Decoding the patterns of ubiquitin recognition by ubiquitin-associated domains from free energy simulations†
Benjamin
Bouvier
Bioinformatics: Structures and Interactions, Bases Moléculaires et Structurales des Systèmes Infectieux, Univ. Lyon I/CNRS UMR 5086, IBCP, 7 passage du Vercors, Lyon 69367, France. E-mail: benjamin.bouvier@ibcp.fr
First published on 25th October 2013
Abstract
Ubiquitin is a highly conserved, highly represented protein acting as a regulating signal in numerous cellular processes. It leverages a single hydrophobic binding patch to recognize and bind a large variety of protein domains with remarkable specificity, but can also self-assemble into chains of poly-diubiquitin units in which these interfaces are sequestered, profoundly altering the individual monomers' recognition characteristics. Despite numerous studies, the origins of this varied specificity and the competition between substrates for the binding of the ubiquitin interface patch remain under heated debate. This study uses enhanced sampling all-atom molecular dynamics to simulate the unbinding of complexes of mono- or K48-linked diubiquitin bound to several ubiquitin-associated domains, providing insights into the mechanism and free energetics of ubiquitin recognition and binding. The implications for the subtle tradeoff between the stability of the polyubiquitin signal and its easy recognition by target protein assemblies are discussed, as is the enhanced affinity of the latter for long polyubiquitin chains compared to isolated mono- or diubiquitin.
Introduction
Ubiquitin (Ubq) is a small (76 amino acids), highly conserved regulatory protein which is found at high concentration (85 μM) in the cell.1 Ubiquitin tagging of protein assemblies is a reversible post-translational modification that modulates numerous cellular processes such as protein degradation, autophagy, endocytosis, DNA repair and transcription.2 It is one of the most frequently occurring regulatory mechanisms in eukaryotes: for example, nearly 5% of the human genome is devoted to the binding and unbinding of Ubq to and from proteins.3Ubiquitin labeling is performed by the consecutive action of ubiquitin-activating (E1), ubiquitin-conjugating (E2) and ubiquitin-protein ligase (E3) enzymes which create an isopeptide bond between a lysine of the tagged protein and the C-terminal glycine of ubiquitin.4 This monoubiquitination is often followed by the grafting of further ubiquitin molecules to one of the seven lysines of the first ubiquitin moiety, resulting in (possibly branched) polyubiquitin chains. K48-connected linear polyubiquitin chains, which tag their host protein for 26S proteasome degradation, are the most commonly observed and studied,5 milestoned by the Nobel prize for chemistry in 2004. However, other linkage types corresponding to different signaling pathways are identified on a regular basis.3,6 The recognition of these complex signals and the implementation of the corresponding cellular response are the subject of intense ongoing research: in particular, polyubiquitination often appears to be a prerequisite for efficient signaling, the efficacy of the recognition process increasing with the number of ubiquitin units. Efficient targeting of a protein for the proteasome has for instance been shown to require at least four moieties.5
The recognition of ubiquitin and ubiquitin-labeled proteins is performed by one or a combination of the more than two dozen ubiquitin-binding domains identified to date.7 Despite very different structural folds and binding modes, they all target a conserved, mostly hydrophobic patch on the ubiquitin surface. Structural studies have shown that the same interface is shared between pairs of ubiquitin monomers in K48-linked polyubiquitin, which consists of a series of diubiquitin (Ubq2) building blocks in a ‘closed’ conformation where the hydrophobic surfaces of both ubiquitin monomers, arranged in quasi-C2h symmetry, shield each other from the solvent.8 This raises the question of how ubiquitin receptors associated with the afferent signaling pathways can effectively gain access to the interface of individual ubiquitin units in polyubiquitin chains for recognition purposes. In a seminal crystallographic study,9 Cook and coworkers found that an ‘open’ conformation of diubiquitin, allowing direct access to the monomers' interfaces, was negligibly populated around physiological pH values, hinting at a sizeable free energy barrier to monomer separation that would need to be overcome for ubiquitin binding to occur. This view has since been confirmed by others, using both X-ray diffraction8 and NMR spectroscopy.10,11 In this context, it is all the more surprising that many proteins of the UBA class (ubiquitin-associated domain, one of the most studied classes of ubiquitin binders) actually have a greater affinity for K48-linked diubiquitin than for ubiquitin itself.12 However, our limited understanding of these phenomena has recently been challenged by NMR/RDC studies of wild-type in vitro-prepared K48-linked Ubq2 showing that the conformational equilibrium between the closed and open states of Ubq2 is in fact displaced in favor of the latter at pH 7 (75% open form).13
Understanding this competitive selectivity calls for additional in-depth information about the mechanistic and energetic aspects of mono- and K48-polyubiquitin recognition by UBA domains, which I address in this study using enhanced sampling, all-atom, explicit-solvent simulations of the controlled unbinding of different experimentally characterized ubiquitin complexes.
Methods
Minimum separation restraint
The minimum distance restraint methodology and its parameters are described in detail in earlier publications14,15 and will only be outlined here. The restraint imposes a minimum distance between two non-overlapping groups of atoms by acting on all atom pairs (with one atom in each group) according to the following biasing potential:where index i identifies atom pairs, di is the Euclidean distance between the atoms of pair i; the minimum separation dmin and force constant k are user-defined parameters. The overall biasing potential acting on the system is the sum of the individual contributions of all possible pairs of atoms. To avoid the two groups of atoms from drifting apart from each other when di ≥ dmin ∀i, a similar quadratic penalty is imposed on the closest pair of atoms only:
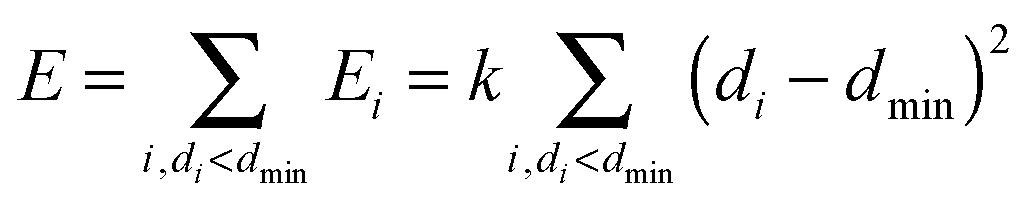
| E = k(dc − dmin)2, dc = min(di) |
In turn, the biasing forces are computed as the negative gradient of the potential and added to those derived from the force field.
A double-cutoff scheme was used for the efficient culling of distant atom pairs (numerous when large groups of atoms are constrained), preserving optimal scalability compared to an equivalent, unbiased molecular dynamics simulation. In this framework, a margin region surrounds the restrained region; atoms in the margin are not restrained (being farther apart than the user-specified minimum distance), but monitored for entry into the restrained region, whereas other atoms are simply ignored. The extension of the margin beyond the restrained region was chosen to be 2 Å, and the list of monitored atom pairs was rebuilt every ten integration steps. These values provided the best balance between accuracy and computational cost (which increases by less than 5% compared to the corresponding unbiased simulation). The minimum distance restraint was implemented in C++ as a dynamically linked library and interfaced to the NAMD 2.8/2.9 molecular dynamics package16 using Tcl bindings. The restraint software is available from the author upon request.
Molecular dynamics simulations – general protocol
Molecular dynamics simulations were performed using NAMD16 2.8 and 2.9 on a local distributed-memory cluster and the Jade scalar parallel supercomputer at the Centre Informatique National de la Recherche Scientifique (CINES) in Montpellier, France. The AMBER parm99 forcefield,17 TIP3P water model18 and Joung–Cheatham monovalent ion parameters were employed.19 A 2 fs integration timestep was made possible by constraining all hydrogen-containing chemical bonds. Constant pressure (1 atm) and temperature (300 K) were imposed using Langevin dynamics (5 ps−1 damping coefficient)20 and the Nosé–Hoover Langevin piston method (period 200 ps, decay 100 ps).21 Boundary conditions were applied, and long-range electrostatics were computed every two steps using the Particle Mesh Ewald method22 with a real-space cutoff of 10 Å inside a multiple-time stepping scheme.System preparation
The structures obtained from the PDB or docking calculations were placed in a truncated octahedral box of sufficient size to ensure a padding of at least 10 Å around the monomers of the fully dissociated complexes. Water molecules and the minimal number of Na+ or Cl− ions necessary to nullify the total electrical charge were added, and the system was minimized to convergence. Positional restraints of 5 kcal mol−1 Å−2 were imposed on the solute heavy atoms, and the temperature of the system was progressively raised from 0 to 300 K over 1 ns. The restraints were then progressively scaled down over 500 ps and the system was simulated without restraints for a further 2.5 ns before production runs were begun. The lengths of the production simulations depended on the system but were no shorter than 20 ns.Biased simulations
When not otherwise mentioned, the separation distance used as a biasing coordinate involved all heavy atoms of the monomers under study. The structures sampled during the production run were averaged after the LSQ fit, and the conformation with the smallest RMSD to the average structure was used as a starting point for the biased simulations. The initial reference value for the interpartner distance was chosen as the average of the distances observed during the production run. From there on, simulation windows with an interpartner distance stride of 0.1 Å (which was checked to provide sufficient overlap between consecutive windows) were performed until a plateau in the free energy profile was reached (or for Ubq2, until the linker region was fully extended). At such large separation distances, the repertoire of relative orientations and positions available to the protein partners becomes increasingly difficult to sample; see ESI† for an estimate of the error involved. The simulation windows were prolonged until additional sampling had no effect on the resulting PMF; the total duration per window depended on the system under study but was no smaller than 3 ns.Potential of mean force
The free energy profile (or potential of mean force) along the separation coordinate was obtained from the combined population densities of the simulation windows and the instantaneous values of the biasing potential, using the weighted histogram analysis method (WHAM).23Accuracy of the simulations
Zhu and Hummer24 have shown that the cumulative statistical error for the potential of mean force G(x) along a reaction coordinate x, sampled using a series of umbrella windows i with harmonic biasing potentials of the form K(x − ri)2 centered at ri = r0 + iΔr, can be expressed as the square root of the variance in the free energy estimator:where var(
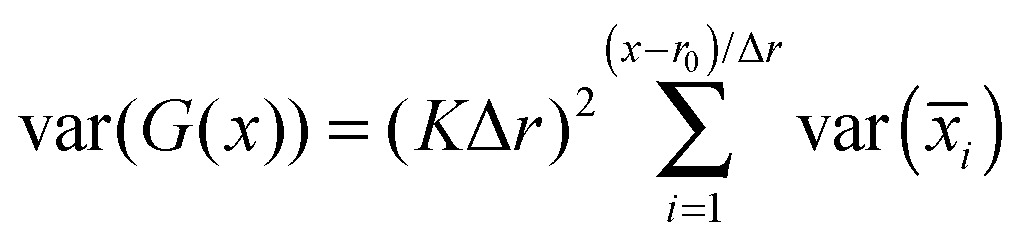
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) i) is the squared error in the estimate of the mean position of the reaction coordinate x in umbrella simulation i. This can be obtained from a straightforward block averaging of the value of x over the corresponding window:
i) is the squared error in the estimate of the mean position of the reaction coordinate x in umbrella simulation i. This can be obtained from a straightforward block averaging of the value of x over the corresponding window: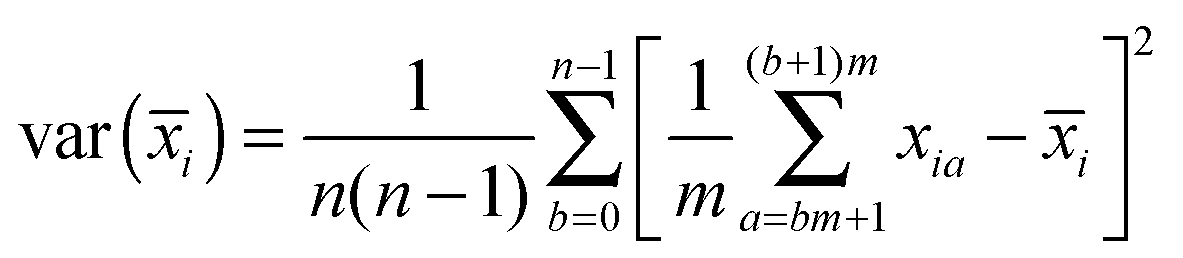
Each umbrella window is split into n blocks of size m such that the averages of x over the blocks can be considered independent from each other. In the present cases, values of n = 5–10 were found to give the best results and an intermediate value n = 8 was selected for the computation of the statistical error. The resulting error bars are shown on the relevant figures.
Selection of representative conformations inside trajectories
For each interdistance value xi, i ∈ [1…n] inside a predefined range, the conformations of umbrella window wj centered at dj, with j chosen such that |dj − xi| = mink∈[1…n](|dk − xi|), were ranked with respect to their interpartner separation compared to xi. The conformations corresponding to the 50 closest matches were selected for further analysis.Voronoi decomposition of hydrated protein–protein interfaces
The characterization of interfaces between the protein complexes under study was performed using Intervor.25 For a complex consisting of two monomers A and B and water molecules W, an approximation of each of the three possible binary interfaces (AB, AW and BW) is obtained as a tessellated surface, by computing the Voronoi diagram (and its mathematical dual, the Delaunay triangulation) of the centers of its constitutive atoms. Unlike more conventional methods based on the loss of solvent accessibility upon binding, the Voronoi/Delaunay decomposition of space is able to detect ‘buried’ interface atoms which have been shown to represent, on average, a non-negligible 13% of an interface.26 The union of the three binary interfaces gives rise to the ternary interface, termed ABW. In this work, the ‘dry’ (AB) and ‘wet’ (ABW) interfaces have been considered. These interfaces can also be shelled to provide burial depth information for their constitutive atoms, defined as the number of Voronoi cells in the shortest path from the location of an atom to the interface rim.The flexibility of protein interfaces and the low residence times of interfacial water molecules (especially close to the interface rim) can modify the two-atom contact maps obtained from Intervor between snapshots of the system of similar overall structure. Thus, meaningful contacts reported in this study were defined as those that appear in 45 or more of the 50-conformation set described above. This value was found to coincide with a plateau of the number of contacts in all cases, providing the best filtration of transient contacts whilst retaining meaningful ones.
Most of the water molecules involved in the ‘wet’ ABW interface border the rim of the interface and do not act as bridges between the binding partners; these were not considered for the study of water-mediated contacts.
Principal component analyses
Cartesian principal component analyses (PCA) were performed on the final windows of the unbinding simulations for all complexes under study, using Carma.27 All Ubq backbone atoms were included both in the preliminary LSQ fit and the PCA. The vector consisting of all eigenvalues was normalized to unity. The contribution of the interface residue backbone atoms (L8, I44, A46, H68, V70, L71) to each eigenvector was computed as the norm of the corresponding subvector, the norm of the complete eigenvector being 1; this was multiplied by the corresponding normalized eigenvalue. The sum of these values over all eigenvectors yielded the overall contribution of interface atoms to the global motion as a number between 0 and 1.Macromolecular docking protocol
Every UBA structure extracted from PDB files 2OOB, 2QHO and 1ZO6 was docked against all Ubq structures encountered in these files, using ClusPro 2.028,29 in the ‘others’ mode (which uses the parameter set designed for the homonymous category in the Protein Docking Benchmark,30 to which Ubq–UBA belongs). All docking poses obtained were kept. They were weighted by their respective docking score (in ClusPro, the size of the corresponding cluster). The similitude of each docking pose to the three classes of Ubq–UBA complex structures (see main text) was assessed by computing the RMSD of Ubq after structural alignment of UBA, dividing this value by the largest RMSD measured over the entire set, and subtracting this from 1. The similitude of a pose was then normalized by the sum of similitudes of this pose to the three references and plotted on a ternary graph using WxTernary.31Miscellaneous
All 3D molecular graphics were generated using VMD.32 All plots and figures were produced with Matplotlib.33Results
Classes and mechanisms of Ubq–UBA recognition
Ubiquitin binds other proteins through a canonical interface patch which has been thoroughly described in the literature ever since the work of Cook.9 It consists of a hydrophobic core (L8, I44, A46, V70, L71) borne by a β-α-β motif and surrounded by a few polar/charged amino acids such as H68. The ubiquitin-associated (UBA) domain is a small motif present in a variety of ubiquitin-binding proteins. Despite a low sequence homology, UBA domains are remarkably conserved from a structural point of view, featuring a bundle of three α-helices and surface patches of hydrophobic residues which contact the similarly hydrophobic binding region on ubiquitin.34,35 However, the mechanism of UBA recognition is still unclear: efficacious binding can be achieved even in the absence of crucial hydrophobic residues36 and using a variety of orientations of the UBA domain. In this part, I attempt to rationalize Ubq–UBA recognition by collating relevant complexes from the Protein Databank (PDB) and comparing their binding–unbinding mechanisms and energetics.The Ubq–UBA complexes in the PDB were found to fall into one of the three distinct structural classes (Fig. 1). In the most frequently encountered complex class, the α1–α2 loop and C-terminal parts of the α1 and α3 helices on UBA contact the hydrophobic residues on the surface of Ubq. This type of binding occurs in the complex between Ubq and the UBA domain of the EDD protein, the E3 ubiquitin ligase ortholog to the tumor suppressor hyperplastic discs protein in Drosophila melanogaster which regulates cell proliferation,35 but is also found in numerous other structures.34,37 The same type of binding is encountered between UBA and the distal Ubq monomer in the Ubq2–HHR23A-UBA complex,38 in which the UBA domain is sandwiched between two K48-linked Ubq monomers termed distal and proximal; as a modulator of polyUbq–proteasome interactions, the human protein HHR23A preferentially binds K48-linked polyUbq chains.
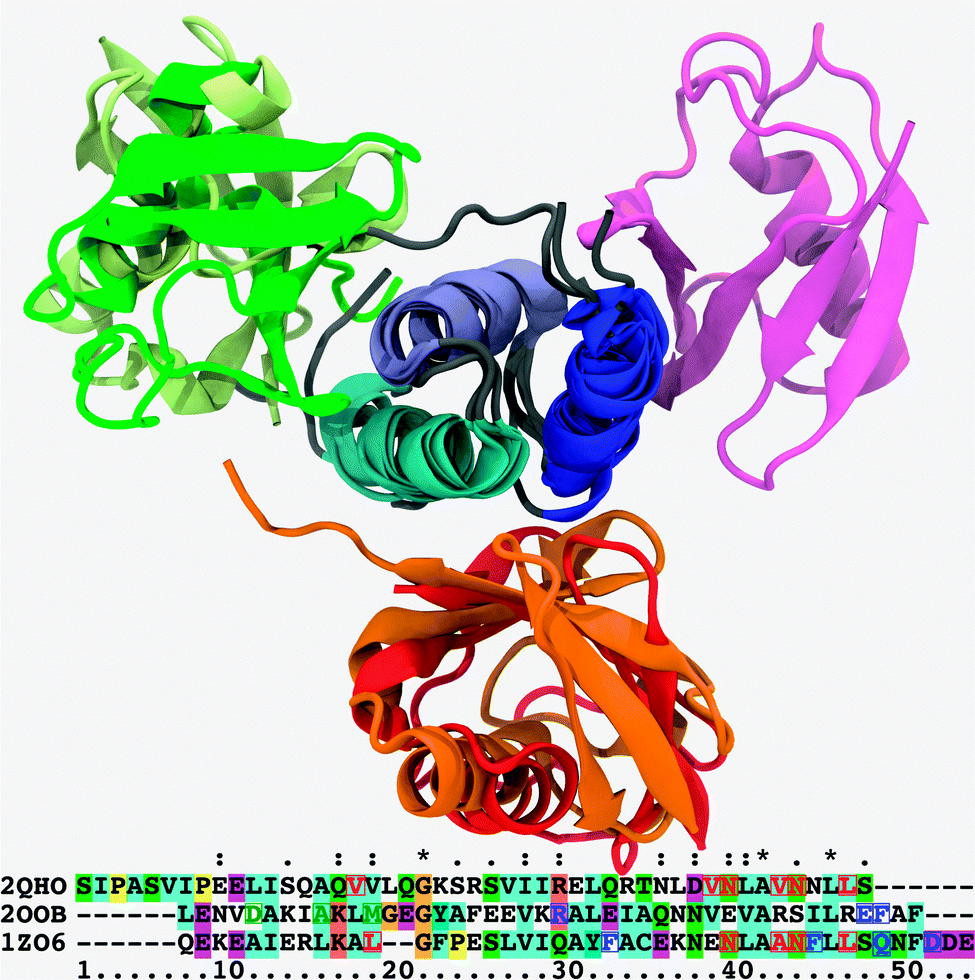 | ||
| Fig. 1 Superposition of cartoon representations for the Ubq–UBA complexes under scrutiny, aligned on the loosely conserved trihelical UBA motif; helix 1: blue, helix 2: indigo, helix 3: cyan. Canonical binding mode: Ubq–EDD-UBA (PDB Id. 2QHO), red; distal monomer of Ubq2–HHR23A-UBA (PDB Id. 1ZO6), orange. Alternate (proximal-type) binding mode: highest-ranking docking pose for the monomers of the Ubq–CBL-B UBA complex (PDB Id. 2OOB), wheat; proximal monomer of Ubq2–HHR23A-UBA (PDB Id. 1ZO6), green. Noncanonical binding mode: Ubq–CBL-B UBA complex (PDB Id. 2OOB), magenta. All cases involve the same conserved, mostly hydrophobic binding patch on Ubq. Relevant UBA sequences are aligned underneath: red, blue and green characters denote key UBA amino acids involved in canonical-type, proximal-type and noncanonical-type bindings, respectively; character background colors are keyed to residue type and conservation patterns as per the standard Clustal X coloring scheme.41 | ||
The second class of structures features an unusual contact surface involving helices α1 and the N-terminal part of α2. Only one structure of this class has been revealed to date: Ubq–CBL-B UBA.39 The CBL protein bearing the UBA motif is an E3 ubiquitin ligase with oncogenic activity, which regulates receptor tyrosine kinases by ubiquitinating them for degradation. However, sequence homologies hint at other UBA-containing systems as members of this structural class.40
The third class is represented by the binding of the proximal monomer to UBA in Ubq2–HHR23A: it is bound to UBA helix α2 and the N-terminal part of α3. To my knowledge, no structures of this type between UBA and monomeric Ubq have been reported to date.
As a preliminary assessment of the relative significance of these three structure types, I generated docking poses from the Ubq and UBA monomers involved in these structures (see Methods). The structural similarity of these poses to the representatives of each of the three structural classes was measured by computing the RMSD of Ubq after structural alignment on the UBA fragment, and the relevance of each pose in the conformational landscape was weighted by its docking score. The resulting score map can be found in Fig. 2. Interestingly, there is a clear preference for proximal-type structures among the high-ranking poses built on partners from all three classes, in striking contrast with the absence of any such experimentally detected structures. Conversely, noncanonical-type structures could not be generated by the docking procedure, despite the existence of an experimental structure and the use of its monomer geometries as input.
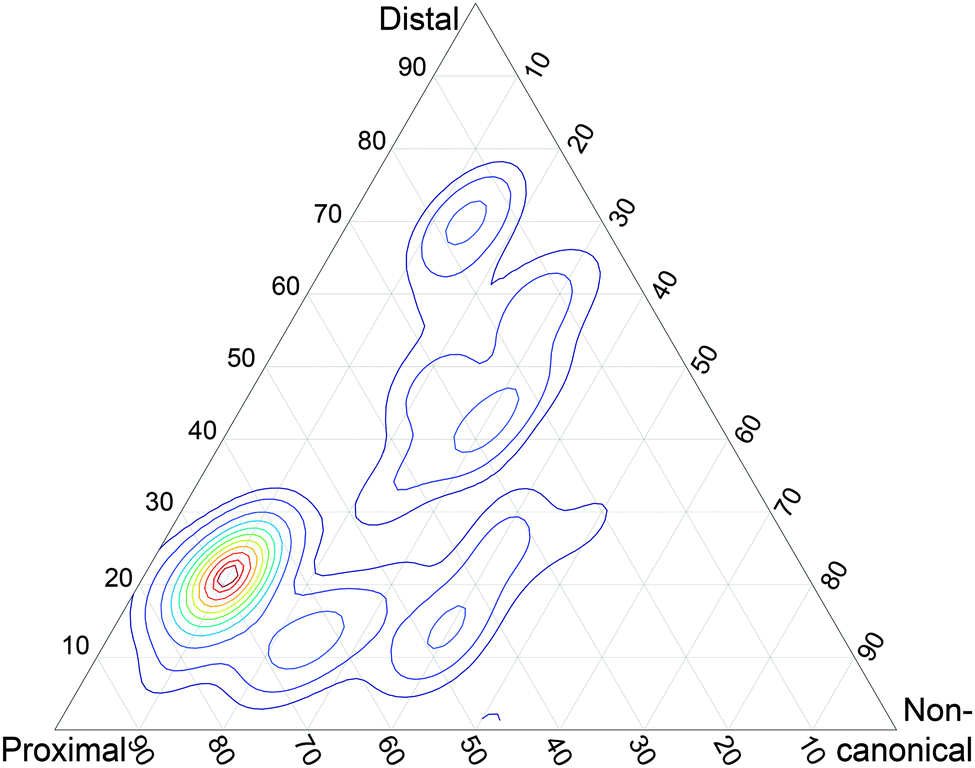 | ||
| Fig. 2 Conformational landscape of Ubq–UBA docking poses, weighted by the poses' docking scores and spanned by their structural similarity to each of the three complex classes (spacing between contour lines: 50 units). | ||
To gain a more quantitative understanding of the relative stability of the complexes, I simulated the separation of the monomers of each of the three complex types using all-atom, explicit solvent molecular dynamics simulations with low-bias intermolecular distance restraints, starting from the corresponding equilibrated PDB structures. The restraint ensures that no atom pair involving both partners is smaller than a user-defined value. By making this value progressively larger, one can simulate the dissociation of a complex inside tractable timescales, overcoming free energy barriers that unbiased molecular dynamics would take many orders of magnitude longer to surmount (see Methods). By potentially considering a very large number of interpartner atom pairs, the restraint does not impose a fixed dissociation pathway, allowing the monomers the liberty to deform or rotate. By thoroughly sampling the unbinding pathway, insight is gained into the binding mechanism; this approach was already proven to be successful for protein–DNA15,42 or drug–DNA14 recognition. Fig. 3–5 present the potentials of mean force (PMF) extracted from the biased simulations using the WHAM procedure (see Methods), as well as the separation mechanisms using residue contact maps. Movies depicting the separation processes are provided as ESI.†
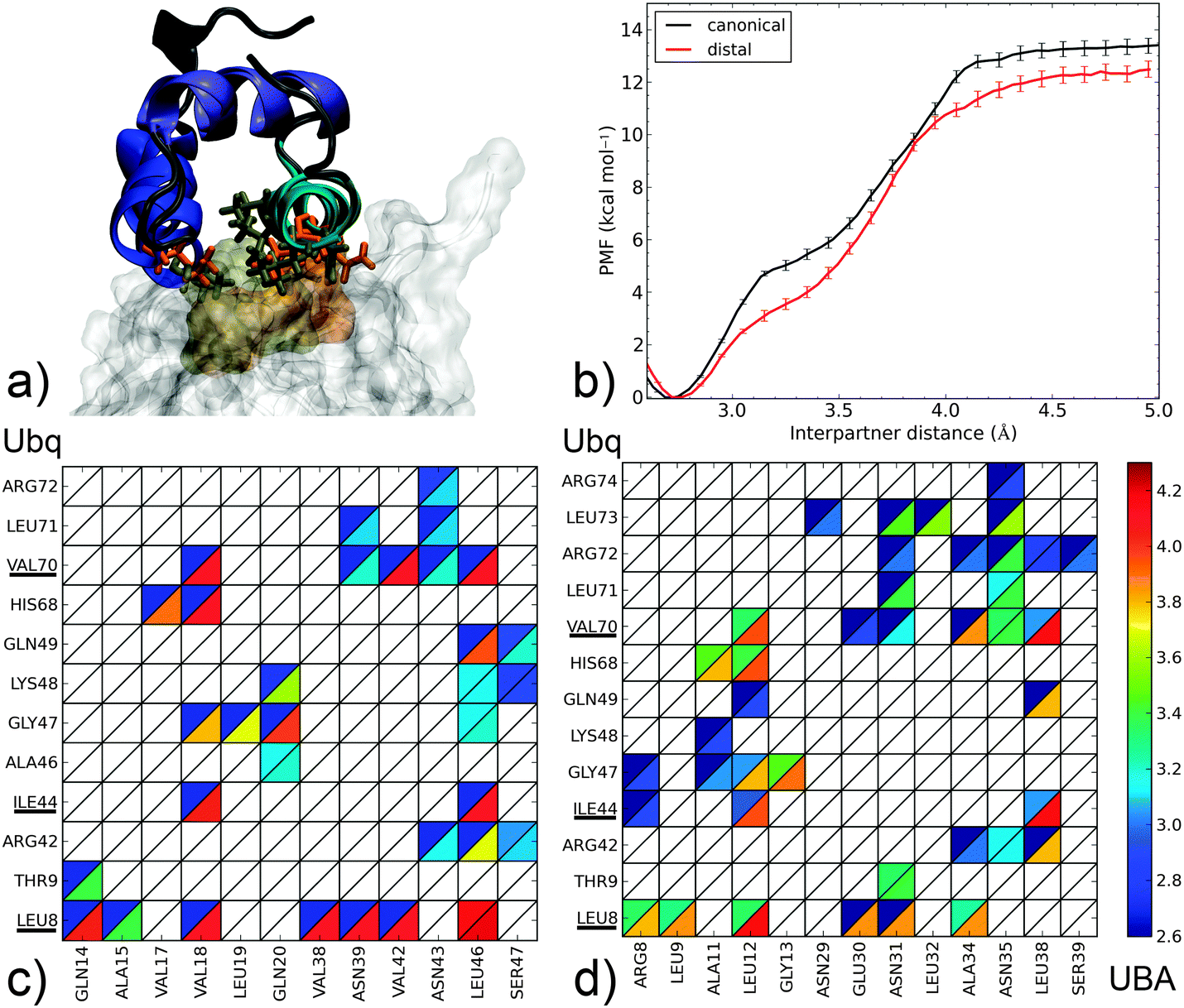 | ||
| Fig. 3 Dissociation mechanism and energetics of two canonical-type Ubq–UBA complexes: (i) the canonical Ubq–EDD-UBA complex and (ii) the distal monomer of the Ubq2–HHR23A-UBA complex. (a) Superposition of bound complex structures aligned on the UBA domain (as cartoons – helix 1: blue, helix 2: indigo, helix 3: cyan); the hydrophobic Ubq interface (surface) and key interacting UBA residues (sticks) are shown in gold for the canonical and orange for the distal complex. (b) PMF as a function of interpartner separation for both complexes. Lower panel: contact maps for interface residues of Ubq (vertical axis) and UBA (horizontal axis) as a function of interpartner separation (color scale, Å). The upper left (resp. lower right) triangle of each cell represents the smallest (resp. largest) distance at which the corresponding contact is detected. Important Ubq interface residues are underlined. (c) Canonical complex; (d) distal complex. | ||
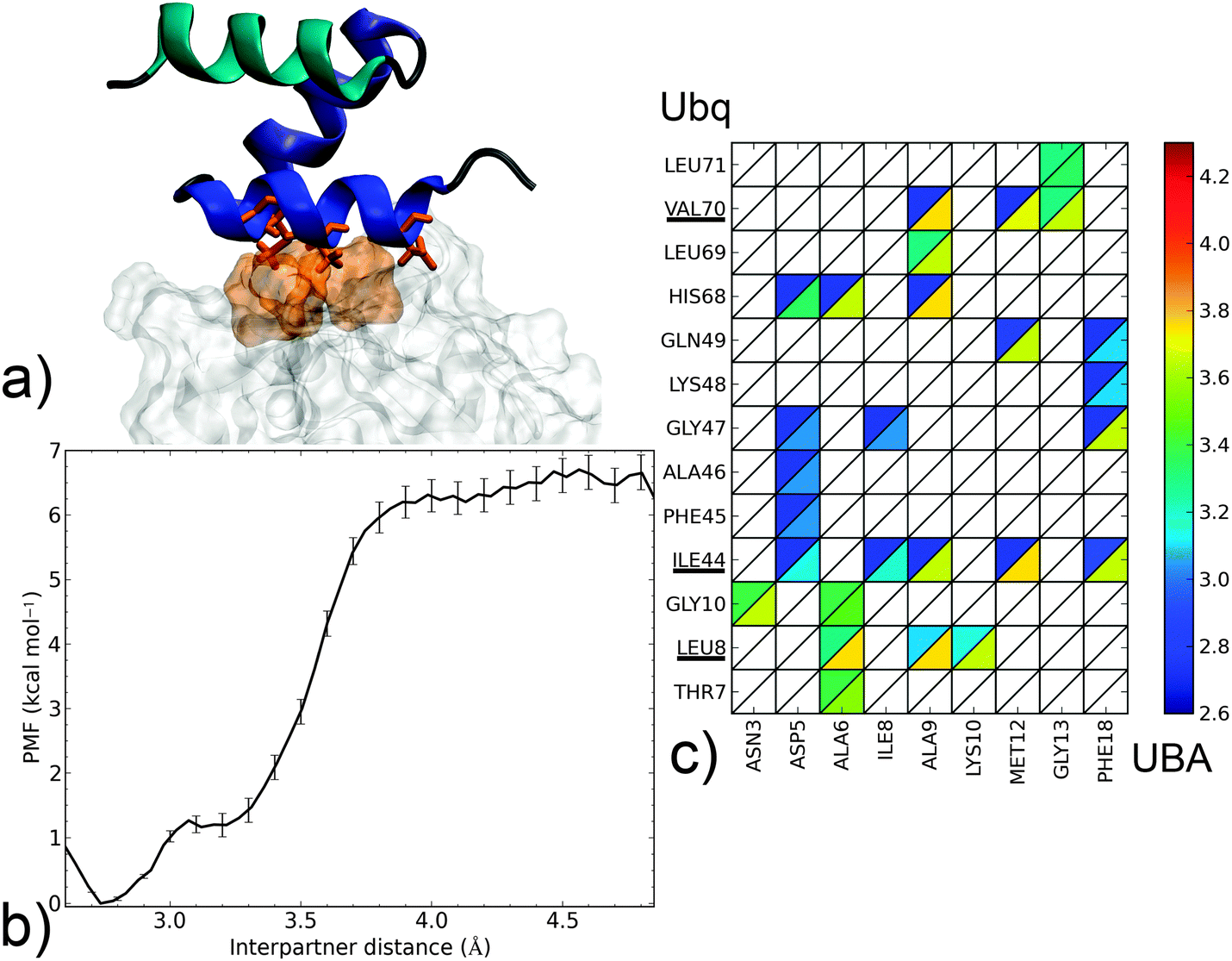 | ||
| Fig. 4 Dissociation mechanism and energetics of the noncanonical Ubq–CBL-B UBA complex. (a) Structure of the bound complex (UBA as cartoon, helix 1: blue, helix 2: indigo, helix 3: cyan; Ubq as surface). Key interface residues on Ubq (surface) and UBA (sticks) are colored orange. (b) PMF as a function of interpartner separation. (c) Contact map along the complex dissociation pathway (see the Fig. 3 caption for details). | ||
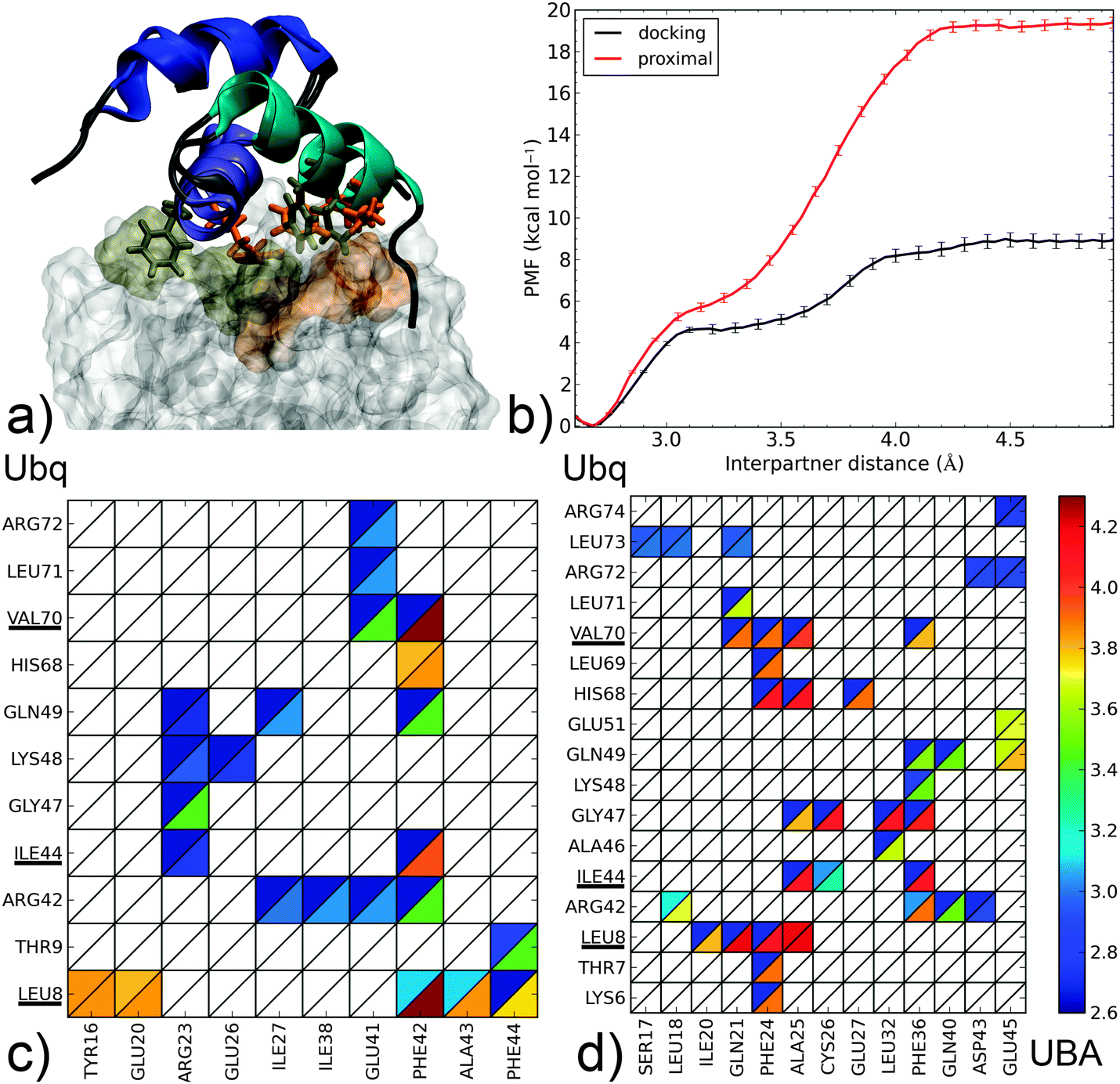 | ||
| Fig. 5 Dissociation mechanism and energetics of two proximal-type Ubq–UBA complexes: (i) highest-ranking docking pose for the monomers of the Ubq–CBL-B UBA complex and (ii) proximal monomer of Ubq2–HHR23A-UBA. (a) Superposition of bound complex structures aligned on the UBA domain (as cartoons – helix 1: blue, helix 2: indigo, helix 3: cyan); the hydrophobic Ubq interface (surface) and key interacting UBA residues (sticks) are shown in gold for the proximal and orange for the docking complex. (b) PMF as a function of interpartner separation for both complexes. Lower panel: contact maps along the dissociation pathways of (c) the docking complex and (d) the proximal complex (see the Fig. 3 caption for details). | ||
The canonical interface between Ubq and UBA (Fig. 3a) is markedly hydrophobic. Amino acids centered around residues L8, I44 and V70 on Ubq interact with residues V18, V42, L46 and their neighbors on UBA. It is interesting to note that contacts through L8, I44 and V70 subsist until complete separation of the proteins, although the nature of the contacted residues on UBA may change and side-chain rotation may occur. Indeed, the formation of an enduring L8–L46 contact at relatively long separation distances, around which the entire UBA domain rotates, constitutes the last event in monomer separation. L8, I44 and V70 thus appear to play a crucial role in long-range recognition, with L8 acting as an anchor and I44, V70 then being used to fine-tune relative monomer orientation. On the other hand, peripheral residues (A46–L48, L71) break contact early on in the separation process. These two distinct regimes of contact breaking–forming, characterized by steep, linear increases in free energy, are separated by a more gently sloping region centered around 3.2 Å. The computed binding free energy of 12–13 kcal mol−1 is larger than the upper range of experimental determinations (8 kcal mol−1)43 but consistent with the numerous favorable interactions detected, and not unreasonable considering the range of experimental and theoretical errors (see ESI† for an assessment of the latter).
The separation mechanism of UBA from the distal Ubq2 monomer (Fig. 3b) is quite similar, with the roles of V18, V42 and L46 taken by similarly placed L12, A34 and L38. Interestingly, polar asparagines N39 and N43 in Ubq–EDD-UBA are strictly conserved to N31 and N35 in Ubq–HHR23A-UBA; they remain in interaction with L8 until the final moments of separation, by transformation of the initial hydrophobic contacts (N Cβ/L Cδ) into electrostatic interactions (N side-chain/L backbone). These asparagines also form conserved polar links to R42 and R72 on Ubq which last until 3.6 Å. Thus, despite the mostly hydrophobic nature of the binding patch core, peripheral electrostatic hotspots play an important role in recognition at both long and short ranges.
The common features in the dissociation mechanisms of both canonical-type complexes translate into a marked similarity for the corresponding free energy profiles, with a binding free energy difference of only 0.8 kcal mol−1, which can be attributed to the lower initial extent of interactions at L8 in the distal Ubq–HHR23A-UBA structure.
In the noncanonical interface between Ubq and UBA (Fig. 4a), interactions appear much less favorable. For instance, contacts at L8 are not initially present but form later on during the separation process. Similarly, the hydrophobic I44–G47 patch is initially involved in weak interactions with polar residue D5 which break early on. The decay of these opposing interactions translates into a low-lying plateau at a short separation distance of 3.1 Å. At this point, the UBA monomer has rotated almost 90°, with the axis of helix α1 perpendicular to the Ubq β-sheet axis: in fact, the complex geometry now closely resembles that of the canonical complex, albeit at a larger interpartner distance. Enduring contacts are formed essentially by UBA residues A9 and M12, the mutation of which has been experimentally shown to be detrimental to binding,39 to Ubq residues I44 and V70. Also noteworthy is an electrostatic contribution from a H68–D5 interaction, absent in canonical-type interfaces. These interactions result in a higher secondary barrier, but the overall binding free energy remains significantly lower than in the canonical case. The good agreement of the computed binding free energy with its experimental determination (5.8 kcal mol−1)35 is quite encouraging despite the impossibility to strictly rule out a cancellation of errors. Such a good performance of the separation restraint method had previously been observed on protein–DNA and drug–DNA systems.14,15
From this separation mechanism, the noncanonical complex appears to be a less stable by-product of its canonical counterpart, with a unified pathway involving UBA recognition via the L8, I44 and V70 regions of Ubq branching off at smaller separation distances when the rupture of the L8 link can be compensated by the formation of alternate contacts.
Finally, we come to proximal-type interfaces. The highest-ranking docking pose of Ubq–CBL-B-UBA identified using ClusPro bears a striking structural similarity to the proximal Ubq monomer in the NMR Ubq2–HHR23A-UBA complex; however, analysis of the interface reveals important differences. The Ubq-binding motif of the proximal HHR23A-UBA domain (Fig. 5b) is markedly hydrophobic: its F24 residue forms an extensive lattice of contacts inside the pocket centered around Ubq residues L8 and V70, while F36 connects with I44 and neighbors. Strong peripheral polar contacts and salt bridges reinforce these interactions: H68–E27 contacts endure until the final instants of unbinding, while R42–Q40/D43 and Q49–Q40 form at close range only. Unsurprisingly, the binding free energy at 19.4 kcal mol−1 is very high, with the large proportion of long-lasting contacts accounting for a secondary barrier that is much higher than the first. It is interesting to note that this apparently very stable complex has no experimental counterpart, and to consider how the addition of the distal monomer present in the NMR complex will affect the binding free energy of UBA (see below).
In contrast, the CBL-B UBA domain (Fig. 5a) is much more polar/charged, with three arginine moieties vs. one in HHR23A and a glutamate residue in place of HHR23A's F24. Consequently, the interface of the docking pose is more polar in nature: R23 on UBA binds K48 and Q49 while R42 on Ubq contacts E41 (however, peripheral contacts via H68 do not occur here). The usual hydrophobic contacts to canonical Ubq residues L8, I44 and V70 are retained, but through a single UBA residue only (F42). The free energy profile features a steep initial rise linked to the attenuation of polar contacts followed by a more progressive phase where the hydrophobic contacts, around which the UBA monomer rotates, are destroyed. The overall binding free energy is only about half of that of the HHR23A-proximal Ubq complex, but still significantly higher than the experimentally identified noncanonical binding of CBL-B discussed above. Interestingly, the initial phase of separation in both proximal-type complexes involves the weakening of similarly placed R/E–D salt bridges, which translates into very similar PMFs for d < 3.1 Å and demonstrate the importance of such contacts for the final stabilization of proximal-type structures, especially when the UBA sequence precludes the formation of extensive hydrophobic contacts (as for CBL-B). The impressive stabilization properties of such an extended hydrophobic patch, conditioned by high geometrical complementarity, are also clearly demonstrated. The striking difference in computed binding free energy between these two ‘hypothetical’ complexes show the versatility that can potentially be achieved by combining polar and hydrophobic contacts.
Ubq2 opening mechanism
The closed form of the diubiquitin complex features two ubiquitin monomers bound in a quasi-C2h arrangement using the same, previously described, canonical binding patch that individual Ubq monomers employ to bind UBA domains or other proteins. The C-terminal extremity of the distal Ubq monomer is connected to the side chain of lysine 48 on the proximal monomer, forming a five-residue, arginine and glycine-rich loop which, in a hypothetical extended state, allows an important translational and rotational conformational freedom for one monomer relative to the other. However, making the canonical interfaces on both monomers accessible to other binders does not require the complete separation of the former: indeed, the formation of an alternate interface between monomers could help stabilize an open conformation. To investigate this, I have simulated the opening of the closed-form ubiquitin dimer starting from its experimental structure,38 by imposing a minimal separation distance between the heavy atoms of the relevant residues on the interface of both ubiquitin moieties (L8, I44, A46, H68, V70, L71). The corresponding free energy profile can be found in Fig. 6. It can be described as the association of three regimes in which the free energy rises almost linearly, separated by plateau regions around 5.7 and 7.5 Å. The first rise corresponds to the symmetrical breaking of the hydrophobic interface between I44/A46/G47 on one monomer and L8/T9, V70/T71 on the other. This is accompanied by a rotation of a monomer with respect to the other around the vertical axis passing through both L48 residues, without any notable extension of the monomer-connecting loop. This loop starts to extend during the second phase and is accompanied by the rupture of salt bonds between K48, G49 on the proximal monomer and R72 on the distal partner. The progressive breaking of the initial symmetry is already apparent here, the distal K48–G49/proximal R72 links being retained until much later in the separation process. The lost contacts are progressively replaced by direct electrostatic interactions between E51 on the distal and D39–Q40 on the proximal monomers, as well as a network of water-mediated interactions which mainly form from 6 to 7.5 Å. The last regime involves further separation of the monomers, with a few enduring, long-range, transiently water-mediated electrostatic interactions involving R42 and R72 generating a much more gradual, almost linear increase in free energy. The free energy barrier at 11.7 Å coincides with the destruction of the alternate electrostatic interface and its replacement by a distal E51-proximal R42 salt bridge. The overall free energy variation associated with the opening of the ubiquitin dimer can be approximated at 18.3 kcal mol−1. I compared representative structures of the open conformation of the complex (at a separation of around 12.7 Å) to the NMR structure proposed by Hirano13 (Fig. 7). These two conformations appear quite similar, although a small difference in the relative rotation of the monomers raises the RMSD to a value of ∼7 Å. However, after 3 ns of free simulation the open complex relaxed to 3.5 Å RMSD from the open NMR structure and remained unchanged for the 10 ns over which it was further simulated. The open structure of the dimer thus appears at least kinetically stable, in no small part due to a loose, water-mediated, alternate interface between monomers.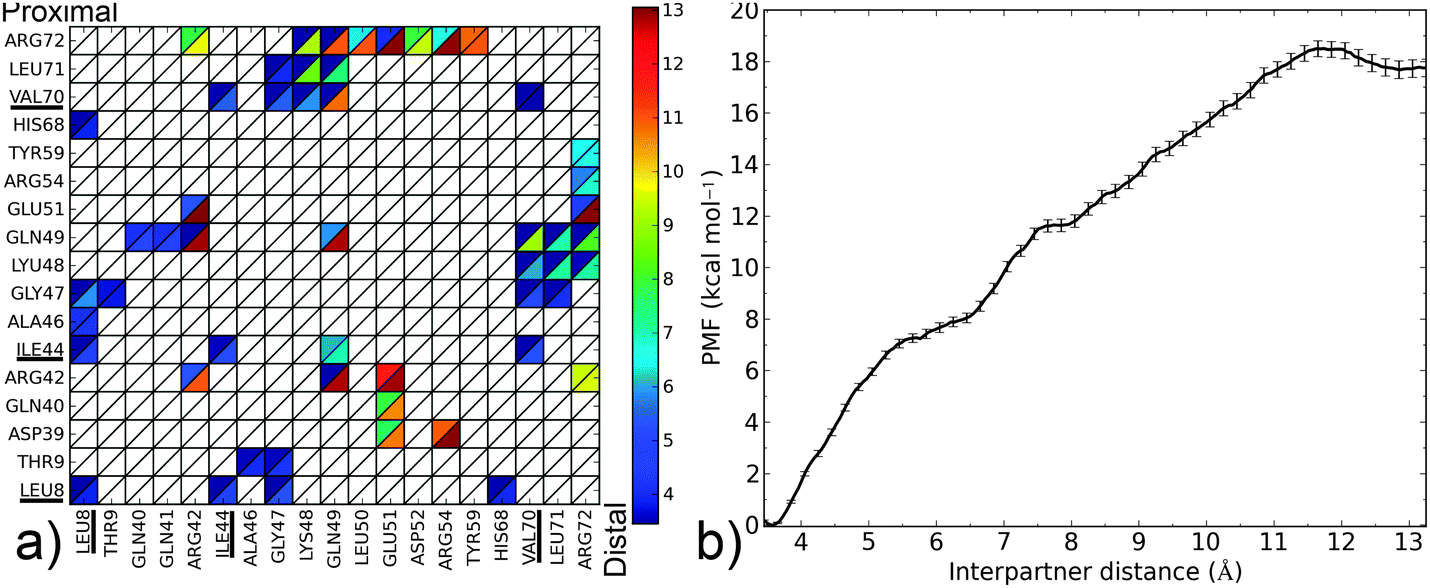 | ||
| Fig. 6 (a) Contact map for the Ubq2 complex (see the Fig. 3 caption for details); the Lys48 residue on the proximal monomer participating in the isopeptide linkage is denoted LYU. (b) PMF as a function of interpartner separation for this complex. | ||
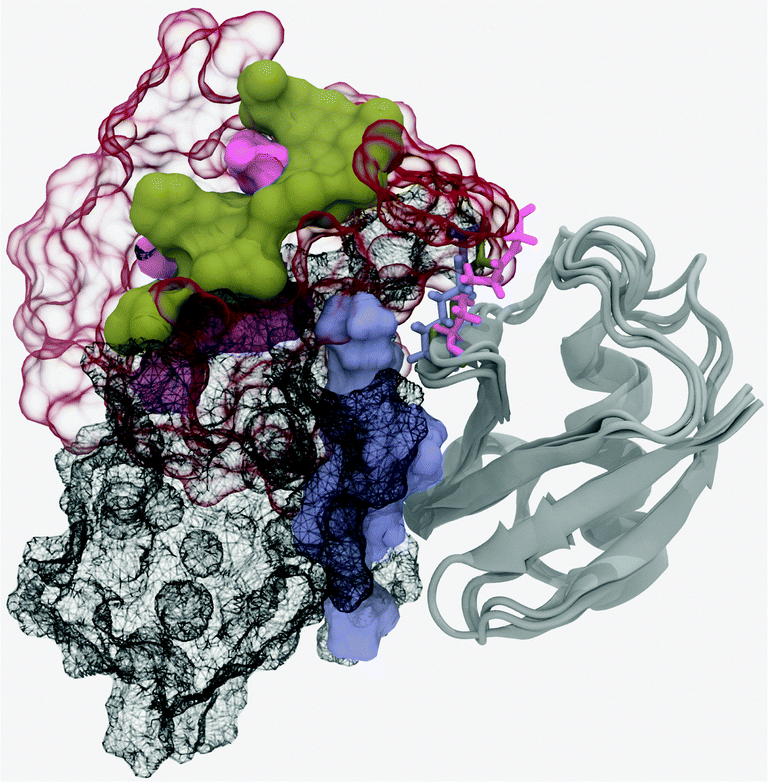 | ||
| Fig. 7 Structural comparison of the open and closed forms of Ubq2. Alignment was performed on the proximal Ubq monomer (grey cartoons). In the closed form, the distal monomer (black wireframe surface) is positioned such that its hydrophobic interface residues (blue surface) contact their counterparts on the proximal monomer. In the experimental open structure, the distal monomer (transparent red surface) has rotated and its interface residues (brown surface) are accessible. The simulated open conformation (interface shown only, as pink surface) is very similar. The connecting Lys48 residue is shown as sticks. | ||
The restriction of the separation restraint's action to a limited subset of atoms on each monomer (the canonical interface) may be seen as a bias, but was deemed necessary to allow the formation of an alternate stabilizing interface whose existence could be inferred from the open NMR structure, and which would have been precluded by the inclusion of all heavy atoms of both monomers in the set of biased atoms. For purposes of comparison, this latter approach was nevertheless attempted (see Fig. 8 and Fig. S2 of ESI† for details). Interestingly, the opening mechanism proved to remain comparable to the one described previously: although the restraint would be most straightforwardly accommodated by a simple translation motion of the monomers away from each other, this motion was found to be coupled to the same relative rotation of monomers encountered in the previous case. The Ubq moieties in the open complex were found to be oriented similar to their counterparts in the previously simulated and experimental structures, but with a higher inter-monomer distance, which a subsequent short unbiased simulation proved sufficient to relax. These findings confirm that the rotation of one monomer relative to the other is the energetically preferred route. The open conformation obtained by biasing the interface residues is only stabilized by 4 kcal mol−1 compared to the fully separated monomers, due to the abrupt initial breaking of the Ubq2 interface in the latter case vs. the progressive replacement of initial contacts by alternate ones in the former. However, the fully open state of diubiquitin with the extended connecting loop is entropically favored, and can diffuse freely over a large volume of conformational space in which the free energy plateaus, reminiscent of the unfolded state of a protein.
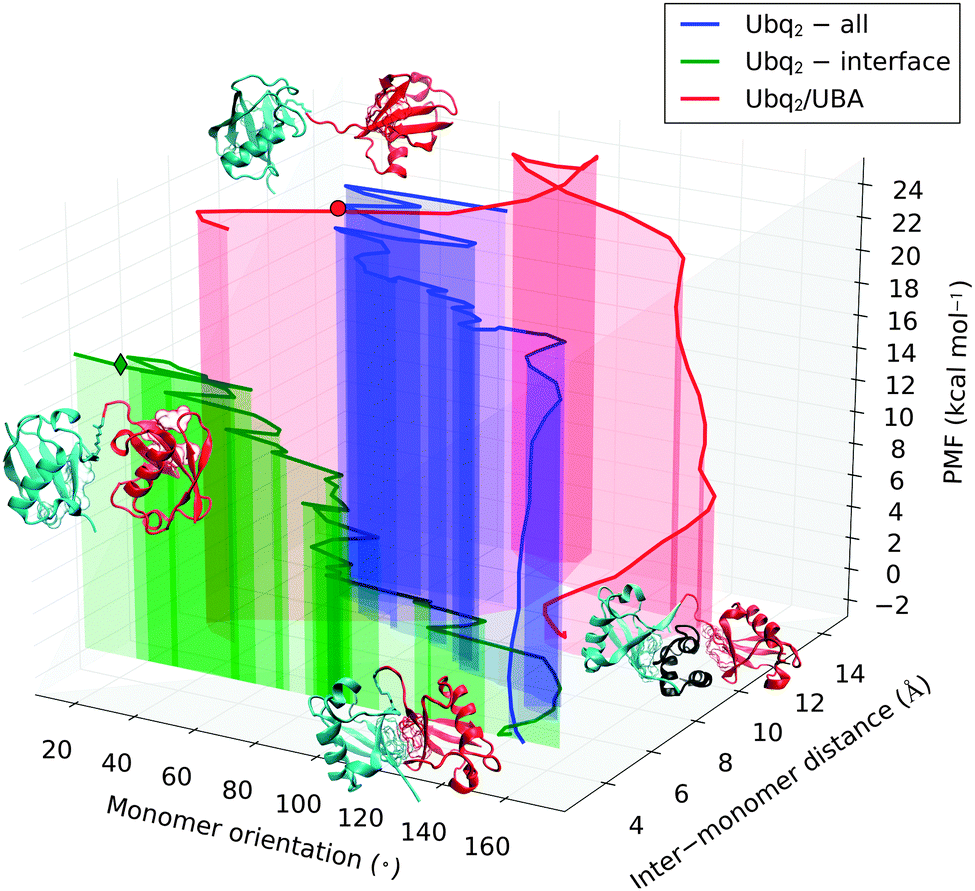 | ||
| Fig. 8 Representation of the dissociation pathways of Ubq2 (green: acting on interface residues only; blue: acting on all residues) and Ubq2–UBA (red) in the conformational subspace spanned by relative monomer orientation, inter-monomer distance, and potential of mean force. The green diamond denotes the experimentally observed open Ubq2 conformation, while the red dot identifies the crossover point between the Ubq2 and Ubq2–UBA dissociation pathway. These structures, as well as the bound Ubq2 and Ubq2–UBA complexes, are also represented as cartoons. | ||
According to these simulations, the free energy cost of monomer separation would appear to preclude any spontaneous opening events without the help of an external factor. This could take the form of a local pH change, triggered for instance by the proximity to acidic membrane components, which would favor histidine protonation and induce same-charge repulsion between H68 on both monomers, in close proximity in the closed complex. The mutation of H68 to a smaller residue has been experimentally shown to favorably impact the binding of Ubq to UBA,44 proving the residue's capacity to impact the Ubq2vs. Ubq2–UBA equilibrium. A more recent NMR study has confirmed this by showing that the H68V mutation of both Ubq monomers did indeed promote the closed Ubq2 conformation over the open one.13 Both these studies, however, measure the pKa of H68 in the subunits of Ubq2 at a low value of 5.5, similar to monomeric Ubq: majorly altering the residue's protonation state would require a drastic change in local pH. The impact of other titratable groups (K6, R42, K48, R72) acting collectively with H68 has been mentioned13 but remains unproven. Indeed, the repulsion between protonated H68 residues as the sole governing mechanism is rendered more unlikely by my observation that both H68 residues only come in close proximity at the very end of the binding process. Interaction with another enzyme might also be responsible for the facilitated opening of closed-form Ubq2. In any case, the simulations presented in this work show that the open conformations can be stabilized either enthalpically by the formation of an alternate Ubq2 interface, or entropically because of the length of the linker joining the monomers.
UBA release mechanism
The previously simulated separation of Lys48-linked Ubq monomers is a preliminary step in the recognition of polyUbq-tagged proteins by proteasome UBA domains, which involves the formation of complexes where UBA is sandwiched between two Ubq monomers. To measure the stability of this structure relative to the open and closed forms of Ubq2, I performed unbinding simulations from the crystal structure of Ubq2 in complex with the C-terminal UBA domain of HHR23A. The restraint operates on all heavy atoms of both the distal and proximal Ubq monomers, imposing that they simultaneously separate from UBA; as a consequence, I was unable to probe a possible sequential binding–unbinding of both monomers. However, the restraint does not prevent the Ubq moieties from getting closer once the UBA domain has been released and should not affect the final value of the binding free energy.Considered individually, the separation of UBA from each monomer in Ubq2 was found to closely resemble the unbinding pathway of UBA and the corresponding isolated monomer (see Fig. S3 in ESI† for details). Fig. 8 represents the separation pathways of Ubq2 and Ubq2–UBA on cross-sections of their conformational landscapes spanned by the minimum distance between monomer heavy atoms and the relative orientation of Ubq monomers (defined as the angle between the normal vectors to the planes of the hydrophobic patches on both monomers). As previously discussed, when acting on the interface residues only, Ubq2 separates by relative rotation of the two moieties with little modification of their separation distance; on the other hand, involving all heavy atoms in the constraint couples this rotation with the translation of monomers away from one another, at a higher free energy cost because of the impossibility to form a stabilizing alternate interface between monomers. By comparison, the separation of UBA from Ubq2 proceeds sequentially: an initial translation of the Ubq monomers partially frees UBA, which is then expelled by the concerted rotation of Ubq monomers. This rotation destroys favorable Ubq–UBA contacts and is associated with the most important free energy penalty. It is followed by a diffusive translation/rotation motion of the monomers, incurring almost no free energy cost, that brings the system into the region of conformational space sampled at the end of the Ubq2 opening pathway. Matching the PMFs of both pathways at this point results in the Ubq2–UBA complex being less than 2 kcal mol−1 stabler than closed-form Ubq2. This explains the experimental coexistence of both species, at least if thermodynamic equilibrium can be achieved.
Role of water
Water is known to play an important role for protein–protein recognition.45 This led me to investigate the role of water during the separation of all Ubq-containing complexes discussed previously, by considering the statistics of the water molecules acting as bridges between the interface of the partners (see Methods for details). The proportion of water-mediated contacts that are present in all analyzed snapshots (separated by a 10 ps stride) at any separation distance quantifies the degree of ordering that is imposed by the protein partners upon the water molecules sandwiched between them. The maximum separation distance at which such immobilized bridging waters are found is a measure of the range of the water-mediated recognition process. I relate these measures of water mobility to the areas of the hydrated interface patches on each partner and their amino acid composition (Fig. 9). Considering the rather similar chemical natures of the interfaces under study, the variety in the behavior of water is quite interesting.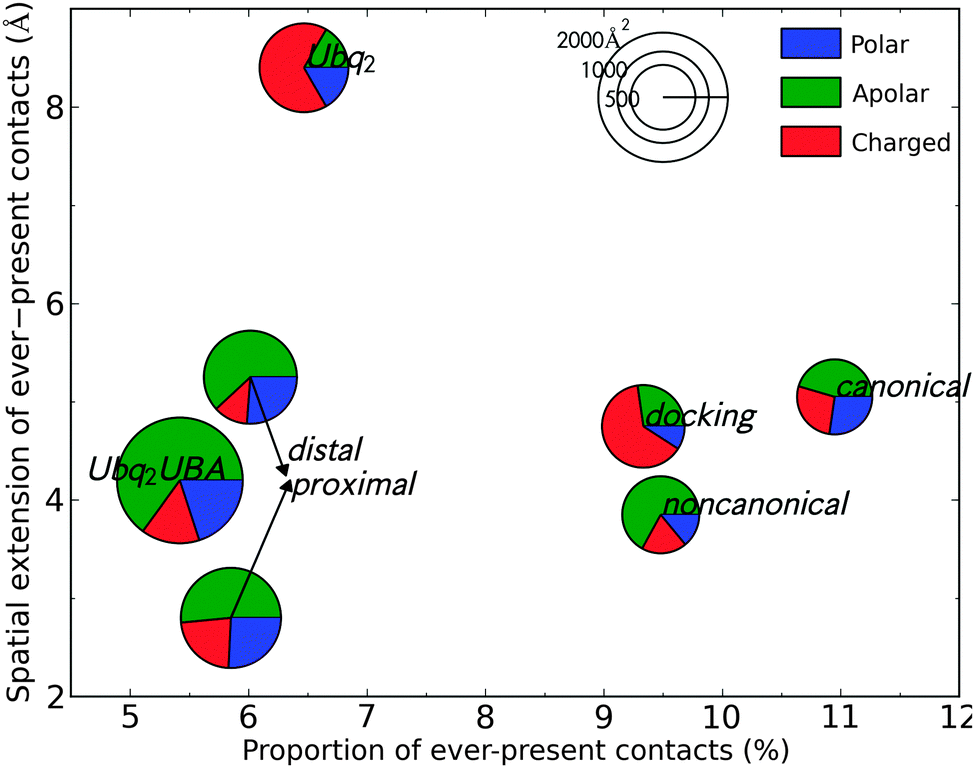 | ||
| Fig. 9 Statistics of water ordering (abscissa), reach of water-mediated interactions (ordinate), wet interface area (pie chart diameter) and composition (pie chart sectors) for the complexes under study; see the text for details. | ||
Despite a rather large hydrated interface, Ubq2–UBA and its distal/proximal monomers individually complexed to UBA feature a few long-lasting water-mediated contacts. These mainly involve hydrophobic residues and trapped solvent molecules which promptly move away as the inter-monomer distance is increased above 4 Å. Ubq2, on the other hand, features opposing arginine residues on both monomers which immobilize nearby water molecules, enabling durable water-mediated intermonomer contacts which are also favored by the presence of additional arginines on the inter-monomer loop. The proportion of durable contacts is not significantly larger than in Ubq2–UBA, but they endure much longer interpartner distances. The proximal-type pose obtained from docking simulations also features a network of charged, water-ordering arginines and glutamines, but most of the corresponding water bridges break earlier on due to relative rotation of the monomers. The strongest structuration of interface water occurs for the canonical Ubq–UBA complex, despite a hydrated interface of mixed polar–apolar-charged character. Ordered water molecules bridge the gap that forms between monomers when UBA rotates around the L8–L46 contact; thus, more than 10% of water-mediated contacts are permanent until relatively large separation distances (>5 Å). Finally, the noncanonical complex has a small, mostly apolar hydrated interface in which water ordering is lost early on (3.8 Å).
The area of the hydrated interface appears to correlate inversely with the proportion of ever-present contacts, implying that a limited number of hotspot residues have a more important effect on the ordering of water than the sandwiching of a large slab of solvent by random amino acids: this hints at a discriminative role of water in the long-range recognition between Ubq and its diverse binding partners. Polar and charged residues seem to be the most likely to give rise to such specific water-mediated contacts, especially at long range, via hydrogen bonding and induced electrostatics. The importance of such contacts is also apparent in the stable open form of Ubq2, as obtained from separation simulations biased on interface residues (not shown in Fig. 9): more than 25% of all water-mediated contacts in this complex (of which 78% involve charged residues) are permanent. However, hydrophobic patches can also contribute to the rigidification of the hydration layer in their vicinity, as was previously stated;46 this happens in the canonical complex.
The presence of strong water-mediated interactions at intermediate separation distances stabilizes intermediate structures but adds a desolvation barrier to the formation of the complex. When the corresponding ‘dry’ interface is not large or specific enough to compensate for this, the resulting free energy profile displays intermediate plateaus or secondary barriers, slowing down conformational diffusion as seen in protein folding.
Discussion
Interface plasticity and binding
Ubiquitin's marked affinity for a variety of binders is linked to the particular flexible/rigid duality of its interface. Most interface residues are borne by a rigid scaffold of two antiparallel β-sheet domains, forming a β-α-β motif ensuring robust specific recognition. Plasticity is introduced by the relative positioning of both sheets, which affects the positions of residues 44–46 with respect to residues 68–71, and by the flexible β1–β2 loop, bearing the important L8 residue, which is involved in a pincer-like collective motion. This correlated motion powers a conformational selection mechanism, enhancing the correlated motion of interface amino acids observed in isolated Ubq and lowering entropic barriers to binding.47 To measure the relative influence of this effect in the complexes under study, I performed Cartesian principal component analyses (PCA) on the structures of Ubq encountered at the end of all studied unbinding pathways, and quantified the participation of interface residues to the correlated motion (see Methods). As a reference, I used the results of a similar analysis of a 15 ns simulation of isolated Ubq (Fig. 10). As can be seen, the residual correlation in interface residues after separation is strongest in the canonical case, followed by both monomers of Ubq2 (whether originally bound to UBA or not), the proximal-type docking solution and the isolated distal monomer (all in close proximity). The lowest correlation occurs for isolated Ubq and the proximal-type binding of mono-Ubq. The observed discrepancy in the behavior of proximal structures is interesting: it could explain the absence of experimentally observed proximal-type mono-Ubq complexes, due to the entropic cost of all interface amino acids assuming the correct relative orientation without the help of correlated motion. In contrast, long-range interactions with the distal monomer in Ubq2 enhance correlations in the proximal monomer interface, facilitating binding; indeed, the proximal monomer in Ubq2–UBA has been experimentally found to be more tightly bound to UBA than its distal counterpart,38,48 in good agreement with the free energy calculations presented here. This high affinity for the proximal monomer of diubiquitin has also been invoked as a reason for the preferential binding of numerous UBA domains to Ubq2 rather than to isolated Ubq.48 Conversely, the dimerization state seems to have no effect on the plasticity of the distal monomer interface which systematically features important correlated motions, facilitating binding (particularly for the distal-like canonical complex); indeed, in stark contrast to proximal-type structures, all distal-like structures discussed in this work have been experimentally observed. Unlike the weak residual correlation induced by UBA in the proximal binding scenario, the correlation of Ubq interface residues does not need to be ‘amplified’ by an additional interaction with another Ubq monomer in the distal case. Finally, the breaking of the initial symmetry between the subunits of Ubq2 upon dissociation (discussed above) is apparent in the collective motion of their interface residues: the longer-lasting contacts via the patch centered around V70 on the proximal monomer result in a marked residual correlation in the latter compared to the distal moiety.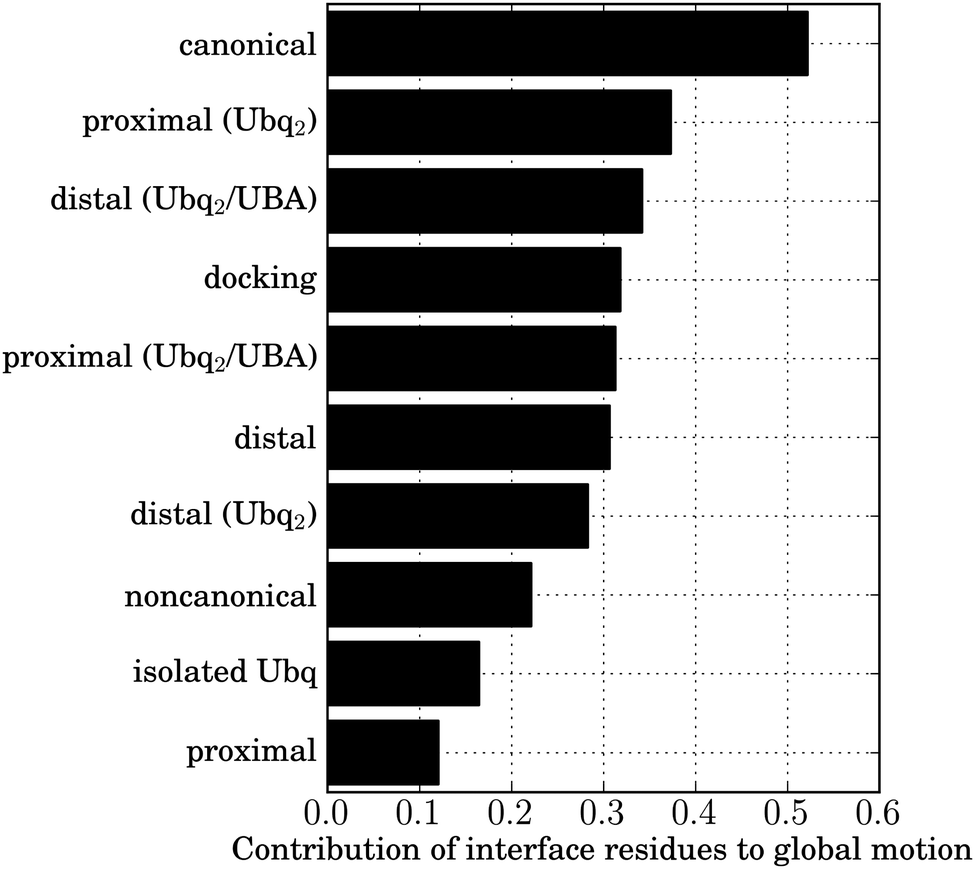 | ||
| Fig. 10 Normalized contribution of the backbone atoms of the canonical interface residues (L8, I44, A46, H68, V70, L71) to the correlated motion of Ubq in the dissociated state of each of the complexes under study. | ||
The existence of entropic barriers could also explain the high values found for the binding free energies of some of the complexes encountered here (proximal Ubq–UBA for instance). In our separation restraint methodology, exhaustive sampling of accessible conformational space becomes more difficult as the separation distance increases – see ESI† for an assessment of the relevance of this effect using WHAM consistency tests. For the complexes under study, plateaus in the computed free energy profiles attest the absence of inter-monomer interactions at the maximum simulated separation, but the differences in interface plasticity compared to isolated Ubq show that complete relaxation of the monomers has not yet been achieved; hence, the endpoints of the free energy profiles could correspond to the top of a broad entropic barrier rather than to the fully dissociated state. This would in turn modulate the binding free energy, destabilizing the bound state for high-barrier, low-correlation cases and providing further arguments for the absence of experimentally detected proximal-type Ubq–UBA complexes despite their marked stability predicted both by docking and all-atom MD simulations.
As a final remark, water-mediated interactions assuredly play an active part in the tuning of the proximal monomer interface plasticity in Ubq2: indeed, the present results show that the water layer separating both monomers in the open-form Ubq2 is highly organized, unlike for the isolated monomers.
Origins of the specificity of UBA for polyUbq and K48 linkage
The proper recognition of Ubq signaling pathways often necessitates a minimum length for the associated polyubiquitin chain;49,50 for instance, two K48-diubiquitin units are required for efficient recognition by the proteasome.5 Such cooperative effects are probably necessary to amplify the small difference in free energy between closed-form and UBA-bound Ubq2, which the free energy calculations I report here estimate at less than 2 kcal mol−1. The specificity for K48-linked diubiquitin motifs stems from the opposition of the two hydrophobic patches which only this type of linkage permits,51 but also from the previously discussed marked affinity of UBA for the proximal Ubq monomer. However, when bound to Ubq2, UBA also forms an interface with the positively charged linker segment via direct and water-mediated contacts to asparagine and serine moieties, which could be involved in the recognition of the correct linkage type. Finally, the UBA motif was found in the present simulations to be highly rigid despite its small size, exhibiting negligible deformations upon Ubq binding or unbinding. This is in line with experimental observations, and has been proposed as an explanation of the capacity of UBA domains to orient the synthesis of polyUbq by exerting selectivity over the linkage type.52 However, evidence that the p62 UBA domain can change conformation upon Ubq binding53 shows that the diversity of UBA–Ubq recognition is still far from completely understood.Specifically binding multiple ligands
The unbinding pathways simulated here, and therefore presumably their binding counterparts, have been shown to comprise recurring phases despite the marked differences in the corresponding UBA sequences. For instance, the binding mechanisms of noncanonical and canonical complexes are identical for large separation distances, involving the interaction of similarly-placed hydrophobic residues on UBA with amino acids L8, I44 and V70 on Ubq, but differ at short range when the interactions through L8 in the noncanonical complex is broken in favor of alternate electrostatic contacts. Conversely, the binding pathways of both proximal-type complexes differ at long range: UBA is locked by the simultaneous interaction of L8, I44 and V70 with multiple residues in one case, whereas it can rotate around the single L8–V70/F42 contact in the other. At close range, however, both complexes follow a similar pathway involving the formation of R/E or R/D salt bridges. This separation of binding pathways into two distance regimes translates into the bimodal free energy profiles which were observed for most complexes under study, with varying ratios of short-range over long-range free energy stabilizations (though the latter usually dominates). The turning point between the two regimes often involves modifications in the binding behavior of residues L8 and V70, which belong to the flexible 7–11 and C-terminal loops and thus have the necessary latitude to modulate the binding affinity of the hydrophobic interface core. However, these hydrophobic interactions are only able to provide limited specificity through geometrical restrictions, unlike hydrogen- or salt bonds through peripheral residues Q49, H68 and R72 which can impose much stricter donor–acceptor geometries. The outcome of the competition between these two types of interaction for the imposition of specificity depends on individual UBA sequences. This competition, which predominantly occurs at close range, explains the ability of Ubq to bind multiple UBA domains of low sequence homology. From the present work, a trend emerges as to the importance of electrostatic interactions for proximal-type complexes; local changes in pH could be used in the cell to disturb these interactions, allowing the fine tuning of Ubq vs. Ubq2 recognition by favoring either the distal or the proximal binding pathways, respectively.Concluding remarks
Ubiquitin recognition is an intricate mechanism which is progressively being revealed, in a piecewise fashion, by intense ongoing research efforts. This work contributes to the overall understanding of the process by applying state-of-the-art free energy methods to a number of key questions. First among these is the relative stability of closed vs. open Ubq2, in the context of recent apparent experimental contradictions; I find the closed state to be very much favored at physiological pH values, but propose a mechanism by which the free energy penalty to opening can be minimized, leading to a stable, well-defined semi-open state which has also been experimentally observed. Secondly, I show that the equilibrium between closed Ubq2 and Ubq2–UBA is only very slightly displaced in favor of the latter, which I relate to the experimental necessity of cooperatively binding two or more K48-linked Ubq2 units for efficient recognition to occur. Finally, I relate the surprising stability of proximal-type Ubq–UBA complexes, observed both using docking and accurate explicit-solvent MD simulations, to the experimentally known ability of most UBA domains to discriminately bind Ubq2 over Ubq. Finally, I explore the mechanisms of Ubq binding by different UBA domains and suggest common trends that can be used to loosely categorize them, although there is a clear need for more inputs on the mechanistical side.K48-linked polyUbq on which I focused here is but one of the signal types recognized by UBA domains, which are also employed to bind K63-connected Ubq chains;12 unlike their K48 counterparts, these favor open conformations of noninteracting Ubq monomers.54 Molecular modeling work is currently underway to fit this new piece into the recognition puzzle.
Acknowledgements
I wish to thank Richard Lavery and Juliette Martin for fruitful discussions and constructive criticism. The financial support of Agence Nationale de la Recherche (MAPPING/ANR Investissement d'Avenir Bioinformatique grant ANR-11-BINF-0003) is gratefully acknowledged. Calculations presented in this work were performed using HPC resources from GENCI-CINES (grant 2012-c201276814).References
- S. E. Kaiser, B. E. Riley, T. A. Shaler, R. S. Trevino, C. H. Becker, H. Schulman and R. R. Kopito, Nat. Methods, 2011, 8, 691 CrossRef CAS PubMed
.
- K. Husnjak and I. Dikic, Annu. Rev. Biochem., 2012, 81, 291 CrossRef CAS PubMed
- E. R. Strieter and D. A. Korasick, ACS Chem. Biol., 2012, 7, 52 CrossRef CAS PubMed
- C. M. Pickart and M. J. Eddins, Biochim. Biophys. Acta, 2004, 1695, 55 CrossRef CAS PubMed
- J. S. Thrower, L. Hoffman, M. Rechsteiner and C. M. Pickart, EMBO J., 2000, 19, 94 CrossRef CAS PubMed
- J. F. Trempe, Curr. Opin. Struct. Biol., 2011, 21, 792 CrossRef CAS PubMed
- I. Dikic, S. Wakatsuki and K. J. Walters, Nat. Rev. Mol. Cell Biol., 2009, 10, 659 CrossRef CAS PubMed
- J. F. Trempe, N. R. Brown, M. E. Noble and J. A. Endicott, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 994 CrossRef CAS PubMed
- W. J. Cook, L. C. Jeffrey, M. Carson, Z. Chen and C. M. Pickart, J. Biol. Chem., 1992, 267, 16467 CAS
- R. Varadan, O. Walker, C. Pickart and D. Fushman, J. Mol. Biol., 2002, 324, 637 CrossRef CAS
- Y. E. Ryabov and D. Fushman, J. Am. Chem. Soc., 2007, 129, 3315 CrossRef CAS PubMed
- J. H. Hurley, S. Lee and G. Prag, Biochem. J., 2006, 399, 361 CAS
- T. Hirano, O. Serve, M. Yagi-Utsumi, E. Takemoto, T. Hiromoto, T. Satoh, T. Mizushima and K. Kato, J. Biol. Chem., 2011, 286, 37496 CrossRef CAS PubMed
- M. Wilhelm, A. Mukherjee, B. Bouvier, K. Zakrzewska, J. T. Hynes and R. Lavery, J. Am. Chem. Soc., 2012, 134, 8588 CrossRef CAS PubMed
- B. Bouvier and R. Lavery, J. Am. Chem. Soc., 2009, 131, 9864 CrossRef CAS PubMed
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781 CrossRef CAS PubMed
- T. E. Cheatham, P. Cieplak and P. A. Kollman, J. Biomol. Struct. Dyn., 1999, 16, 845 CAS
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS
- I. S. Joung and T. E. Cheatham, J. Phys. Chem. B, 2008, 112, 9020 CrossRef CAS PubMed
-
A. T. Brünger, X-PLOR: A System for X-ray Crystallography and NMR, Yale University Press, New Haven, CT, USA, 1993 Search PubMed
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177 CrossRef CAS
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011 CrossRef CAS
- F. Zhu and G. Hummer, J. Comput. Chem., 2012, 33, 453 CrossRef CAS PubMed
- B. Bouvier, R. Grünberg, M. Nilges and F. Cazals, Proteins, 2009, 76, 677 CrossRef CAS PubMed
- F. Cazals, F. Proust, R. P. Bahadur and J. Janin, Protein Sci., 2006, 15, 2082 CrossRef CAS PubMed
- N. M. Glykos, J. Comput. Chem., 2006, 27, 1765 CrossRef CAS PubMed
- S. R. Comeau, D. W. Gatchell, S. Vajda and C. J. Camacho, Nucleic Acids Res., 2004, 32, W96 CrossRef CAS PubMed
- D. Kozakov, D. R. Hall, D. Beglov, R. Brenke, S. R. Comeau, Y. Shen, K. Li, J. Zheng, P. Vakili, I. C. h. Paschalidis and S. Vajda, Proteins, 2010, 78, 3124 CrossRef CAS PubMed
- H. Hwang, T. Vreven, J. Janin and Z. Weng, Proteins, 2010, 78, 3111 CrossRef CAS PubMed
-
T. Heijboer, WxTernary, 2012 Search PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33 CrossRef CAS
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90 CrossRef
- A. Ohno, J. Jee, K. Fujiwara, T. Tenno, N. Goda, H. Tochio, H. Kobayashi, H. Hiroaki and M. Shirakawa, Structure, 2005, 13, 521 CrossRef CAS PubMed
- G. Kozlov, L. Nguyen, T. Lin, G. De Crescenzo, M. Park and K. Gehring, J. Biol. Chem., 2007, 282, 35787 CrossRef CAS PubMed
- S. Raasi, R. Varadan, D. Fushman and C. M. Pickart, Nat. Struct. Mol. Biol., 2005, 12, 708 CAS
- Y. G. Chang, A. X. Song, Y. G. Gao, Y. H. Shi, X. J. Lin, X. T. Cao, D. H. Lin and H. Y. Hu, Protein Sci., 2006, 15, 1248 CrossRef CAS PubMed
- R. Varadan, M. Assfalg, S. Raasi, C. Pickart and D. Fushman, Mol. Cell, 2005, 18, 687 CrossRef CAS PubMed
- P. Peschard, G. Kozlov, T. Lin, I. A. Mirza, A. M. Berghuis, S. Lipkowitz, M. Park and K. Gehring, Mol. Cell, 2007, 27, 474 CrossRef CAS PubMed
- N. Chim, W. E. Gall, J. Xiao, M. P. Harris, T. R. Graham and A. M. Krezel, Proteins, 2004, 54, 784 CrossRef CAS PubMed
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Bioinformatics, 2007, 23, 2947 CrossRef CAS PubMed
- B. Bouvier, K. Zakrzewska and R. Lavery, Angew. Chem., Int. Ed., 2011, 50, 6516 CrossRef CAS PubMed
- L. Hicke, H. L. Schubert and C. P. Hill, Nat. Rev. Mol. Cell Biol., 2005, 6, 610 CrossRef CAS PubMed
- K. Fujiwara, T. Tenno, K. Sugasawa, J. G. Jee, I. Ohki, C. Kojima, H. Tochio, H. Hiroaki, F. Hanaoka and M. Shirakawa, J. Biol. Chem., 2004, 279, 4760 CrossRef CAS PubMed
- Y. Levy and J. N. Onuchic, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 389 CrossRef CAS PubMed
- H. Xu and B. J. Berne, J. Phys. Chem. B, 2001, 105, 11929 CrossRef CAS
- O. F. Lange, N. A. Lakomek, C. Farès, G. F. Schröder, K. F. A. Walter, S. Becker, J. Meiler, H. Grubmüller, C. Griesinger and B. L. de Groot, Science, 2008, 320, 1471 CrossRef CAS PubMed
- D. Zhang, S. Raasi and D. Fushman, J. Mol. Biol., 2008, 377, 162 CrossRef CAS PubMed
- S. Raasi and C. M. Pickart, J. Biol. Chem., 2003, 278, 8951 CrossRef CAS PubMed
- S. Raasi, I. Orlov, K. G. Fleming and C. M. Pickart, J. Mol. Biol., 2004, 341, 1367 CrossRef CAS PubMed
- J. F. Trempe, N. R. Brown, E. D. Lowe, C. Gordon, I. D. Campbell, M. E. Noble and J. A. Endicott, EMBO J., 2005, 24, 3178 CrossRef CAS PubMed
- R. C. Wilson, S. P. Edmondson, J. W. Flatt, K. Helms and P. D. Twigg, Biochem. Biophys. Res. Commun., 2011, 405, 662 CrossRef CAS PubMed
- J. Long, T. R. Gallagher, J. R. Cavey, P. W. Sheppard, S. H. Ralston, R. Layfield and M. S. Searle, J. Biol. Chem., 2008, 283, 5427 CrossRef CAS PubMed
- D. Komander, F. Reyes-Turcu, J. D. Licchesi, P. Odenwaelder, K. D. Wilkinson and D. Barford, EMBO Rep., 2009, 10, 466 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Complementary analyses and validation of separation methodology (umbrella sampling consistency analyses, evaluation of residual translational and rotational entropy losses at maximum simulated complex separations); PMF for the opening of Ubq2 with bias on all heavy atoms; contact map and PMF for the dissociation of HHR23A UBA from Ubq2; multi-frame PDB files containing representative conformations along the dissociation pathways for all complexes under study. See DOI: 10.1039/c3cp52436a |
| This journal is © the Owner Societies 2014 |