial for the epitaxial growth of silicene on a Ag(111) surface, including the nucleation and edge growth of silicene. To understand the role of surface diffusion in the nucleation of silicene, the possible diffusion behavior of Si feedstock is studied. In most of the experiments of silicene growth on a Ag(111) surface,17–22 the concentration of Si feedstock is very low in order to maintain the single-layer 2D growth of silicene. Hence, Si monomers should be the main Si species during the nucleation and growth of silicene. Certainly, there is a probability of forming small Si clusters by the combination of Si atoms. Kinetically, the concentration of SiN clusters should be proportional to the rate of N Si atoms merging on the Ag(111) surface or CN ∝ (C1)N, where CN and (C1)N are the concentrations of SiN clusters and Si atoms, respectively. It can be seen that the concentration of Si clusters is far lower than that of Si atoms on Ag(111). Here we mainly focus on the diffusion of Si monomers on the Ag(111) surface.As shown in Fig. 2a, we consider four different adsorption sites, including the top (T), bridge (B), fcc hollow (F), and hcp hollow (H), to investigate the stability of a Si monomer on the Ag(111) surface. It is found that the F site is the most stable adsorption site. The H and B sites are also low-energy sites, their formation energies are only 0.03 eV and 0.01 eV higher than that of the F site (see Fig. 2b), respectively. In contrast, a Si atom at the T site is unstable and it will spontaneously move to the hollow sites after structural relaxation. The diffusion path of a Si atom on Ag(111) is thus set to F → B → H.
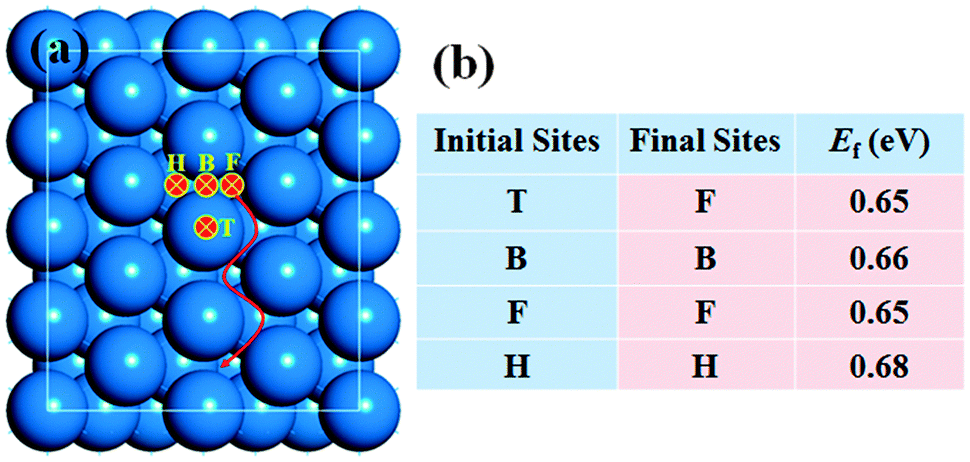 | ||
| Fig. 2 (a) Potential adsorption sites of a Si monomer on a Ag(111) surface and (b) their corresponding formation energies. The adsorption sites include the hcp hollow (H), bridge (B), fcc hollow (F), and top (T) sites. The arrow indicates the possible diffusion path of Si atoms on the Ag(111) surface. | ||
Fig. 3 shows the diffusion barriers and the atomic structures of the low-energy and transition states (TS) of a Si atom on a Ag(111) surface. The barrier for Si atom diffusion along the F → B path is 0.14 eV which is slightly lower than the activation barrier for Si diffusion along the B → H path (0.15 eV) on the Ag(111) surface. Therefore, the threshold barrier for the diffusion of a Si atom on a Ag(111) surface is 0.16 eV (see Fig. 3) which is far lower than that of C diffusion on TM surfaces during graphene CVD growth. The low diffusion barrier implies that the Si atoms can migrate rapidly to the edges of large Si clusters or silicene nuclei, which contributes to the fast nucleation and growth of silicene.
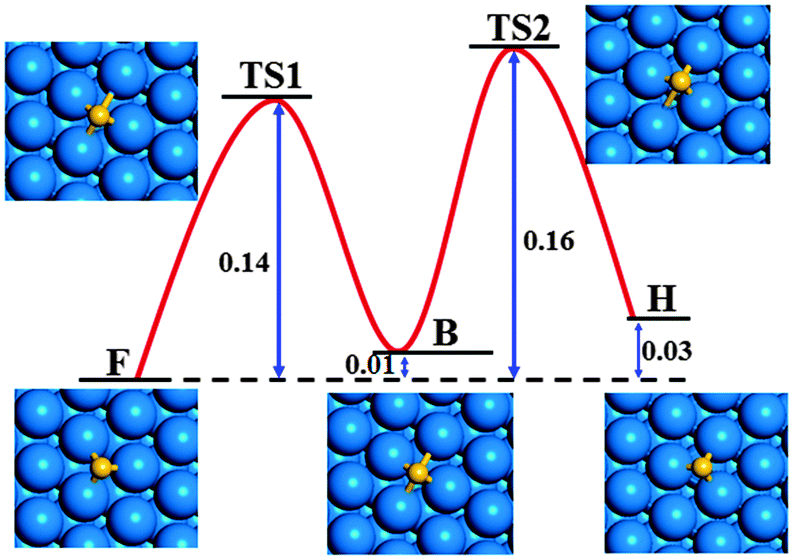 | ||
| Fig. 3 The diffusion barriers and atomic structures of low-energy and transition states of a Si atom on a Ag(111) surface. All energies have the unit eV. | ||
3.3 Nucleation of silicene on a Ag(111) surface
During the nucleation and growth process of silicene, the change in Gibbs free energy ΔG as a function of the number of atoms in the crystalline structure can be expressed as follows,42| ΔG(N) = Ef(N) − ΔμN | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnC/Ceq, where kB is the Boltzmann constant, C is the Si concentration on the Ag substrate surface during growth and Ceq is the equilibrium concentration of Si atoms at the growth temperature), and it dominates the behavior of both nucleation and growth. To obtain the ΔG of Si clusters on a Ag(111) surface, the formation energies Ef(N) need to be defined first. Based on the above results, the formation energies of GS Si clusters as a function of the cluster size N are shown in Fig. 4a. It is found that the formation energies of GS Si clusters increase with the increasing cluster size. The increasing formation energies can be attributed to the contribution of two things: one is the contribution of edge Si atoms and the other is the energy difference ΔE of Si atoms in bulk and Ag(111)-supported silicene. Therefore, the formation energies of the Si clusters can be written as follows,
lnC/Ceq, where kB is the Boltzmann constant, C is the Si concentration on the Ag substrate surface during growth and Ceq is the equilibrium concentration of Si atoms at the growth temperature), and it dominates the behavior of both nucleation and growth. To obtain the ΔG of Si clusters on a Ag(111) surface, the formation energies Ef(N) need to be defined first. Based on the above results, the formation energies of GS Si clusters as a function of the cluster size N are shown in Fig. 4a. It is found that the formation energies of GS Si clusters increase with the increasing cluster size. The increasing formation energies can be attributed to the contribution of two things: one is the contribution of edge Si atoms and the other is the energy difference ΔE of Si atoms in bulk and Ag(111)-supported silicene. Therefore, the formation energies of the Si clusters can be written as follows,| Ef(N) = aN1/2 + ΔE × N | (3) |
Our calculated result shows that the energy difference between the Si atoms in bulk and Ag(111)-supported silicene is very small (∼0.001 eV), thus eqn (3) can be simplified into Ef(N) = aN1/2. By fitting the DFT data points, we obtain the formation energies of Si clusters on a Ag(111) surface
| Ef(N) = 0.8N1/2 | (4) |
Taking this expression into eqn (2), the ΔG as a function of cluster size N in a specified Δμ can be determined. The nucleus size (N*) and nucleation barrier (ΔG*) are defined as the maximum of the ΔG curve (Fig. 4b). According to the definition, we can easily determine the N* and ΔG* as a function of Δμ for silicene nucleation on Ag(111) from eqn (2) and (4).
 | ||
| Fig. 4 (a) Formation energies of Si clusters on a Ag(111) surface as a function of cluster size N. The circles denote the calculated values and the solid curve is fitted by eqn (4). (b) The change of Gibbs free energy ΔG of Si clusters as a function of cluster size N at Δμ = 0, 0.05, 0.10, 0.15, and 0.20 eV, respectively. The nucleus size N* and nucleation barrier ΔG* of silicene under four different Δμ have been labeled by arrows. | ||
Fig. 5a indicates the nucleus size N* and nucleation barrier ΔG* of silicene as a function of Δμ. It can be found that both the N* and ΔG* of silicene decrease with increasing Δμ, suggesting that Δμ is an important parameter for controlling the nucleation of silicene on a Ag(111) surface. For the Δμ in the range from 0 to 0.05 eV, the ΔG* shows a nearly linear decrease but the nucleation barrier of silicene is still higher than 3.2 eV. Therefore, it needs a long incubation time to produce a silicene nucleus. As the Δμ increases from 0.05 eV to 0.15 eV, the ΔG* correspondingly decreases from 3.2 to 1.0 eV. In this regime of Δμ, the silicene nucleates readily due to the relatively low ΔG*. When the Δμ is beyond 0.15 eV, the nucleation barrier is lower than 1.0 eV and the nucleus size drops to N* < 6. The ultralow nucleation barrier will result in a rapid nucleation of silicene on the Ag(111) surface, thus the density of silicene nuclei will increase sharply.
 | ||
| Fig. 5 (a) The nucleus size N* and nucleation barrier ΔG* of silicene on a Ag(111) surface as a function of Δμ, (b) the nucleation rate of silicene as a function of Δμ at temperatures of 200, 300, 400, 500 and 600 K. | ||
To quantify the relationship between the nucleation of silicene and the temperature and Δμ, the 2D nucleation rate Rn of silicene on Ag(111) is estimated based on the classic nucleation theory,42
| Rn = R0 exp(ΔG*/KBT) | (5) |
Hence, the high temperature (>500 K) and large Δμ (>0.1 eV) facilitate a high nucleation rate that results in the spontaneous formation of numerous silicene nuclei on the Ag(111) surface. Owing to the different crystal orientations and edge structures, the high-density nuclei produce large amounts of defects, such as grain boundaries and linear defects when these growing nuclei merge. To reduce the defects in silicene, the growth conditions should be chosen to favour a low nucleation rate, such as by reducing the growth temperature or the concentration of the Si source. This result elucidates why the synthesis temperature of silicene is generally maintained in the range of 450–500 K14–24 which is far lower than the nucleation temperature of CVD-grown graphene (900–1100 °C).43–45 From Fig. 5b, the temperature dependence of nucleation rate indicates that there exists the possibility to grow silicene on Ag(111) at a lower temperature (e.g. 300 K) by adjusting the Δμ. For instance, the nucleation rate of silicene is over 1 cm−2 s−1 at room temperature when the Δμ is larger than 0.12 eV.
4. Conclusions
In summary, we have performed a detailed theoretical investigation on the structural evolution of Si clusters and the nucleation of silicene on a Ag(111) surface. The geometries and stability of SiN clusters (N from 1 to 25) with different configurations on the Ag(111) surface were carefully examined by DFT calculations. Unlike graphene nucleation on TM surfaces, the ground states of small Si clusters are not dominated by Si chains and favour the formation of planar network structures on the Ag(111) surface in the initial nucleation of silicene. As the cluster size increases, the GS Si clusters undergo a significant structural transition from non-hexagonal planar structures to fully-hexagonal ones, which is a crucial step for growing high-quality silicene. On the basis of the 2D crystal nucleation theory, the nucleation barrier, nucleus size, and nucleation rate of silicene on a Ag(111) surface as a function of Δμ were further explored. The results show that the vapor-grown silicene has a high nucleation rate on Ag(111) compared to graphene nucleation on TM surfaces, which originates from the low diffusion barrier of Si atoms (0.16 eV) and the low nucleation barrier (e.g. ∼3.2 eV for Δμ = 0.05 eV). The high nucleation rate favors the production of high-density nuclei on the Ag(111) surface, which will result in a large number of grain boundaries in silicene due to the coalescence of growing nuclei. To obtain a high-quality silicene nanosheet, the growth temperature should be maintained at ∼500 K or lower, which is in agreement with the experimental observations. The present theoretical study for the atomistic nucleation mechanism of silicene is expected to guide the growth of high-quality silicene on Ag(111) surfaces in experiments.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- J. U. Park, S. W. Nam, M. S. Lee and C. M. Lieber, Nat. Mater., 2012, 11, 120 CrossRef CAS PubMed.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147 CrossRef CAS PubMed.
- H. L. Zeng, J. F. Dai, W. Yao, D. Xiao and X. D. Cui, Nat. Nanotechnol., 2012, 7, 490 CrossRef CAS PubMed.
- A. Castellanos-Gomez, M. Barkelid, A. M. Goossen, V. E. Calado, H. S. J. van der Zant and G. A. Steele, Nano Lett., 2012, 12, 3187 CrossRef CAS PubMed.
- K. H. Lee, H. J. Shin, J. Lee, I. Y. Lee, G. H. Kim, J. Y. Choi and S. W. Kim, Nano Lett., 2012, 12, 714 CrossRef CAS PubMed.
- K. Takeda and K. Shiraishi, Phys. Rev. B: Condens. Matter, 1994, 50, 14916 CrossRef CAS.
- G. G. Guzmán-Verri and L. C. Lew Yan Voon, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 075131 CrossRef.
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Sahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS.
- C. C. Liu, W. X. Feng and Y. G. Yao, Phys. Rev. Lett., 2011, 107, 076802 CrossRef.
- B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, S. Vizzini, B. Ealet and B. Aufray, Appl. Phys. Lett., 2010, 97, 223109 CrossRef.
- P. Vogt, P. De Padova, C. Quaresima, J. Avila, E. Frantzekakis, M. C. Asensio, A. Resta, B. Ealet and G. Le Lay, Phys. Rev. Lett., 2012, 108, 155501 CrossRef.
- C.-L. Lin, R. Arafune, K. Kawahara, M. Kanno, N. Tsukahara, E. Minamitani, Y. Kim, M. Kawai and N. Takagi, Phys. Rev. Lett., 2013, 110, 076801 CrossRef.
- R. Arafune, C.-L. Lin, R. Nagao, M. Kawai and N. Takagi, Phys. Rev. Lett., 2013, 110, 229701 CrossRef CAS.
- B. J. Feng, Z. J. Ding, S. Meng, Y. G. Yao, X. Y. He, P. Cheng, L. Chen and K. H. Wu, Nano Lett., 2012, 12, 3507 CrossRef CAS PubMed.
- C. L. Lin, R. Arafune, K. Kawahara, N. Tsukahara, E. Minamitani, Y. Kim, N. Takagi and M. Kawai, Appl. Phys. Express, 2012, 5, 045802 CrossRef.
- D. Chiappe, C. Grazianetti, G. Tallarida, M. Fanciulli and A. Molle, Adv. Mater., 2012, 24, 5088 CrossRef CAS PubMed.
- H. Jamgotchian, Y. Colington, N. Hamzaouri, B. Ealet, J. Hoarau, B. Aufray and J. P. Biberian, J. Phys.: Condens. Matter, 2012, 24, 172001 CrossRef CAS PubMed.
- L. Chen, H. Li, B. J. Feng, Z. J. Ding, J. L. Qiu, P. Cheng, K. H. Wu and S. Meng, Phys. Rev. Lett., 2013, 110, 085504 CrossRef.
- L. Chen, C. C. Liu, B. Feng, X. He, P. Cheng, Z. Ding, S. Meng, Y. G. Yao and K. H. Wu, Phys. Rev. Lett., 2012, 109, 056804 CrossRef.
- A. Fleurence, R. Friedlein, T. Ozaki, H. Kawai, Y. Wang and Y. Yamada-Takamura, Phys. Rev. Lett., 2012, 108, 245501 CrossRef.
- L. Meng, Y. L. Wang, L. Z. Zhang, S. X. Du, R. T. Wu, L. F. Li, Y. Zhang, F. Li, H. T. Zhou, W. A. Hofer and H. J. Gao, Nano Lett., 2013, 13, 685 CrossRef CAS PubMed.
- G. Le Lay, P. De Padova, A. Resta, T. Bruhn and P. Vogt, J. Phys. D: Appl. Phys., 2012, 45, 392001 CrossRef.
- D. Kaltsas, L. Tsetseris and A. Dimoulas, J. Phys.: Condens. Matter, 2012, 24, 442001 CrossRef CAS PubMed.
- J. F. Gao and J. J. Zhao, Sci. Rep., 2012, 2, 861 Search PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- X. Zhang, Z. Xu, L. Hui, J. Xin and F. Ding, J. Phys. Chem. Lett., 2012, 3, 2822 CrossRef CAS.
- M. Fuentes-Cabrera, M. I. Baskes, A. V. Melechko and M. L. Simpson, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 035405 CrossRef.
- C. Gong, G. Lee, B. Shan, E. M. Vogel, R. M. Wallace and K. Cho, J. Appl. Phys., 2010, 108, 123711 CrossRef.
- Q.-M. Zhang, J. C. Wells, X. G. Gong and Z. Y. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 205413 CrossRef.
- F. Abild-Pedersen and J. K. Nøskov, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 115419 CrossRef.
- G. Graziano, J. Klimeš, F. Fernandez-Alonso and A. Michaelides, J. Phys.: Condens. Matter, 2012, 24, 424216 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- J. F. Gao, J. Yip, J. J. Zhao, B. I. Yakobson and F. Ding, J. Am. Chem. Soc., 2011, 133, 5009 CrossRef CAS PubMed.
- T. C. Niu, M. Zhou, J. L. Zhang, Y. P. Feng and W. Chen, J. Am. Chem. Soc., 2013, 135, 8409 CrossRef CAS PubMed.
- D. J. Cheng, G. Barcaro, J. C. Charlier, M. Hou and A. Fortunelli, J. Phys. Chem. C, 2011, 115, 10537 CAS.
- Q. H. Yuan, J. F. Gao, H. B. Shu, J. J. Zhao, X. S. Chen and F. Ding, J. Am. Chem. Soc., 2012, 134, 2970 CrossRef CAS PubMed.
- I. V. Markov, Crystal Growth for Beginners: Fundamentals of Nucleation, Cystal Growth and Epitaxy, World Scientific Publishing Co. Pte Ltd, Singapore, 2nd edn, 2006 Search PubMed.
- H. Wang, G. Z. Wang, P. F. Bao, S. L. Yang, W. Zhu, X. Xie and W.-J. Zhang, J. Am. Chem. Soc., 2012, 134, 3627 CrossRef CAS PubMed.
- S. Bhaviripudi, X. T. Jia, M. S. Dresselhaus and J. Kong, Nano Lett., 2010, 10, 4128 CrossRef CAS PubMed.
- L. Gao, J. R. Guest and N. P. Guisinger, Nano Lett., 2010, 10, 3512 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53933d |
| This journal is © the Owner Societies 2014 |