Electronically excited states and photochemical reaction mechanisms of β-glucose†
Deniz Tuna*a,
Andrzej L. Sobolewskib and
Wolfgang Domckea
aDepartment of Chemistry, Technische Universität München, 85747 Garching, Germany. E-mail: deniz.tuna@ch.tum.de; Fax: +49 8928913622; Tel: +49 8928913610
bInstitute of Physics, Polish Academy of Sciences, 02668 Warsaw, Poland
First published on 20th August 2013
Abstract
Carbohydrates are important molecular components of living matter. While spectroscopic and computational studies have been performed on carbohydrates in the electronic ground state, the lack of a chromophore complicates the elucidation of the excited-state properties and the photochemistry of this class of compounds. Herein, we report on the first computational investigation of the singlet photochemistry of β-glucose. It is shown that low-lying singlet excited states are of nσ* nature. Our computations of the singlet vertical excitation energies predict absorption from 6.0 eV onward. Owing to a dense manifold of weakly-absorbing states, a sizable and broad absorption in the ultraviolet-C range arises. We have explored two types of photochemical reaction mechanisms: hydrogen-detachment processes for each of the five O–H groups and a C–O ring-opening process. Both types of reactions are driven by repulsive nσ* states that are readily accessible from the Franck–Condon region and lead to conical intersections in a barrierless fashion. We have optimized the geometries of the conical intersections involved in these photochemical processes and found that these intersections are located around 5.0 eV for the O–H hydrogen-detachment reactions and around 4.0 eV for the C–O ring-opening reaction. The energies of all conical intersections are well below the computed absorption edge. The calculations were performed using linear-response methods for the computation of the vertical excitation energies and multiconfigurational methods for the optimization of conical intersections and the computation of energy profiles.
1 Introduction
Glucose is a constituent of many forms of biological matter found in nature, for example, sucrose (table sugar), lactose (milk sugar), amylose (starch), and cellulose, the biopolymer found in the cell walls of green plants and the most abundant organic material on earth. Carbohydrates (also known as saccharides or sugars) fulfill a multitude of biological functions. They are part of nucleic acids (DNA and RNA), glycoproteins, proteoglycans, glycolipids, glycoside hydrolases, and glycosyltransferases.1For decades, carbohydrates have been the focus of many experimental, spectroscopic, and computational investigations. Studies include the exploration of the conformational space and structural properties of mono-,2–12 di-,13–16 oligo-, and polysaccharides,17–20 chemical reactions (condensation and isomerization,21–26 hydrogen abstraction27 and (photo-)fragmentation28–31) and the computation of vibrational spectra.32–34 Progress has also been made in the computational simulation of the structure of biopolymers such as cellulose35,36 as well as computations of their physical and chemical properties.37–39
Computational and experimental studies of carbohydrates were reviewed by Imberty and Péres.40 A review on joint spectroscopic and computational studies of carbohydrates in the gas phase was given by Simons et al.,41 and a review on the merits of using quantum chemistry to investigate various aspects of carbohydrate chemistry by da Silva.42 Considerable efforts were put into the development and evaluation of suitable force-field,43,44 semi-empirical,45 and ab initio methods46–48 for the study of carbohydrates.
Despite a number of computational studies published to date, we are not aware of any computational study on the excited states and photochemical reaction mechanisms of carbohydrates. Herein, we report on results for the singlet vertical excitation energies, the absorption spectrum and photochemical reaction mechanisms of β-glucose (more precisely, β-D-glucopyranose), which we chose as a representative of this huge class of biomolecules. The present investigation is the first of its kind for a carbohydrate. Previous research on the possible conformations of β-glucose in the ground state2–9 was helpful for the present work.
The study of the photochemical and photophysical properties of the “molecules of life”—bioorganic molecules that constitute living matter—has been a very active and fruitful field of research for more than a decade. The combination of gentle laser evaporation of biomolecules with sophisticated double-resonance spectroscopic techniques in supersonic jets or molecular traps allows highly precise spectroscopic investigations of biomolecules with aromatic chromophores, for example, the DNA bases and the aromatic amino acids.49–52 The electronic and vibrational spectra can be assigned to specific conformers or tautomers of these flexible biomolecules by matching the experimental spectra to first-principles calculations.52 Extensive spectroscopic and computational studies have been performed for the five nucleobases53–57 and the nucleosides and nucleotides,50,53,57 as well as hydrogen-bonded base pairs.58,59 The determination of the conformational preferences of aromatic amino acids and of peptides with aromatic chromophores is of relevance for the understanding of protein folding.60–62 While excited-state lifetimes of biomolecules or supramolecular complexes could be measured only in exceptional cases,63 the excited-state lifetime is implicit in the observed spectra. A sufficiently long excited-state lifetime is required for the detection of resonant two-photon ionization or laser-induced fluorescence. The fact that the spectra of the energetically most stable conformers or tautomers are often not observed is a clear indication that ultrafast (sub-picosecond) radiationless excited-state deactivation processes prevail.50–53,63 It has been argued that these ultrafast excited-state quenching processes provide biological matter with a particularly high degree of UV photostability.52,53,64–66
Carbohydrates, like non-aromatic amino acids, lack a chromophore and therefore do not exhibit sharp, intense and sparse UV spectra suitable for double-resonance spectroscopy. Simons and co-workers tagged carbohydrates with a phenyl chromophore, which allowed them to obtain structural information on monomeric and oligomeric carbohydrates in the gas phase.10,14,41 Owing to their flexible nature and the lack of any symmetry, carbohydrates also are a challenge for computational investigations. For these reasons, very little is known on the mechanistic details of the photochemistry of even the simplest carbohydrates. In the present work, we aim at closing this gap of knowledge by computational investigations of the photochemistry of glucose.
It is a widely accepted paradigm that photochemical reactions are governed by critical points on the excited-state potential-energy surfaces, so-called conical intersections or photochemical funnels.67,68 These photochemical funnels are believed to be of comparable significance for excited-state chemistry as are transition states for ground-state chemistry.67,69 The unique features of conical intersections are a complete breakdown of the Born–Oppenheimer approximation and an exceptionally strong local anharmonicity of the potential-energy surfaces, which leads to a very efficient energy exchange between vibrational degrees of freedom.68 The conical intersection can act as a funnel that directs the photochemical reaction to specific photoproducts.70 The analysis of the topography of the lower adiabatic energy surface near a conical intersection can provide qualitative predictions of the relative yields of photoproducts. Photochemical reactions that are aborted at conical intersections of the S1 and S0 energy surfaces are a very efficient mechanism for ultrafast internal conversion, that is, recovery of the reactant after photoexcitation.67–71 The quenching of deleterious photochemical reactions by ultrafast internal conversion is believed to be decisive for a high intrinsic photostability of DNA bases as well as amino acids and peptides with aromatic chromophores.64–66
2 Results
2.1 Vertical excitation energies and absorption spectra
We consider in the present work a conformer of β-D-glucopyranose (for simplicity called β-glucose in the following) that is, according to calculations,2–9 one of the most stable conformers in the gas phase. This conformer is characterized by the following structural properties (cf. Fig. 3 for the numbering of atoms): (1) a 4C1 chair conformation, that is, all OH groups and the CH2OH group occupying an equatorial position; (2) a counterclockwise orientation of the four OH groups connected to carbon atoms C1 to C4; (3) a gauche–gauche orientation of the CH2OH group with respect to the C4–C5 and C5–O5 bonds; and (4) a syn-orientation of the C6–OH group with respect to the C5–C6 bond.While glucose can be found in numerous conformations regarding the torsion angles of the OH groups, the barriers for the conversion of the conformation described in the previous paragraph into rotamers range from around 9 to 20 kJ mol−1 (at the DFT level). For comparison, the harmonic vibrational frequencies of the OH torsional modes of this conformer are found in the range 400–600 cm−1 (4.8–7.2 kJ mol−1). It is therefore reasonable to assume that the OH torsional angles can be considered as fixed for a given conformer. Herein, we investigate the photochemistry of the lowest-energy conformer with respect to the OH torsional angles.
According to the Franck–Condon principle, excitation of a molecule by absorption of a photon proceeds in a vertical manner, that is, without a change of the nuclear geometry during the electronic transition. The trajectory of nuclear motion on the excited-state potential-energy surface starts in the Franck–Condon region vertically above the minimum of the ground-state potential well. The calculation of the vertical electronic excitation energies of the ground-state equilibrium geometry is the starting point for the computational simulation of the absorption spectrum as well as the photochemical reaction dynamics.
To our knowledge, neither the vertical excitation energies nor the absorption spectra of any carbohydrate have been calculated using ab initio methods so far. The most popular method for the computation of excitation energies of organic molecules is the time-dependent density functional theory (TDDFT) method. We compared the performance of this method with the CC2 method, which is an approximate second-order coupled cluster method (cf. Computational methods). The vertical excitation energies (cf. Table 1) predict the location of the lowest singlet excited state at 6.52 eV (CC2) or at 6.22 eV (TDDFT). Glucose thus absorbs only in the UV-C range of the electromagnetic spectrum. The oscillator strengths of all individual excitations are lower than 0.09 (cf. Fig. 2), which reflects the lack of a chromophore. Although the individual excitations possess only low oscillator strengths, the latter add up to a sizable magnitude: summation of the oscillator strengths for the first 50 excited states yields 0.4164 (CC2) or 0.3066 (TDDFT).
| State | CC2/eV | f | TDDFT/eV | f |
|---|---|---|---|---|
| 2 1A | 6.52 | 0.0087 | 6.22 | 0.0047 |
| 3 1A | 6.88 | 0.0028 | 6.44 | 0.0035 |
| 4 1A | 6.90 | 0.0151 | 6.58 | 0.0007 |
| 5 1A | 7.04 | 0.0119 | 6.65 | 0.0124 |
| 6 1A | 7.05 | 0.0094 | 6.70 | 0.0027 |
| 7 1A | 7.18 | 0.0080 | 6.72 | 0.0089 |
| 8 1A | 7.30 | 0.0344 | 6.77 | 0.0015 |
The dipole moment of the ground state is 3.42 D at the CC2 level. The dipole moments of the first 50 excited states range from as low as 0.94 to as high as 9.91 D.
Fig. 1 shows the self-consistent-field (SCF) orbitals at the HOMO–LUMO frontier. The HOMO − 1 and the HOMO are mainly lone-pair orbitals of the oxygen atoms O5 and O6 with some σ-bonding contributions. The LUMO and LUMO + 1 are σ* orbitals of partial Rydberg character of the O–H bonds connected to the carbon atoms C1 and to C2 and C3, respectively (cf. Fig. 3 for the numbering of atoms). These SCF orbitals look very similar to the Kohn–Sham orbitals used in the TDDFT computation. Basically, all low-lying excited states are generated by excitation of electrons from lone-pair orbitals of one or several oxygen atoms to diffuse antibonding σ* orbitals of one or several O–H or C–H bonds (the σ* orbitals of the O–H bonds are lower in energy than the σ* orbitals of the C–H bonds). Therefore, all these low-lying excited states can be classified as nσ* states of partial Rydberg character. This nσ* nature is further illustrated by the differences in the electron densities of the excited states and the ground state shown in Fig. S1 in the ESI.†
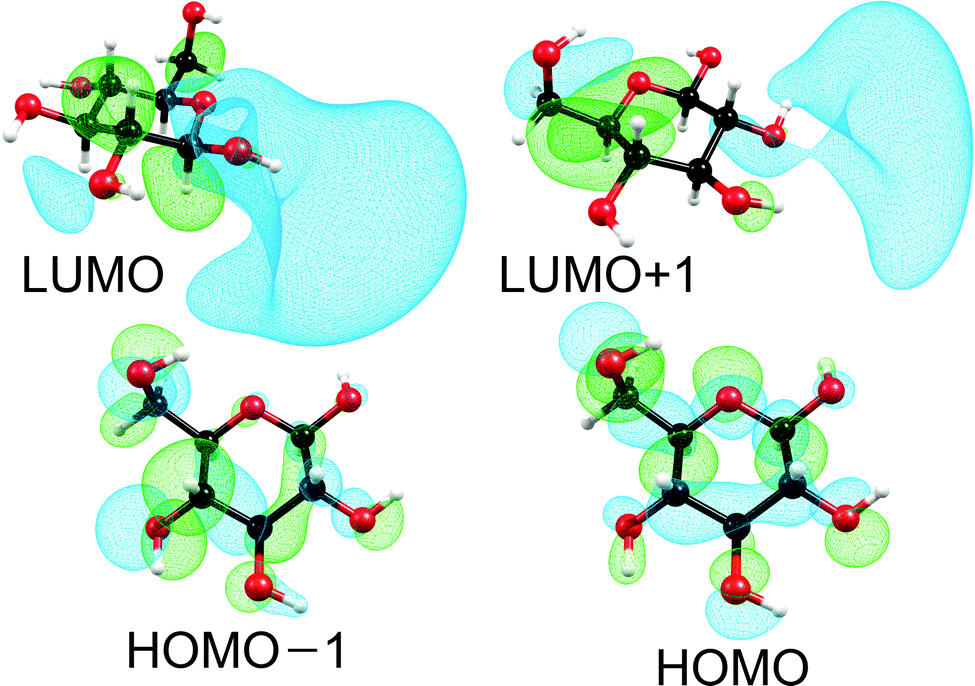 | ||
| Fig. 1 Self-consistent-field orbitals at the HOMO–LUMO frontier. For the HOMOs an isosurface value of 0.03 was used, while for the LUMOs a value of 0.015 was used. | ||
The absorption spectra computed at the CC2 and the TDDFT levels obtained by convolution of the stick spectra with Gaussian envelopes are shown in Fig. 2. Despite the low oscillator strengths of individual transitions, a sizable absorption occurs due to the high density of excited electronic states. The absorption edge is located around 6.1 eV (CC2) or 5.9 eV (TDDFT), a local peak is located around 7.8 eV (CC2) or 7.5 eV (TDDFT), and a shoulder is located near 7.0 eV (CC2) or 6.8 eV (TDDFT). The absorption profiles continue to increase toward higher energies. We cannot compare our computed spectra to spectroscopic results, since we were unable to find an experimentally recorded gas-phase absorption spectrum of glucose.
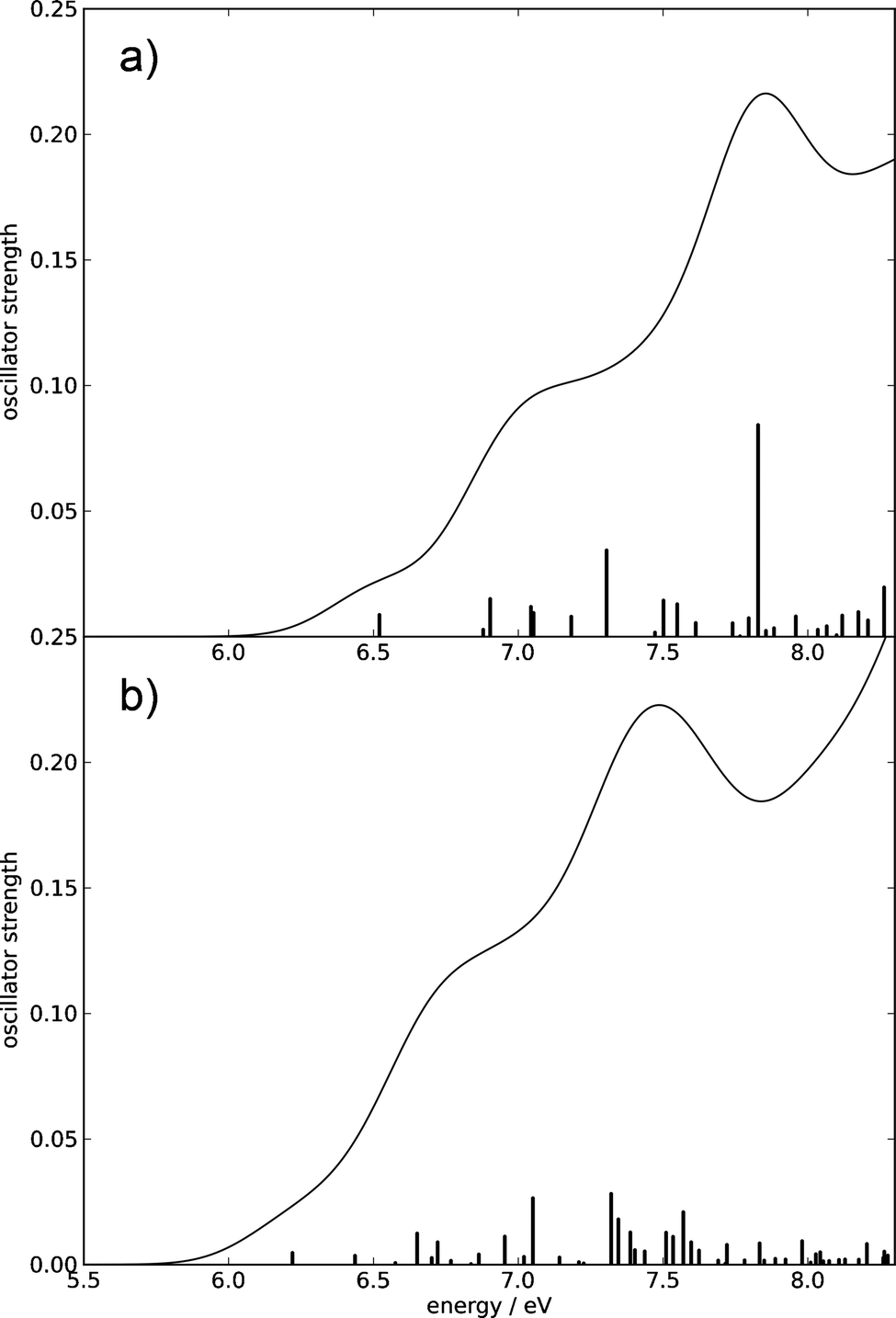 | ||
| Fig. 2 Absorption spectra of β-glucose computed using the CC2 method (a) and the TDDFT method (b) up to 8.3 eV. The spectral envelopes were obtained by convolution of the stick spectra using Gaussian functions of 0.4 eV FWHM. (For details, consult the section Computational methods.) | ||
2.2 Potential-energy surfaces and conical intersections: hydrogen-detachment reactions
The exploration of the lowest singlet excited-state potential-energy surface (S1) from the Franck–Condon region to the conical intersection with the electronic ground state (S0), at which the system can convert its excess electronic energy into vibrational energy and deactivate to the electronic ground state, is required for the understanding of the kinetic feasibility of a particular deactivation pathway. In order to describe a photochemical reaction one has to explore the reaction path from the Franck–Condon region to the relevant conical intersection connecting the excited state with the ground state.72–75 This strategy was dubbed the “pathway approach” by Fuß et al.76Fig. 3 shows the potential-energy profiles of the ground state and the nσ* excited state obtained for the linearly interpolated reaction path (cf. Computational methods) from the ground-state equilibrium geometry (full black circle at the lower left) to the conical intersection (full black and red circles on the right) for the hydrogen-detachment process of the O–H group located at the carbon atom C1. Clearly, the reaction path on the nσ* potential-energy surface is barrierless from the Franck–Condon region to the conical intersection. The conical intersection is located at around 5.0 eV, about 1.5 eV below the lowest vertical excitation energy.
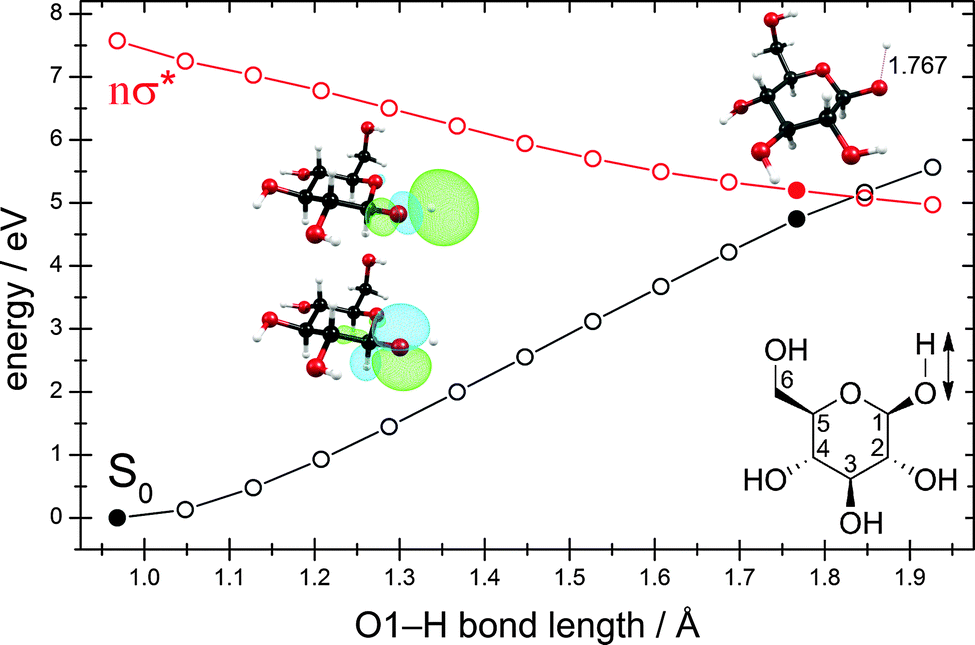 | ||
| Fig. 3 Energy profiles of the ground state and the nσ* state along the stretching coordinate of the O–H group located at the carbon atom C1. The profiles were obtained by linear interpolation between the ground-state equilibrium geometry (full black circle at the lower left) and the conical intersection (full black and red circles at the right). The last two geometries were obtained by rigid scan originating from the conical intersection. The insets show the lone-pair orbital of the oxygen atom and the σ* orbital of the O–H bond. Also shown is the structure of the conical intersection with the value of the O–H internuclear distance and the structural formula with the numbering of the C atoms. (For details, consult the section Computational methods.) | ||
As one can see from the molecular orbitals shown in the insets, the nσ* state corresponds to the excitation of an electron from the lone-pair orbital of the oxygen atom to the antibonding σ* orbital of the O–H bond. At short O–H distances, the σ* orbital exhibits a partial Rydberg character. As the distance between the two nuclei is increased, this orbital contracts and at longer distances collapses to the 1s orbital of the hydrogen atom.
At a point of degeneracy of two potential-energy surfaces of like multiplicity, that is, a point of conical intersection, only two nuclear degrees of freedom, the so-called branching-space vectors, are able to lift the degeneracy. The other 3N − 8 degrees of freedom describe the motion of the system along the multidimensional intersection seam, along which degeneracy is preserved.68 Fig. 4 shows the nuclear displacements of the orthogonalized branching-space vectors of the conical intersection shown in Fig. 3, namely, the gradient-difference vector g and the non-adiabatic-coupling vector h. The gradient-difference vector g describes the molecular distortion of maximal splitting of the two intersecting potential-energy surfaces. The non-adiabatic-coupling vector h describes the molecular distortion that leads to the strongest non-adiabatic coupling between the two adiabatic electronic states.68 Fig. 4 shows that the effective reaction coordinate depicted in Fig. 3, the elongation of the O–H bond length, corresponds to the nuclear displacements of the non-adiabatic-coupling vector h. The gradient-difference vector g, on the other hand, is a combination of bond-length and bond-angle alterations involving mainly the nuclei O1, C1 and C2 (cf. Fig. 3 for the numbering of atoms). The two most pronounced distortions are the C1–C2 elongation and the C2–C1–O1 bending.
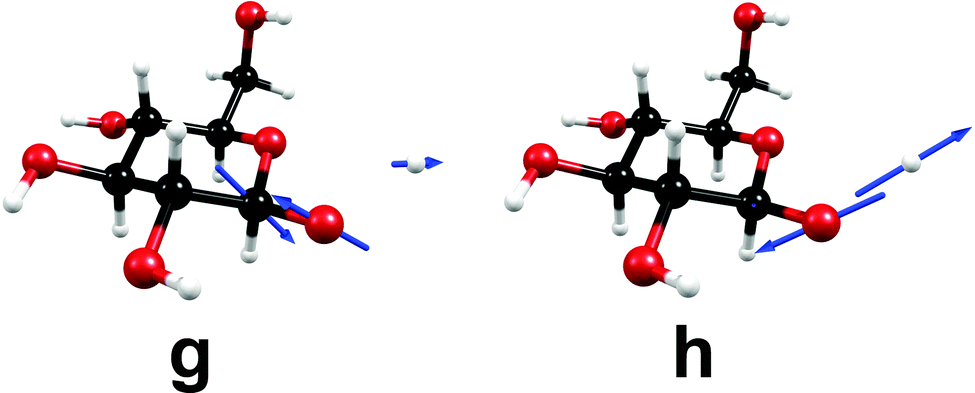 | ||
| Fig. 4 Nuclear-displacement vectors of the gradient-difference vector g and the non-adiabatic-coupling vector h of the conical intersection shown in Fig. 3. (For details, consult the section Computational methods.) | ||
A linear approximation of the potentials in the branching space,77 that is, the shape of the intersecting potential-energy surfaces in close proximity to the conical intersection, is shown in Fig. 5. The slope of the ground-state potential-energy surface towards the negative direction of the h vector (which corresponds to the recombination of the hydrogen atom and the glucosyl radical and thus to an aborted photochemical reaction), is steeper than the slope towards the positive direction (which corresponds to the fragmentation yielding a glucosyl radical and a hydrogen atom). This topography classifies this conical intersection, according to Atchity et al.,78 as a sloped one, that is, the gradient of the ground-state potential-energy surface towards the O–H recombination is steep, whereas towards the fragmentation products it is only weakly sloped. The topography of the conical intersection thus suggests that the probability of an aborted photoreaction regenerating the reactant could be substantial.
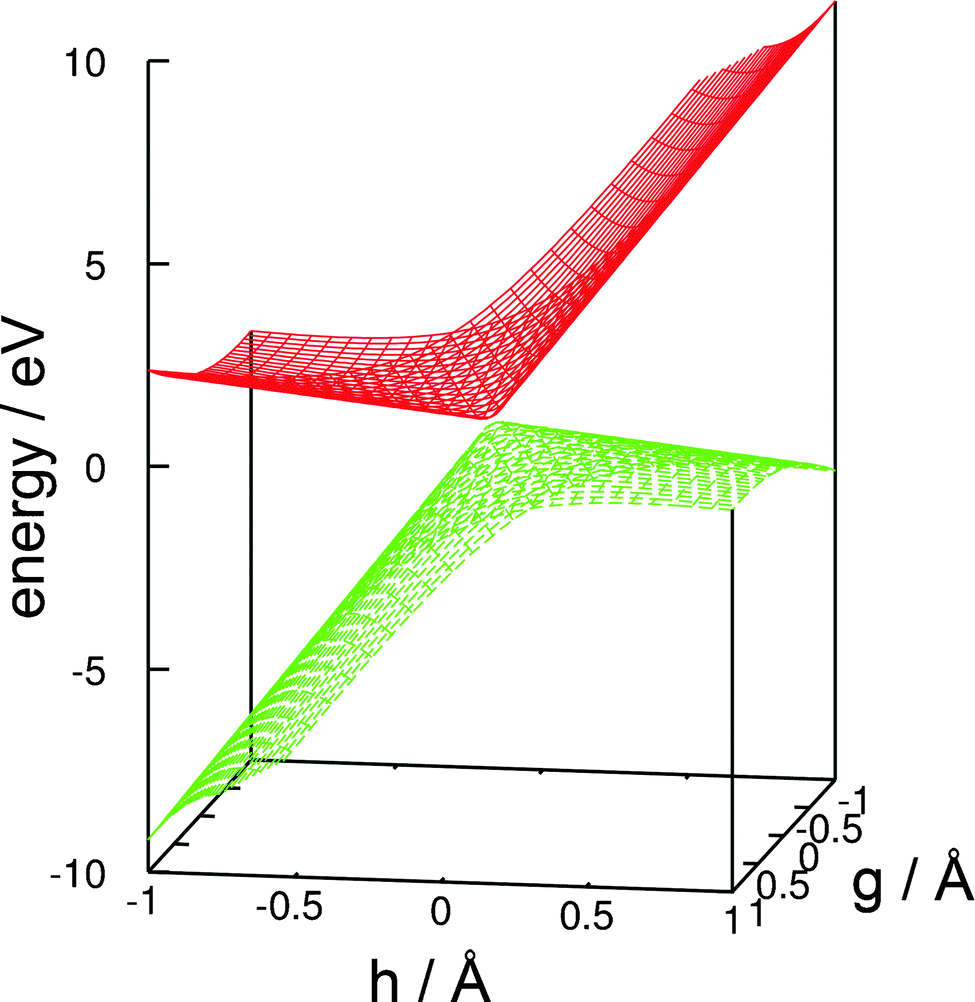 | ||
| Fig. 5 Linear approximation of the potential-energy surfaces of the ground state and the nσ* state in the branching space of the conical intersection shown in Fig. 3. (For details, consult the section Computational methods.) | ||
In order to determine the most suitable second internal coordinate for a more extended two-dimensional scan of the potential-energy surfaces of the ground state and the nσ* state (the first coordinate is the O–H bond length), we computed the extent of the splitting of the energy between the intersecting states for several internal coordinates approximating the nuclear displacements of the gradient-difference vector g (cf. Fig. 4) of the conical intersection shown in Fig. 3. Fig. S2 in the ESI† shows the extent of the splitting of the energy of the ground-state and nσ* potential-energy surfaces near the point of conical intersection. The figure shows that the splitting of the energy along the C2–C1–O1 bending coordinate is more pronounced than the splitting along the C1–C2 elongation coordinate. We therefore chose the C2–C1–O1 bending coordinate as an approximation for the nuclear displacements of the gradient-difference vector g for the computation of the extended potential-energy surfaces shown in Fig. 6.
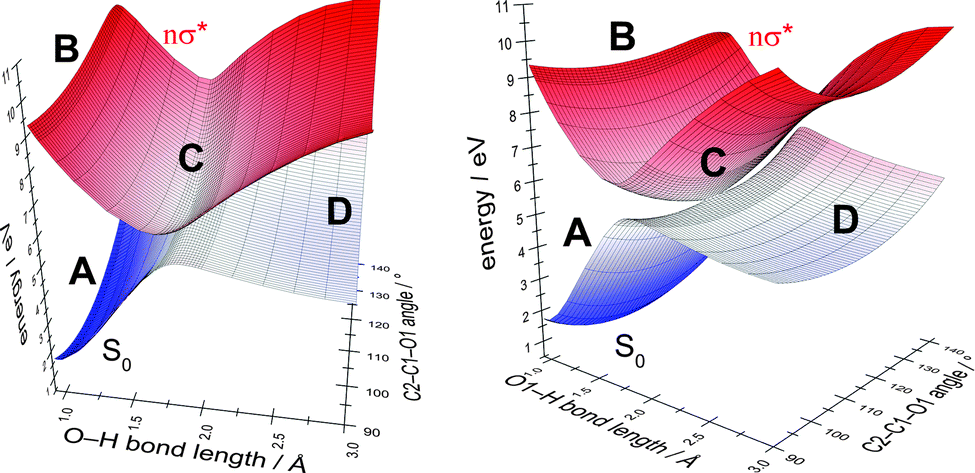 | ||
| Fig. 6 Two-dimensional adiabatic potential-energy surfaces of the ground state and the nσ* excited state for the O1–H elongation and the C2–C1–O1 bending coordinate of the C1–OH group (shown from two perspectives). Four distinct regions of the energy surfaces are shown: the ground-state equilibrium geometry (A), the Franck–Condon region on the excited-state potential-energy surface (B), the conical intersection (C) and the photoproducts (D). In the case of an aborted photoreaction, the reactant (A) is regenerated after passage through the conical intersection. | ||
In Fig. 6, we highlight four distinct regions. The first region, (A), is the ground-state equilibrium region. The second region, (B), is the excited-state Franck–Condon region that the molecule is promoted to by absorption of a photon. The third region, (C), is the region of conical intersection between the potential-energy surfaces of the ground state and the nσ* state. As mentioned before, the system can move from the Franck–Condon region along the potential-energy surface of the nσ* state towards this conical intersection without having to overcome any barriers. At the conical intersection, (C), the photoreaction can proceed along at least two routes. One route is the regeneration of the reactant (A). In this case, the process is an aborted photochemical reaction and the outcome is radiationless deactivation of the excited state (also known as internal conversion). The other possible route is dissociation towards the photoproducts (region D), which correspond to a glucosyl radical and a hydrogen atom.
The reaction paths, conical intersections and branching-space vectors for the hydrogen-detachment reactions of the other four O–H groups of β-glucose are shown in Fig. S3–S7 in the ESI.†
2.3 Potential-energy surfaces and conical intersections: ring-opening reactions
We were able to optimize the conical intersection between the electronic ground state and the lowest nσ* state for the ring-opening reaction breaking the C1–O5 bond. Given the ground-state equilibrium geometry and the conical intersection, a linearly interpolated reaction path (cf. Computational methods) was constructed between these two structures. The resulting energy profiles are shown in Fig. 7. Interestingly, this conical intersection is located at around 4.0 eV with respect to the ground-state equilibrium geometry and is thus about 1.0 eV lower than the conical intersection for the hydrogen-detachment reactions of the O–H groups (cf. Fig. 3 and Fig. S3–S6 in the ESI†).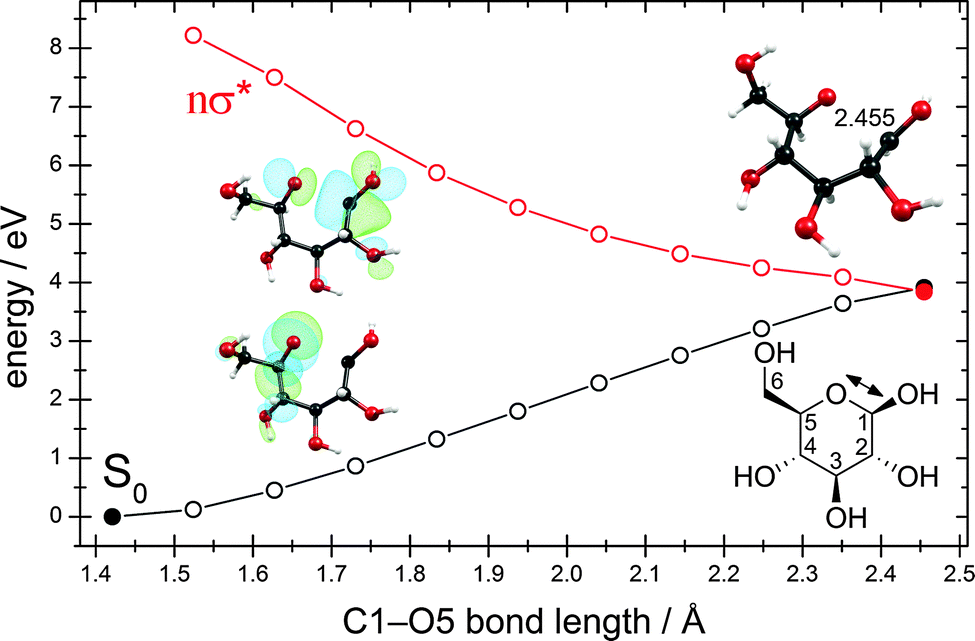 | ||
| Fig. 7 Energy profiles of the ground state and the nσ* state along the C1–O5 ring-opening coordinate. The profiles were obtained by linear interpolation between the ground-state equilibrium geometry (full black circle at the lower left) and the optimized conical intersection (full black and red circles at the right). The insets show the lone-pair orbital of the oxygen atom and the σ* orbital of the C1–O5 bond. Also shown is the structure of the conical intersection with the value of the C1–O5 internuclear distance and the structural formula with the numbering of the C atoms. (For details, consult the section Computational methods.) | ||
The nuclear-displacement vectors of the gradient-difference vector g and the non-adiabatic-coupling vector h of the conical intersection related to C1–O5 bond-breaking are shown in Fig. 8. In this case, the gradient-difference vector g is the effective reaction coordinate shown in Fig. 7, that is, the ring-opening/ring-closing motion. The non-adiabatic-coupling vector h is a combination of bending coordinates mainly involving the nuclei C5, C4 and C1.
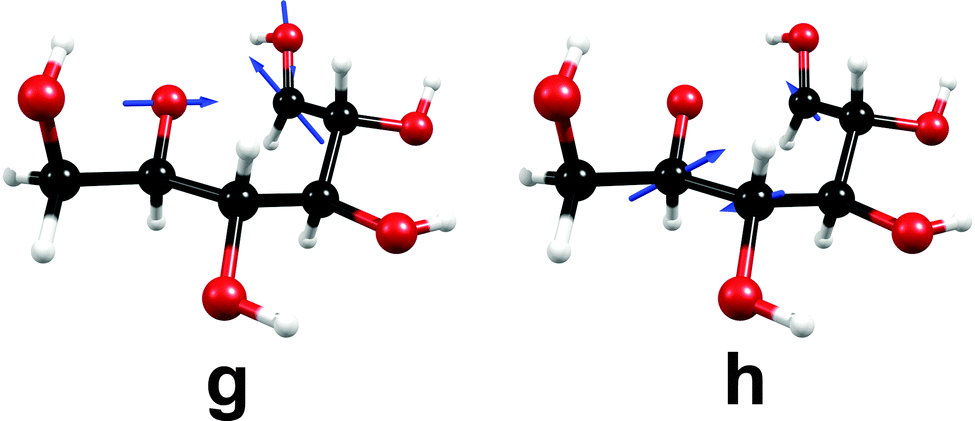 | ||
| Fig. 8 Nuclear-displacement vectors of the gradient-difference vector g and the non-adiabatic-coupling vector h of the conical intersection shown in Fig. 7. (For details, consult the section Computational methods.) | ||
The linear approximation of the potentials in the branching space in close proximity to the conical intersection is shown in Fig. S8 in the ESI.† Again, the ground-state potential-energy surface exhibits the steeper slope for the ring-closing reaction than for the direction leading to a biradical open-chain sugar, which classifies this conical intersection as a sloped one.78 The topography of this conical intersection seems to favor the aborted photochemical reaction in which the reactant (the cyclic form of β-glucose) is regenerated. The successful photoreaction yielding the biradical open-chain sugar appears less favorable due to the small gradient of the ground-state potential-energy surface in the direction leading to this photoproduct.
While the optimization of the conical intersection for the C1–O5 ring-opening process was unproblematic, this was not the case for the four possible C–C ring-opening reactions. We were unsuccessful in optimizing the conical intersection for any of these ring-opening processes. In principle, conical intersections should exist for these reactions. This is shown by Fig. S9 in the ESI.† In a series of calculations we were able to obtain two points of the reaction path for the C2–C3 ring-opening reaction, one with a C2–C3 distance of 2.49 Å, the other with a distance of 2.75 Å. Although the energies of the points are too high due to the use of geometries optimized for the ground state, this figure nevertheless reveals that between these two geometries a crossing of the energies of the ground state and the ring-opening nσ* state must exist. From this figure, the (non-optimized) energy of this conical intersection can be estimated to be about 6.0 eV.
3 Discussion and conclusions
We have performed the first detailed computational investigation of the singlet vertical excitation energies, the absorption spectrum and possible photochemical reaction pathways of β-glucose in the gas phase. We have shown that the lowest excited electronic states are of nσ* character, originating from excitation of lone-pair electrons of oxygen into O–H antibonding σ* orbitals of partial Rydberg character. The estimated absorption profile shows an onset at around 6.0 eV and a local peak near 7.5 or 7.8 eV. Although individual excitations exhibit only very low oscillator strengths, the sum of all oscillator strengths leads to a sizable absorption in the UV-C region.Our results on photochemical pathways indicate that two generic types of reaction processes dominate the photochemistry of β-glucose: hydrogen-detachment processes, which have been known for many systems of aromatic and non-aromatic character, and ring-opening processes. Both are potential photochemical reactions (which means they may yield photoproducts, namely, a glucosyl radical and a hydrogen atom in the case of a hydrogen-detachment reaction, and a biradical open-chain sugar in the case of a ring-opening reaction), but are also potential radiationless deactivation channels regenerating the reactant. We have shown that the relevant conical intersections are readily accessible via barrierless reaction paths from the Franck–Condon region. The topography of the involved conical intersections indicates that the probability of aborted photoreactions could be higher than the probability of the generation of photoproducts. The conical intersections appear to be highly efficient channels for internal conversion. The one- and two-dimensional energy profiles of the ground-state and nσ* potential-energy surfaces for the hydrogen-detachment channels (cf. Fig. 3 and 6) show the typical behavior of an X–H detachment reaction (X being an oxygen, nitrogen or sulfur atom) as previously reported for the πσ* and nσ* states of phenol,79–81 pyrrole80–83 and several other aromatic and non-aromatic molecules.80,81,83–85
While the conical intersections for the hydrogen-detachment reactions are located around 5.0 eV (cf. Fig. 3 and Fig. S3–S6 in the ESI†), the conical intersection for the C–O ring-opening reaction is located near 4.0 eV (cf. Fig. 7). The energies of both types of conical intersections are well below the computed absorption edge of glucose and should thus be readily accessible for radiationless deactivation of electronically excited molecules. Although the conical intersection for the C–O ring-opening reaction is about 1.0 eV lower in energy than the conical intersections for the hydrogen-detachment processes, we expect the latter to prevail due to the faster motion of the light hydrogen atom. This hypothesis, however, can only be proven by photofragmentation spectroscopy81,84,85 or by computational studies of the non-adiabatic nuclear-wavepacket or mixed quantum-classical surface-hopping dynamics.63,79,86–94
Although we were unable to optimize the conical intersections for the C–C ring-opening reactions, we can confirm based on the results of test calculations that they exist and are located below 6.0 eV (cf. Fig. S9 in the ESI†). Our results indicate that the C–O ring-opening channel should be energetically favored compared to the C–C ring-opening channel.
While no photodissociation experiments have been performed on carbohydrates so far, numerous experiments have been performed on methanol. Methanol can be regarded as a “methylene-hydrate”, and as such, as the simplest building block of carbohydrates. The absorption spectrum of methanol reveals the lowest-lying excited states to be of n → 3s and n → 3p Rydberg character.95 We predict a UV absorption for glucose with an onset of around 200 nm which is also very similar to the UV absorption behavior of methanol.95 It has been concluded that a transition to a state of valence nσ* character occurs upon elongation of the C–O or the O–H bonds of methanol.81,96 A study of the photodissociation dynamics of methanol revealed that the hydrogen-detachment process occurs on a repulsive excited-state potential-energy surface96 and exhibits a higher energy threshold than the C–O bond fission,97 but is, nevertheless, the dominating channel due to the lower mass of the hydrogen atom.96 For glucose, we have found that the conical intersections for the hydrogen-detachment reactions are located higher in energy than the intersections for the C–O ring-opening reaction, which seems to be analogous to the theoretical findings on methanol.81 Nevertheless, it can be expected that the hydrogen-detachment reactions will dominate over the C–O ring-opening processes due to the lower mass of the hydrogen atom.
It is likely that other carbohydrates will exhibit the same photochemical reaction mechanisms that we have analyzed for glucose in this work. The hydrogen-detachment channels for O–H groups and the ring-opening channels for C–O and C–C ring-opening should be generic for the entire class of compounds.
The present study could be the starting point for further investigations of the excited-state chemistry of carbohydrates. It remains to be explored what kind of photophysical and photochemical properties of carbohydrates are the underlying cause for the diversity with which nature has incorporated this class of compounds into a multitude of molecular systems in the course of biological evolution. It also remains to be investigated how these photochemical reaction mechanisms of carbohydrates are modified when they are bound to typical chromophores, for example, in nucleosides and nucleotides. Furthermore, it is possible that the conversion of carbohydrates from cyclic forms, that is, hemiacetals or hemiketals, into open-chain forms, that is, hydroxy-aldehydes or hydroxy-ketons, could also be accomplished via a photochemical mechanism.
Another challenge is the understanding of the excited-state deactivation mechanisms in carbohydrate oligomers and polymers, for example, cellobiose (the glucose dimer) or cellulose (the glucose polymer). The vast number of hydrogen bonds in these structures may offer a multitude of radiationless deactivation channels via intramolecular proton-transfer reactions along hydrogen bonds.
4 Computational methods
The ground-state equilibrium geometry of the conformer of β-glucose described in Section 2.1 was optimized at the MP2 level (Møller–Plesset perturbation theory of second order) using the correlation-consistent split-valence polarized double-zeta basis set (cc-pVDZ)98 and the resolution-of-the-identity (RI) approximation. The energy of the ground-state equilibrium geometry at the respective levels of theory served as the reference energy for the determination of vertical excitation energies and relative energies of potential-energy surfaces. The barriers for the conversion of the conformer into rotamers and the harmonic vibrational frequencies were determined at the DFT level using the hybrid functional B3LYP99,100 and the basis sets cc-pVDZ and aug-cc-pVTZ,98 respectively.For the computation of vertical excitation energies and excited-state properties we used three linear-response methods: the CC2 (approximate second-order coupled cluster) method,101 the ADC(2) (algebraic diagrammatic construction of second order) method,102 and the LR-TDDFT (linear-response time-dependent density functional theory) method.103 The RI approximation was employed in the CC2 and ADC(2) calculations.104 For the TDDFT calculations we used the hybrid functional B3LYP.99,100 For the calculation of vertical excitation energies we used the augmented cc-pVTZ basis set (aug-cc-pVTZ).98 All of these calculations, as well as the MP2 geometry optimization mentioned in the previous paragraph, were carried out using the Turbomole 6.3.1 program package.105
Linear-response methods are particularly suitable for the computation of the absorption spectrum of carbohydrates due to the distribution of the oscillator strength over a dense manifold of excited states. When using standard linear-response methods, one has to pay attention to the limitations of single-configuration methods, that is, a potential multiconfigurational character of the ground state and the weight of doubly excited configurations. The D1 diagnostic of 0.0464 at the CC2 level shows that the multiconfigurational character of the ground state is negligible.106 The contributions of doubly excited configurations to the excited states range from 8.94 to 10.84% for the first 50 excited states at the CC2 level, which is also within the acceptable range. At the ADC(2) level, we obtained practically the same results for the excitation energies, oscillator strengths and convoluted spectrum as at the CC2 level.
For the geometry optimization of minimum-energy conical intersections (minima on the conical intersection seam), the calculation of the nuclear displacements of the orthogonalized branching-space vectors, and linear approximations of potentials in the branching space we used the state-averaged complete-active-space self-consistent-field wavefunction (SA-CASSCF) method with the cc-pVDZ98 basis set. These calculations were carried out using Columbus 7.0.107
The reaction paths for hydrogen-detachment processes and for the ring-opening process were constructed by linear interpolation of internal coordinates between the initial geometry (the ground-state equilibrium geometry) and the final geometry (the respective optimized conical intersection). The energy profiles of the reaction paths were obtained by single-point energy calculations along the interpolated path, using single-state second-order perturbation theory on top of an SA-CASSCF wavefunction (SS-CASPT2//SA-CASSCF) with a level-shift of 0.2. The two-dimensional scan shown in Fig. 6 was obtained by a rigid scan. For the computation of the hydrogen-detachment reaction paths we used a partially-augmented basis set that we denote as “aug(2)-cc-pVDZ”. The cc-pVDZ basis was employed for all atoms except for the oxygen and hydrogen atoms of the O–H group involved in the hydrogen-detachment process (the “2” in aug(2)-cc-pVDZ indicates the use of the augmented basis on only two atoms of the molecule). The use of this partial augmentation helps to describe the short bond-length regions of the reaction paths (where Rydberg-valence mixing is important). The partial augmentation lowers the energy of the nσ* state belonging to the O–H group under investigation. The SS-CASPT2//SA-CASSCF single-point calculations were carried out using Molpro 2006.1.108
It is also possible to use an aug(1)-cc-pVDZ basis, that is, augmenting only the oxygen atom of the O–H group with diffuse basis functions. Test calculations showed that using diffuse basis functions on both the oxygen and the hydrogen atom leads to a better and smoother description of short bond-length regions of the potential-energy profiles of the nσ* states. For the computation of reaction paths of ring-opening processes we used the cc-pVDZ98 basis for all atoms, since a partial augmentation is not helpful in this case.
For the computation of photochemical hydrogen-detachment and ring-opening energy profiles as well as for the optimization of conical intersections we used a compact active space of two electrons in two active orbitals. The averaging of the energy included only the electronic ground state and the nσ* state of interest. This recipe is denoted as SA2-CASSCF(2,2). We found that this (2,2) active space works well for a qualitative description of the reactions under investigation. The use of a larger active space (e.g., four electrons in four orbitals) introduces active electrons and orbitals that barely contribute to the wavefunction of the lowest nσ* state.
Acknowledgements
D.T. is grateful for a PhD fellowship granted by the International Max Planck Research School of Advanced Photon Science (IMPRS-APS) and for support by the TUM Graduate School. This work was partially supported by a research grant of the Deutsche Forschungsgemeinschaft (DFG) and by the DFG Cluster of Excellence “Munich-Centre for Advanced Photonics”.References
- R. V. Stick and S. J. Williams, Carbohydrates. The Essential Molecules of Life, Elsevier, Amsterdam, The Netherlands, 2nd edn, 2009 Search PubMed.
- P. L. Polavarapu and C. S. Ewig, J. Comput. Chem., 1992, 13, 1255–1261 CrossRef CAS.
- C. J. Cramer and D. G. Truhlar, J. Am. Chem. Soc., 1993, 115, 5745–5753 CrossRef CAS.
- G. I. Csonka, I. Kolossváry, P. Császár, K. Éliás and I. G. Csizmadia, THEOCHEM, 1997, 395–396, 29–40 CrossRef CAS.
- B. Ma, H. F. Schaefer III and N. L. Allinger, J. Am. Chem. Soc., 1998, 120, 3411–3422 CrossRef CAS.
- S. E. Barrows, J. W. Storer, C. J. Cramer, A. D. French and D. G. Truhlar, J. Comput. Chem., 1998, 19, 1111–1129 CrossRef CAS.
- M. Appell, G. Strati, J. L. Willet and F. A. Momany, Carbohydr. Res., 2004, 339, 537–551 CrossRef CAS PubMed.
- J. C. Corchado, M. L. Sánchez and M. A. Aguilar, J. Am. Chem. Soc., 2004, 126, 7311–7319 CrossRef CAS PubMed.
- N. Miura, T. Taniguchi, K. Monde and S.-I. Nishimura, Chem. Phys. Lett., 2006, 419, 326–332 CrossRef CAS PubMed.
- P. Çarçabal, R. A. Jockusch, I. Hünig, L. C. Snoek, R. T. Kroemer, B. G. Davis, D. P. Gamblin, I. Compagnon, J. Oomens and J. P. Simons, J. Am. Chem. Soc., 2005, 127, 11414–11425 CrossRef PubMed.
- A. F. Jalbout, L. Adamowicz and L. M. Ziurys, Chem. Phys., 2006, 328, 1–7 CrossRef CAS PubMed.
- E. J. Cocinero, A. Lesarri, P. Écija, F. J. Basterretxea, J.-U. Grabow, J. A. Fernández and F. Castaño, Angew. Chem., Int. Ed., 2012, 51, 3119–3124 CrossRef CAS PubMed.
- G. L. Strati, J. L. Willett and F. A. Momany, Carbohydr. Res., 2002, 337, 1833–1849 CrossRef CAS.
- E. J. Cocinero, D. P. Gamblin, B. G. Davis and J. P. Simons, J. Am. Chem. Soc., 2009, 131, 11117–11123 CrossRef CAS PubMed.
- F. A. Momany and U. Schnupf, Carbohydr. Res., 2011, 346, 619–630 CrossRef CAS PubMed.
- A. D. French, G. P. Johnson, C. J. Cramer and G. I. Csonka, Carbohydr. Res., 2012, 350, 68–76 CrossRef CAS PubMed.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- K. Mazeau and L. Heux, J. Phys. Chem. B, 2003, 107, 2394–2403 CrossRef CAS.
- S. Queyroy, F. Müller-Plathe and D. Brown, Macromol. Theory Simul., 2004, 13, 427–440 CrossRef CAS.
- Y. Nishiyama, G. P. Johnson, A. D. French, V. T. Forsyth and P. Langan, Biomacromolecules, 2008, 9, 3133–3140 CrossRef CAS PubMed.
- A. M. Silva, E. C. da Silva and C. O. da Silva, Carbohydr. Res., 2006, 341, 1029–1040 CrossRef CAS PubMed.
- D. Liu, M. R. Nimlos, D. K. Johnson, M. E. Himmel and X. Qian, J. Phys. Chem. A, 2010, 114, 12936–12944 CrossRef CAS PubMed.
- X. Qian and X. Wei, J. Phys. Chem. B, 2012, 116, 10898–10904 CrossRef CAS PubMed.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Catal., 2012, 295, 122–132 CrossRef CAS PubMed.
- R. S. Assary and L. A. Curtiss, Energy Fuels, 2012, 26, 1344–1352 CrossRef CAS.
- V. Seshadri and P. R. Westmoreland, J. Phys. Chem. A, 2012, 116, 11997–12013 CrossRef CAS PubMed.
- N. Luo, A. Litvin and R. Osman, J. Phys. Chem. A, 1999, 103, 592–600 CrossRef CAS.
- I. Baccarelli, F. A. Gianturco, A. Grandi, N. Sanna, R. R. Lucchese, I. Bald, J. Kopyra and E. Illenberger, J. Am. Chem. Soc., 2007, 129, 6269–6277 CrossRef CAS PubMed.
- G. Vall-Ilosera, M. A. Huels, M. Coreno, A. Kivimäki, K. Jakubowska, M. Stankiewicz and E. Rachlew, ChemPhysChem, 2008, 9, 1020–1029 CrossRef CAS PubMed.
- J.-W. Shin, F. Dong, M. E. Grisham, J. J. Rocca and E. R. Bernstein, Chem. Phys. Lett., 2011, 506, 161–166 CrossRef CAS PubMed.
- D. Ghosh, A. Golan, L. K. Takahashi, A. I. Krylov and M. Ahmed, J. Phys. Chem. Lett., 2012, 3, 97–101 CrossRef CAS.
- W. B. Bosma, U. Schnupf, J. L. Willet and F. A. Momany, THEOCHEM, 2009, 905, 59–69 CrossRef CAS PubMed.
- B. Brauer, M. Pincu, V. Buch, I. Bar, J. P. Simons and R. B. Gerber, J. Phys. Chem. A, 2011, 115, 5859–5872 CrossRef CAS PubMed.
- L. Jin, J. P. Simons and R. B. Gerber, J. Phys. Chem. A, 2012, 116, 11088–11094 CrossRef CAS PubMed.
- X. Qian, S.-Y. Ding, M. R. Nimlos, D. K. Johnson and M. E. Himmel, Macromolecules, 2005, 38, 10580–10589 CrossRef CAS.
- Y. Li, M. Lin and J. W. Davenport, J. Phys. Chem. C, 2011, 115, 11533–11539 CAS.
- M. Bergenstråhle, L. A. Berglund and K. Mazeau, J. Phys. Chem. B, 2007, 111, 9138–9145 CrossRef PubMed.
- T. Shen, P. Langan, A. D. French, G. P. Johnson and S. Gnanakaran, J. Am. Chem. Soc., 2009, 131, 14786–14794 CrossRef CAS PubMed.
- S. Barsberg, J. Phys. Chem. B, 2010, 114, 11703–11708 CrossRef CAS PubMed.
- A. Imberty and S. Péres, Chem. Rev., 2000, 100, 4567–4588 CrossRef CAS PubMed.
- J. P. Simons, R. A. Jockusch, P. Çarçabal, I. Hünig, R. T. Kroemer, N. A. Macleod and L. C. Snoek, Int. Rev. Phys. Chem., 2005, 24, 489–531 CrossRef CAS.
- C. O. da Silva, Theor. Chem. Acc., 2006, 116, 137–147 CrossRef CAS PubMed.
- S. Pérez, A. Imberty, S. B. Engelsen, J. Gruza, K. Mazeau, J. Jimenez-Barbero, A. Poveda, J.-F. Espinosa, B. P. van Eyck, G. Johnson, A. D. French, M. L. C. E. Kouwijzer, P. D. J. Grootenuis, A. Bernardi, L. Raimondi, H. Senderowitz, V. Durier, G. Vergoten and K. Rasmussen, Carbohydr. Res., 1998, 314, 141–155 CrossRef.
- C. A. Stortz, G. P. Johnson, A. D. French and G. I. Csonka, Carbohydr. Res., 2009, 344, 2217–2228 CrossRef CAS PubMed.
- J. Y. Mane and M. Klobukowski, Chem. Phys. Lett., 2010, 500, 140–143 CrossRef CAS PubMed.
- J.-H. Lii, B. Ma and N. L. Allinger, J. Comput. Chem., 1999, 20, 1593–1603 CrossRef CAS.
- G. I. Csonka, A. D. French, G. P. Johnson and C. A. Stortz, J. Chem. Theory Comput., 2009, 5, 679–692 CrossRef CAS.
- W. M. C. Sameera and D. A. Pantazis, J. Chem. Theory Comput., 2012, 8, 2630–2645 CrossRef CAS.
- E. G. Robertson and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1–18 RSC.
- E. Nir, C. Plützer, K. Kleinermanns and M. de Vries, Eur. Phys. J. D, 2002, 20, 317–329 CrossRef CAS.
- H. Saigusa, J. Photochem. Photobiol., C, 2006, 7, 197–210 CrossRef CAS PubMed.
- M. S. de Vries and P. Hobza, Annu. Rev. Phys. Chem., 2007, 58, 585–612 CrossRef CAS PubMed.
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Kohler, Chem. Rev., 2004, 104, 1977–2019 CrossRef PubMed.
- S. Perun, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2005, 127, 6257–6265 CrossRef CAS PubMed.
- L. Blancafort, J. Am. Chem. Soc., 2006, 128, 210–219 CrossRef CAS PubMed.
- L. Serrano-Andrés, M. Merchán and A. C. Borin, J. Am. Chem. Soc., 2008, 130, 2473–2484 CrossRef PubMed.
- Radiation Induced Molecular Phenomena in Nucleic Acids, ed. M. Shukla and J. Leszczynski, Springer, New York, 2008 Search PubMed.
- A. Abo-Riziq, L. Grace, E. Nir, M. Kabelac, P. Hobza and M. S. de Vries, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 20–23 CrossRef CAS PubMed.
- A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2004, 6, 2763–2771 RSC.
- H. Valdes, V. Spiwok, J. Rezac, D. Reha, A. G. Abo-Riziq, M. S. de Vries and P. Hobza, Chem.–Eur. J., 2008, 14, 4886–4898 CrossRef CAS PubMed.
- D. Shemesh, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2009, 131, 1374–1375 CrossRef CAS PubMed.
- E. Gloaguen, B. de Courcy, J.-P. Piquemal, J. Pilmé, O. Parisel, R. Pollet, H. S. Biswal, F. Piuzzi, B. Tardivel, M. Broquier and M. Mons, J. Am. Chem. Soc., 2010, 132, 11860–11863 CrossRef CAS PubMed.
- M. Mališ, Y. Loquais, E. Gloaguen, H. S. Biswal, F. Piuzzi, B. Tardivel, V. Brenner, M. Broquier, C. Jouvet, M. Mons, N. Došlić and I. Ljubić, J. Am. Chem. Soc., 2012, 134, 20340–20351 CrossRef PubMed.
- A. L. Sobolewski and W. Domcke, Europhys. News, 2006, 37, 20–23 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2010, 12, 4897–4898 RSC.
- W. Domcke and A. L. Sobolewski, Nature Chem., 2013, 5, 257–258 CrossRef CAS PubMed.
- M. Klessinger and J. Michl, Excited States and Photochemistry of Organic Molecules, VCH Publishers, New York, 1995 Search PubMed.
- Conical Intersections: Electronic Structure, Dynamics & Spectroscopy, ed. W. Domcke, D. R. Yarkony and H. Köppel, World Scientific Publishing, Toh Tuck Link, Singapore, 2004 Search PubMed.
- I. Schapiro, F. Melaccio, E. N. Laricheva and M. Olivucci, Photochem. Photobiol. Sci., 2011, 10, 867–886 CAS.
- F. Bernardi, M. Olivucci and M. A. Robb, Chem. Soc. Rev., 1996, 25, 321–328 RSC.
- T. J. Martínez, Nature, 2010, 467, 412–413 CrossRef PubMed.
- S. Kato, J. Chem. Phys., 1988, 88, 3045–3056 CrossRef CAS.
- A. L. Sobolewski, C. Woywod and W. Domcke, J. Chem. Phys., 1993, 98, 5627–5641 CrossRef CAS.
- I. J. Palmer, I. N. Ragazos, F. Bernardi, M. Olivucci and M. A. Robb, J. Am. Chem. Soc., 1993, 115, 673–682 CrossRef CAS.
- J. Dreyer and M. Klessinger, J. Chem. Phys., 1994, 101, 10655–10665 CrossRef CAS.
- W. Fuß, S. Lochbrunner, A. M. Müller, T. Schikarski, W. E. Schmid and S. A. Trushin, Chem. Phys., 1998, 232, 161–174 CrossRef.
- D. R. Yarkony, J. Chem. Phys., 2001, 114, 2601–2613 CrossRef CAS.
- G. J. Atchity, S. S. Xantheas and K. Ruedenberg, J. Chem. Phys., 1991, 95, 1862–1876 CrossRef.
- Z. Lan, W. Domcke, V. Vallet, A. L. Sobolewski and S. Mahapatra, J. Chem. Phys., 2005, 122, 224315 CrossRef PubMed.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- M. N. R. Ashfold, G. A. King, D. Murdock, M. G. D. Nix, T. A. A. Oliver and A. G. Sage, Phys. Chem. Chem. Phys., 2010, 12, 1218–1238 RSC.
- V. Vallet, Z. Lan, S. Mahapatra, A. L. Sobolewski and W. Domcke, J. Chem. Phys., 2005, 123, 144307 CrossRef PubMed.
- A. L. Sobolewski and W. Domcke, Chem. Phys., 2000, 259, 181–191 CrossRef CAS.
- T. A. A. Oliver, G. A. King and M. N. R. Ashfold, Chem. Sci., 2010, 1, 89–96 RSC.
- T. A. A. Oliver, G. A. King and M. N. R. Ashfold, J. Chem. Phys., 2010, 133, 194303 Search PubMed.
- M. Barbatti and H. Lischka, J. Am. Chem. Soc., 2008, 130, 6831–6839 CrossRef CAS PubMed.
- G. Groenhof, L. V. Schäfer, M. Boggio-Pasqua, M. Goette, H. Grubmüller and M. A. Robb, J. Am. Chem. Soc., 2007, 129, 6812–6819 CrossRef CAS PubMed.
- G. Tomasello, M. Wohlgemuth, J. Petersen and R. Mitrić, J. Phys. Chem. B, 2012, 116, 8762–8770 CrossRef CAS PubMed.
- T. S. Venkatesan, S. G. Ramesh, Z. Lan and W. Domcke, J. Chem. Phys., 2012, 136, 174312 CrossRef CAS PubMed.
- P. R. L. Markwick and N. L. Doltsinis, J. Chem. Phys., 2007, 126, 175102 CrossRef PubMed.
- E. Fabiano and W. Thiel, J. Phys. Chem. A, 2008, 112, 6859–6863 CrossRef CAS PubMed.
- H. R. Hudock and T. J. Martínez, ChemPhysChem, 2008, 9, 2486–2490 CrossRef CAS PubMed.
- F. Plasser, M. Barbatti, A. J. A. Aquino and H. Lischka, Theor. Chem. Acc., 2012, 131, 1073–1087 CrossRef.
- M. Barbatti, Z. Lan, R. Crespo-Otero, J. J. Szymczak, H. Lischka and W. Thiel, J. Chem. Phys., 2012, 137, 22A503 CrossRef PubMed.
- J. B. Nee, M. Suto and L. C. Lee, Chem. Phys., 1985, 98, 147–155 CrossRef CAS.
- R. J. Buenker, G. Olbrich, H.-P. Schuchmann, B. L. Schürmann and C. von Sonntag, J. Am. Chem. Soc., 1984, 106, 4362–4368 CrossRef CAS.
- S. Harich, J. J. Lin, Y. T. Lee and X. Yang, J. Phys. Chem. A, 1999, 103, 10324–10332 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS.
- J. Schirmer, Phys. Rev. A, 1982, 26, 2395–2416 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- C. Hättig and F. Weigend, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef.
- TURBOMOLE V6.3.1 2011, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- C. L. Janssen and I. M. B. Nielsen, Chem. Phys. Lett., 1998, 290, 423–430 CrossRef CAS.
- H. Lischka, R. Shepard, I. Shavitt, R. M. Pitzer, M. Dallos, T. Müller, P. G. Szalay, F. B. Brown, R. Ahlrichs, H. J. Böhm, A. Chang, D. C. Comeau, R. Gdanitz, H. Dachsel, C. Ehrhardt, M. Ernzerhof, P. Höchtl, S. Irle, G. Kedziora, T. Kovar, V. Parasuk, M. J. M. Pepper, P. Scharf, H. Schiffer, M. Schindler, M. Schüler, M. Seth, E. A. Stahlberg, J.-G. Zhao, S. Yabushita, Z. Zhang, M. Barbatti, S. Matsika, M. Schuurmann, D. R. Yarkony, S. R. Brozell, E. V. Beck, J.-P. Blaudeau, M. Ruckenbauer, B. Sellner, F. Plasser and J. J. Szymczak, COLUMBUS, an ab initio electronic structure program, release 7.0, 2012 Search PubMed.
- H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schütz, P. Celani, T. Korona, G. Rauhut, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, A. W. Lloyd, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, R. Pitzer, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni and T. Thorsteinsson, MOLPRO, version 2006.1, a package of ab initio programs, see http://www.molpro.net Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Differences in the electron densities of the excited states and the ground state. Comparison of scans along the C1–C2 elongation and the C2–C1–O1 bending coordinate originating from the conical intersection for the O1–H hydrogen-detachment process shown in Fig. 3. Linearly interpolated reaction paths between the ground-state equilibrium geometry and the conical intersections for the remaining four O–H groups (O2–H, O3–H, O4–H, O6–H). Nuclear-displacement vectors of the branching-space vectors of the four remaining conical intersections. Linear approximation of the potential-energy surfaces of the ground state and the nσ* state in the branching space of the conical intersection for the C1–O5 ring-opening process shown in Fig. 7. Potential energies of the ground state and the nσ* state in the vicinity of a conical intersection along the C2–C3 ring-opening reaction coordinate. Cartesian coordinates of the ground-state equilibrium geometry and all conical intersections. See DOI: 10.1039/c3cp52359d |
| This journal is © the Owner Societies 2014 |