Ground state spectroscopy of hydroxyquinolines: evidence for the formation of protonated species in water-rich dioxane–water mixtures†
Osama K.
Abou-Zied
*a,
John
Husband
a,
Najla
Al-Lawatia
a and
Thomas B.
Steinbrecher
b
aDepartment of Chemistry, Faculty of Science, Sultan Qaboos University, P.O. Box 36, Postal Code 123, Muscat, Sultanate of Oman. E-mail: abouzied@squ.edu.om; Fax: +968-2414-1469; Tel: +968-2414-1468
bDepartment for Theoretical Chemical Biology, Institute for Physical Chemistry, Karlsruhe Institute of Technology, Kaiserstr. 12, 76131 Karlsruhe, Germany
First published on 10th October 2013
Abstract
We have recently used 6-, 7-, and 8-hydroxyquinolines (HQs) as fluorescent probes to study the binding mechanism in one of the drug binding sites of human serum albumin. In the present work we study the absorption spectra of the HQ molecules in neat and binary mixtures of dioxane and water in order to identify the different tautomeric species in the ground state. This study should help in identifying the environment in nanocavities of macromolecules when HQs are used as local reporters. The enol form is shown to be the only tautomer for the three HQs in dioxane and water, with the exception of 7HQ in which both the enol and the zwitterion tautomers exist in equilibrium in water. The results are confirmed by the density functional theory (DFT) calculations using the B3LYP method with a 6-311++G(2d,p) basis set. In water-rich dioxane mixtures, all HQs are protonated. The results were confirmed by comparing the absorption spectra in binary solvents with those in acidic and basic aqueous solutions, and by DFT calculations of the Franck–Condon S1 ← S0 transitions. The number of water molecules solvating the polar sites in each HQ molecule was estimated from the spectral change in the binary solvent mixtures, and structures were calculated by DFT. Mapping the water density around the polar sites in each HQ using molecular dynamics (MD) simulations shows well-defined hydrogen bonds around the N-heteroatom in each HQ molecule. Water density is only well-defined around the hydroxyl group in 8HQ. The MD simulations indicate free rotation of the OH group in 6HQ and 7HQ, and the stability of the cis-isomer in 8HQ. The results point to the unique spectral signatures of 7HQ in water which make this molecule a potential probe to detect the presence of water in nanocavities of macromolecules, and to the ability of the three HQs to detect acidic media in binding sites.
1 Introduction
Characterization of the binding sites in biological systems is important in correlating their interior properties to their structure and functionality.1 These properties play a crucial role in the specificity and affinity of protein–ligand binding which is important to understand critical biological functions such as the role of drugs when bound to enzymes and receptors.2,3 Such clarification is an essential step towards optimizing drug formulae and maximizing drug potency and efficacy.One effective way to study binding sites in proteins is by using small molecular probes as ligands. Several small organic molecules containing both acidic and basic functional groups show high sensitivity in their spectra to the change in polarity and the hydrogen bonding nature of their immediate environment.4,5 They can thus be used to report on their local environment after binding to a protein, and may serve as models to study proton transfer in enzymes and other proteins. We have recently used three hydroxyquinolines (6-hydroxyquinoline (6HQ), 7-hydroxyquinoline (7HQ) and 8-hydroxyquinoline (8HQ)) as local reporters to examine one of the drug binding sites of human serum albumin.6–8 The results point to the existence of water inside the binding site and the role of water in molecular recognition and ligand binding. HQs are useful in this regard due to the high sensitivity of their spectra to protic solvents. The small size of the HQ backbone is not expected to induce large perturbations at the protein binding sites. The absorption spectra of most HQs extend to a region which is transparent to proteins. This fact is expected to provide selective excitation of the probes while encapsulated in the macromolecular binding sites, and the ability to study the effect of binding on the spectra of the probes.
In the present work, we characterize the spectra of 6-, 7-, and 8HQ (structures of all mono-hydroxyquinolines are shown in Fig. 1) in different solvents, particularly in aqueous medium, in order to understand the photophysics of the reversible interaction of HQs with water molecules. The current study is an extension to our recent report on the spectroscopy of 7HQ.9
 | ||
| Fig. 1 Structures of mono-hydroxyquinolines. | ||
The chemistry of HQs has attracted special interest due to their therapeutic properties and the fact that they are well-known bidentate ligands.10 HQs belong to the class of molecules that can exist in enol as well as keto forms by enol–keto tautomerization. The most stable structure of HQs in their ground state is the enolic form.11 It has been shown that direct intramolecular proton transfer is possible only in 2HQ and 8HQ, whereas solvent-assisted proton transfer is clearly needed for the other HQs owing to the large distance between the hydroxyl group and the N-heteroatom.12 The different OH locations in HQs provide an opportunity to study the mechanisms of intramolecular versus intermolecular proton transfers. These two different mechanisms may be useful in detecting water in binding sites of macromolecules.
HQs can exist in five different forms, the enol molecule (E), the protonated (C), the deprotonated (A), the keto (K), and the zwitterion (Z) species.13,14 Aqueous solutions of 7HQ, as well as of 3HQ and 6HQ, are reported to undergo stepwise excited-state tautomerization by forming a deprotonated intermediate species. Photoinduced excited state proton transfer (ESPT) in 7HQ is a unique reaction system.11 In a neutral aqueous solution, 7HQ exists mostly as E (67%) and Z (29%) with minor species of C (3%) and A (1%).15 In other solvents, 7-HQ exists in equilibrium between the E and the K tautomers which is solvent dependent. Z is stabilized in aqueous medium only which represents a valence bond resonance structure of the K tautomer that emphasizes its large dipole moment. Considering the structure of 7HQ, direct intramolecular proton transfer is not possible, and thus solvent-assisted proton transfer is the only mechanism linking together different tautomers. Solvent-assisted ESPT was proposed in protic solvents16,17 in which the enol and imino groups become more acidic and basic, respectively, in the excited state than in the ground state.13 As a result, the K resonance species was reported to form in nonaqueous protic solvents through a solvent chain,18 whereas the Z resonance species was proposed to form in aqueous solvents via a deprotonated intermediate species.19,20 Fluorescence was observed from the E tautomer only in nonprotic solvents, whereas in aqueous solution fluorescence was observed from the Z tautomer only.7,9,21,22 The formation of the Z tautomer was proposed to be through a cyclical complex between 7HQ and water molecules.23 We found the number of water molecules in this network to be three.8 Dual fluorescence was observed in alcohols.16,22,24 In the ground state, the E tautomer is the only species that exists in nonprotic and nonaqueous protic solvents, while both the E and the Z tautomers are stable in comparable amounts in aqueous solution.9,21
2HQ is also reported to exist in two tautomeric forms, which are nearly equal in energy. The two tautomers are the lactim (E) and the lactam (K) forms and are interconvertable by simple hydrogen atom transfer between the OH group and the ring nitrogen. Nimlos et al. studied the time of flight mass spectra, fluorescence excitation and dispersed emission spectra for 2HQ and could not observe any evidence of ESPT (K ← E).25 Rautela et al. concluded that in non-polar solvents, both the E and K forms of 2HQ exist while in polar protic solvents the K form is predominant. In water, the E along with the Z/K forms are proposed to exist.26
In 3HQ, the photoacidic and photobasic groups are in close proximity to each other, making this molecule a suitable candidate for the study of proton migration in water. 3HQ in water exists as one of the four prototropic species: (E, C, A, and Z). The Z species undergoes fast electron rearrangement in the excited singlet state to form the quinonoid tautomer E. The ground-state equilibrium constants indicate that 3HQ in water exists almost exclusively as C, E, and A at pH values of 2, 6, and 10, respectively; in addition to that, 6% of 3HQ exists as Z at pH 6.13 The A species transforms rapidly into Z in the pH region of 4–6. The proton migration is suggested to proceed by the continuous formation and breakage of hydrogen bonds involved in the hydrated proton cluster. However, the Z tautomer is found to undergo rearrangement immediately to form the more stable E tautomer.27
The 6HQ molecule is reported to exist in the trans-E form in neutral medium, the C form in acidic solution, and in the A form in basic solution.13,28 Similar to 7HQ, the OH group in 6HQ is more acidic and the ring nitrogen atom is more basic in the excited state than in the ground state.13 García-Ochoa et al. reported the absorption and emission spectra of 6HQ and 7HQ in ethylene glycol and glycerol.29 Unlike 7HQ, 6HQ is unable to form the K species in ethylene glycol via a hydrogen bond bridge, whereas in glycerol, a solvent molecule that is longer than the ethylene glycol molecule, the fluorescence spectra exhibit two bands, one for the E tautomer (380 nm) and one for the Z tautomer (575 nm). Both 6HQ and 7HQ have the acidic (H-bond-donating) and the basic (H-bond-accepting) sites at a relatively large distance. Thus a single solvent molecule cannot form a H-bonded bridge between the donor and the acceptor.28
In 8HQ, similar to 2HQ, the close proximity between the OH group and the N-heteroatom enhances the efficiency of intramolecular proton transfer and the ability of a single protic solvent molecule to bind to both sites simultaneously. Previous studies on 8HQ have shown that this molecule is fluorescent in concentrated acidic media but not in dilute acidic, neutral, or basic aqueous solutions.30–33 8HQ has a low fluorescence quantum yield in many solvents which can be explained as a consequence of the quenching action of the strong hydrogen bond that leads to active intramolecular proton transfer in the excited state.31,34 The fluorescence quantum yield is slightly higher in solvents of intermediate polarity such as THF and CHCl3.35 Water can be a possible environment for 8HQ isomerization to trans-E as predicted by molecular dynamics and ab initio computations. The presence of trans-E in H2O suggests an alternative fluorescence quenching reaction path in which a direct participation of solvent molecules induces intermolecular proton transfer.35
We examine in this paper the spectra of 6HQ, 7HQ, and 8HQ in different solvents with an emphasis on aqueous solvents in order to better understand the change in their spectra when incorporated into macromolecules. We investigate the mechanism of water solvation of the polar centers in HQs, experimentally in mixtures of binary solvents, and theoretically by density functional theory (DFT) and molecular dynamics (MD) simulations.
2 Experimental and theoretical methods
6HQ (98%) and 7HQ (99%) were obtained from Acros Organics. 8HQ (99%) was purchased from Sigma-Aldrich. All HQs were recrystallized from ether and the purity was checked via thin layer chromatography and from their absorption and fluorescence spectra in different solvents. Anhydrous dioxane was obtained from Sigma-Aldrich Chemical Co. Deionized water (Millipore) was used. For acidic media, fuming HCl (36.5–38.0%, analytical grade from Sigma-Aldrich) was used to adjust the pH of the solution, while 1.0 M NaOH (Pellets, >99%, Sigma-Aldrich) was used for the basic media. The concentration of HQs in all solvents was 0.01 mM. For the study in binary mixtures, a stock solution of HQ in dioxane (5 mM) was prepared. Equivolume amounts (50.0 μL) of HQ in dioxane were pipetted into separate test tubes and diluted with the appropriate amounts of dioxane–water to make volume:volume (v/v) mixtures.Absorption spectra were obtained using an HP 845× Diode Array spectrophotometer. In all the experiments, samples were contained in a 1 cm path-length quartz cell and the measurements were conducted at 23 ± 1 °C.
Geometry optimization and time-dependent calculations for the different tautomers of HQs uncomplexed and complexed with water molecules were carried out using the GAMESS program.36 Polarizable continuum model (PCM) calculations were carried out using Gaussian 03.37 MD simulations were performed using the AMBER 11 program.38 The gaff forcefield with AM1BCC charges and the TIP4PEW water model were used.39,40 Free energies of solvation were computed using the thermodynamic integration formalism in Amber, using 19 evenly spaced λ-points with independent 1 ns equilibration and 2 ns data collection phases for each sub-step. The simulation setup represents the removal of the solute from a box of water molecules, using soft core potentials41 to facilitate an efficient single step transformation.
3 Results and discussions
3.1 Absorption spectra and structures of different tautomers
The absorption spectra of HQs in water and dioxane are shown in Fig. 2. The peak in the region 310–340 nm is due to transition to the lowest 1(π,π*) excited state of the E tautomer.18 This peak is structured in dioxane for 6HQ and 7HQ which can be attributed to weak solute–solvent interaction. The peak is broad in water indicating a strong interaction between the solvent and each of the OH group and the N heteroatom of the HQ molecules. For 8HQ, the E peak is broad in both solvents which can be explained by the presence of a strong intramolecular hydrogen bond. In water, a second peak at 400 nm is observed in 7HQ which is due to absorption of the Z tautomer that gains its intensity at the expense of the E tautomer.9,23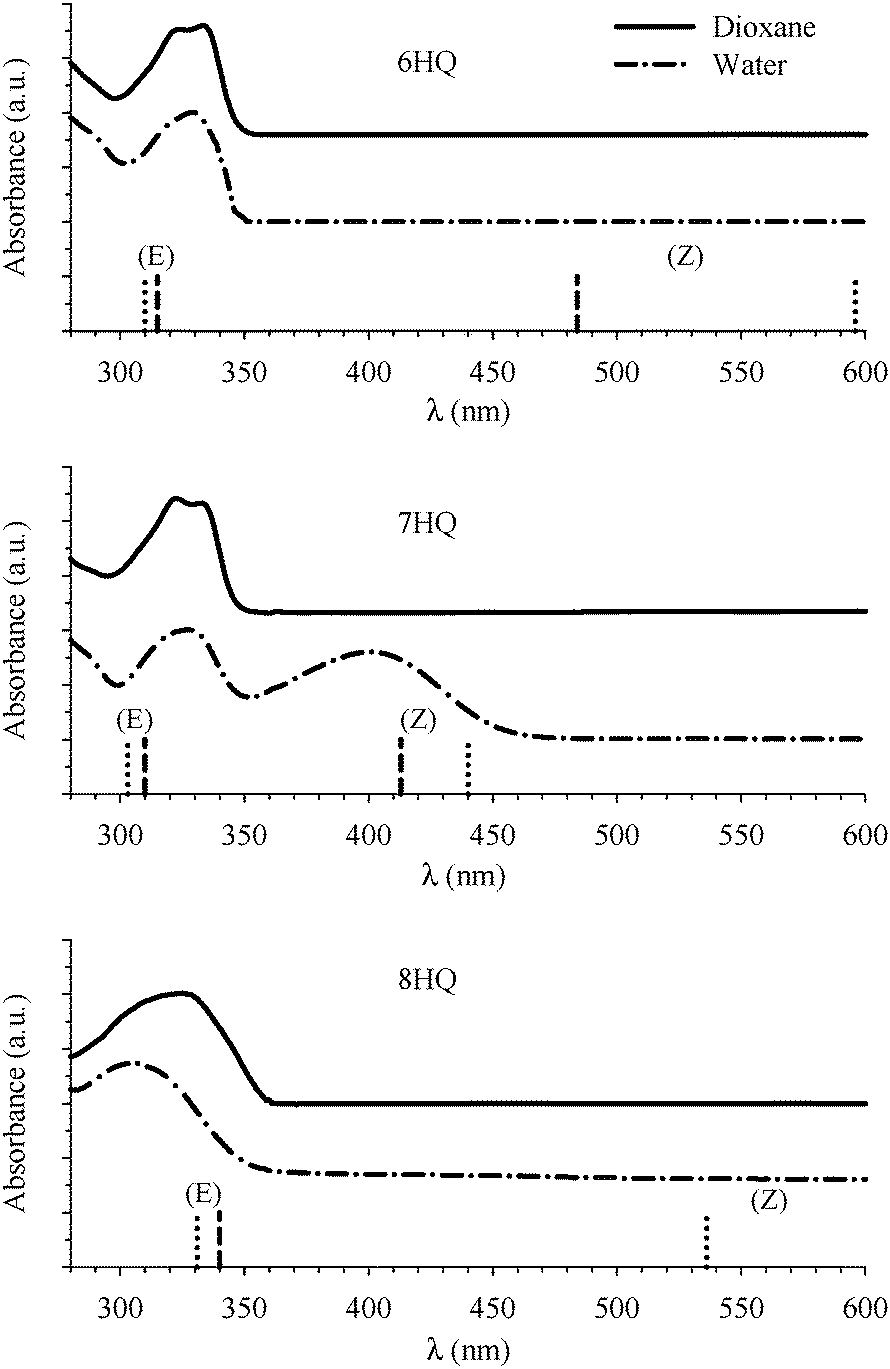 | ||
| Fig. 2 Absorption spectra of HQs in water and dioxane. The spectra are vertically spaced for clarity. Vertical lines represent the DFT calculations for the Franck–Condon S1 ← S0 transitions (dashed lines for gas phase, dotted lines for PCM). | ||
The structures of the E and Z tautomers in each HQ molecule were calculated in their ground states using the DFT-B3LYP method with a 6-311++G(2d,p) basis set. The calculations were carried out for the bare molecules (gas phase) and including the PCM with water as the solvent. The structures were fully optimized without any symmetry constraint. The results are summarized in Table 1. For the gas phase tautomers, the optimized structures were found to be planar in all cases, with the cis-E tautomers more stable than the Z tautomers in 7HQ and 8HQ. The trans-E isomer (in which the O–H bond points away from the N heteroatom) is calculated to be 1.1 and 8.0 kcal mol−1 less stable than the cis-E isomer in 7HQ and 8HQ, respectively. Similar results were obtained at the HF/6-31G(d,p) level of calculations for 7HQ.42 Experimental results in supersonic jets indicate that the trans-rotamer is barely populated for the free 7HQ molecule, as well as for 7HQ complexed with one water molecule.42 In 6HQ, although our calculated total energy for the trans-E rotamer is more stable than the cis one (only by 0.4 kcal mol−1), the difference is such that both rotamers are expected to contribute to the total population through a near free-rotation of the OH group (kT = 0.6 kcal mol−1 at 298 K). The existence of both rotamers was observed in jet-cooled spectra of the bare 6HQ molecule, as well as for 6HQ complexed with one H2O molecule.28
| 6HQ | 7HQ | 8HQ | ||||
|---|---|---|---|---|---|---|
| Bare | PCM | Bare | PCM | Bare | PCM | |
| Normalized ΔE (kcal mol−1) | ||||||
| cis-E | 0 | 0 | 0 | 0 | 0 | 0 |
| trans-E | −0.43 | −0.34 | 1.10 | 0.35 | 8.00 | 1.82 |
| Z | 20.32 | 6.22 | 11.56 | 1.63 | 15.36 | 4.66 |
| S1 ← S0 (nm) | ||||||
| cis-E | 310 | 315 | 303 | 310 | 340 | 331 |
| Z | 596 | 484 | 440 | 413 | 655 | 536 |
It is observed that the energy difference between the cis and the trans isomers decreases when including the PCM. In the case of 8HQ, a dramatic decrease in the energy of the trans-E narrows the gap between the two isomers which may indicate that the trans isomer gains stability in water. This is as expected with the breaking of the intramolecular hydrogen bond, somewhat compensated for by the solvation of the polar centers. Table 1 also shows that the energy difference between the cis-E and the Z tautomers in each HQ decreases dramatically when applying the PCM (compared to gas phase). The relative population of the Z tautomer to the cis-E tautomer is estimated from the calculated energies for each tautomer using the PCM to be >0.003% for 6HQ, 6.4% for 7HQ, and >0.04% for 8HQ. The values are consistent with the observation of the Z tautomer in 7HQ (as evidenced by the absorbance at 400 nm in Fig. 2) and the absence of any absorption due to the Z tautomer in 6HQ and 8HQ. While the Z resonance form is likely to be favored over that of the K form in water, the contribution of the K resonance form appears to remain an important factor in the relative stability of the Z/K tautomer vs. the E tautomer. DFT and MD calculations for 2-, 3-, 4-, 6-, 7-, and 8HQ, summarized in Table 2, show two distinct sets of behaviors. For 3-, 6-, and 8HQ, hydrogen transfer from oxygen to nitrogen can only yield the Z-tautomer, and the energy differences between the E and Z forms are very large. For 2-, 4-, and 7HQ, both Z and K resonance structures can be drawn. In this case the energy difference between the E and Z/K forms is calculated to be considerably smaller for 7HQ and reversed (Z/K more stable than E) for 2HQ and 4HQ. When the PCM is applied the Z(/K) forms are all preferentially stabilized with respect to the E, with 3-, 6-, and 8HQ (charged-separated Z form only) stabilized to a greater extent. However, without the extra stability imparted by the contribution of the K form, 3-, 6-, and 8HQ are not sufficiently stabilized to expect any significant population of the Z tautomer in water. Calculation of the nitrogen charge (DFT-PCM or AM1BCC atomic partial charges from MD parameterization) and ΔGsolv (MD) also support the premise that the contribution of the K form to the resonance is not insignificant: the changes in charge (Z–E) and ΔGsolv (Z–E) are consistently larger when the K form cannot be drawn.
| E | |EPCM − EBare| | |ΔGsolv| | N charge | N charge | |
|---|---|---|---|---|---|
| DFT | DFT | MD | DFT-PCM | AM1BCC | |
| (kcal mol−1) | (au) | ||||
| 2HQ | −4.19 | 3.65 | 4.29 | 0.47 | 0.30 |
| 4HQ | −3.40 | 4.19 | 5.41 | 0.50 | 0.22 |
| 7HQ | 11.56 | 9.93 | 5.77 | 0.52 | 0.45 |
| 3HQ | 14.67 | 10.53 | 10.10 | 0.55 | 0.62 |
| 6HQ | 20.32 | 14.10 | 9.92 | 0.58 | 0.65 |
| 8HQ | 15.36 | 10.70 | 10.59 | 0.64 | 0.69 |
Returning to our discussions of the spectra of 6-, 7-, and 8HQ, the calculated Franck–Condon S1 ← S0 transitions in the cis-E and Z tautomers are included in Table 1 and are displayed in Fig. 2 as vertical lines. The values are in good agreement with the measured absorption spectra shown in Fig. 2. The results also indicate that the peak at ∼330 nm in each HQ is due only to the corresponding cis-E tautomer with no contribution from the Z tautomer. The peak at ∼400 nm for 7HQ is also due solely to the Z tautomer.
3.2 Solvation of HQs by water
The above results indicate a strong interaction between HQs and water. The selective stabilization of the Z tautomer in 7HQ in the presence of water is due to solute–solvent interactions that not only determine the relative stability of the tautomeric forms but can also influence the interconversion mechanism (E ⇌ Z). A protic solvent like water, a strong hydrogen bond donor–acceptor, can accept a proton from the donor site of the solute molecule and transfer a different proton to the acceptor site of the solute. Water-assisted proton transfer mechanism studies have shown that the assistance of a water molecule significantly lowers the free energy barriers in proton-transfer-related reactions.43,44 The dynamics of such proton transfer reactions can be greatly influenced by the presence of water molecules through short-range hydrogen-bonding interactions. In this case, an explicit interaction with a limited number of water molecules could influence the whole reaction path by lowering the energy barrier due to the direct participation of water molecules in the proton transfer process. The absence of the Z absorption in other protic solvents such as methanol and ethanol7,9,15,21,22 may indicate that, unlike water, short range interaction between solvent molecules and the polar centers in 7HQ is not enough to stabilize the Z tautomer in the ground state.The tendency of water molecules to strongly associate with each other through intermolecular hydrogen bonds allows more than one molecule of water to form a solvent network or a solvent wire along which proton transfers can take place to and from the solute. Such proton transfer mechanisms through a water bridge have been formulated theoretically.44–47 We have recently estimated the number of water molecules solvating the hydrogen bonding centers of 7HQ and several other systems experimentally in binary mixtures of dioxane–water48–50 and theoretically using ab initio methods.48,49 The choice of dioxane is convenient because it is miscible with water in all proportions and thus provides an opportunity to study the effect of a broad range of solvent polarity. Mixtures of dioxane and water were also proposed as media to study probes in nanoenvironments similar to those encountered in vesicles and at interfaces.48,50–53
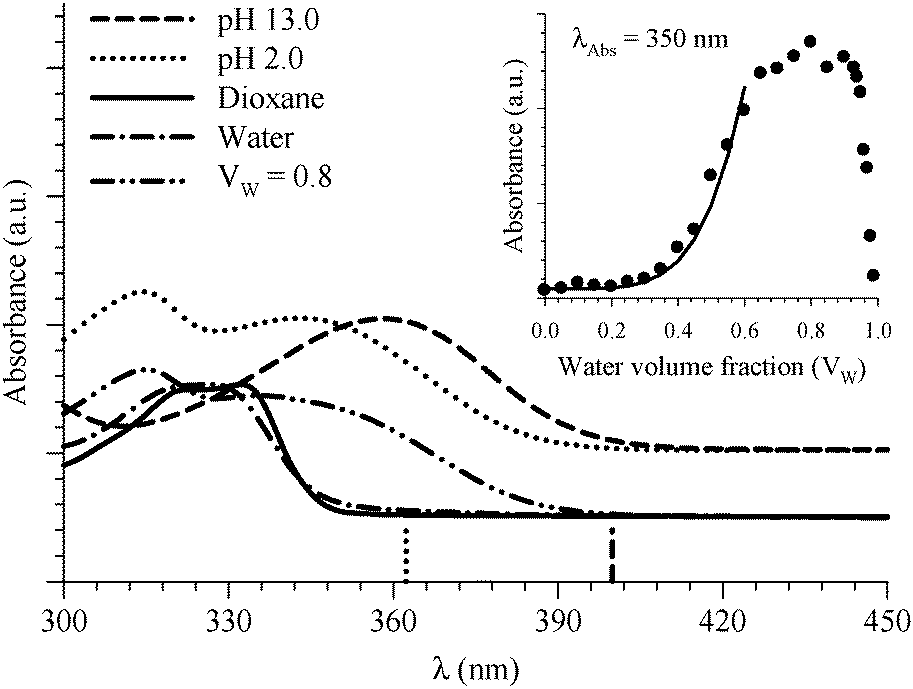 | ||
| Fig. 3 Absorption spectra of 6HQ in different media as indicated in the graph. Vertical lines represent the DFT calculations (PCM) for the Franck–Condon S1 ← S0 transitions in the protonated (dotted line) and deprotonated (dashed line) species. The inset shows the absorbance change of the peak at 350 nm as a function of water volume fraction in the binary mixtures of dioxane and water. The solid line represents the best nonlinear regression fit to eqn (3). | ||
Formation of the protonated species is a consequence of specific 6HQ–water H-bonding interaction that is absent in neat water solvent. (Dioxane alone is not a hydrogen bond donor solvent according to its Kamlet–Taft solvatochromic parameter (α = 0).54) The formation of the protonated form in a water-rich dioxane mixture must then stem from the local solvation of the N-heteroatom of 6HQ by water molecules that behave in an acidic manner in the presence of dioxane. We have shown recently that a molecule such as 2-(2′-hydroxyphenyl)benzoxazole experiences a basic environment in its excited state when dissolved in dioxane or methanol containing high water contents.48 Dioxane and methanol solvents that contain high water contents are reported to have higher ionization strength than solvents containing low water contents.55 To our knowledge, there have been no reports on either acidic or basic effects on the ground state of any dye dissolved in binary mixtures of dioxane or methanol with water. This may be due to undetectable or no effects on the ground state because of little to no molecular response. The present study may be the first to report an acid-like influence by a dioxane–water binary mixture on the ground state of a solute molecule. This effect could be a consequence of the amphoteric character of water that can behave as either an acid or a base.54 In order to confirm this behavior, we studied 6HQ in binary mixtures of dioxane–methanol and methanol–water (data are included in the ESI†). The results show no acidic or basic effect of the binary solvents, which emphasizes the unique behavior of water in the presence of dioxane.
The acidic effect of the binary mixtures of dioxane and water on 6HQ can be explained by selective solvation of the polar sites of 6HQ by water. In pure dioxane no absorption for the protonated species is observed. As the water content is increased the absorbance at 350 nm steadily increases until dropping-off sharply as the solvent mixture approaches pure water. This behavior can be used to investigate the solvation of the protonated species. Increasing the water content leads to local solvation of the polar sites by n water molecules which can be expressed by the following equilibrium:
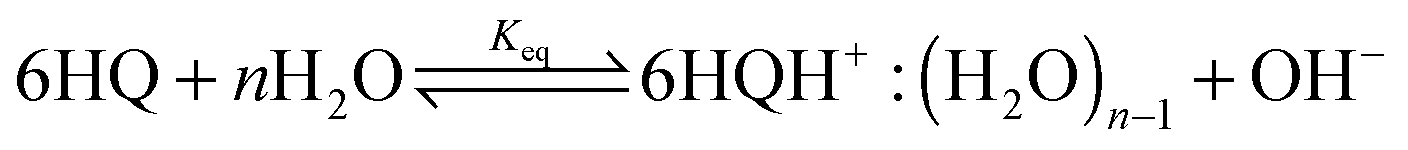 | (1) |
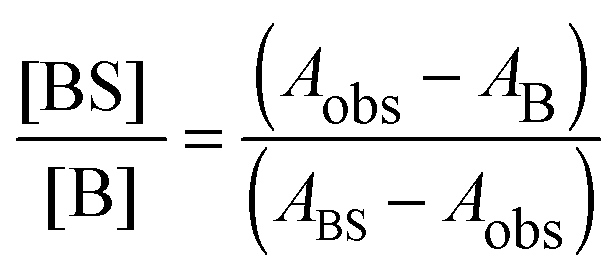 | (2) |
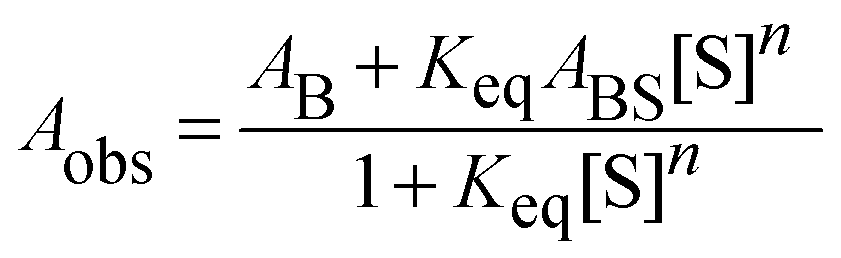 | (3) |
The inset of Fig. 3 indicates that the absorbance of 6HQH+ drops sharply after water volume fraction >80% (equivalent to [H2O] > 44 M). The behavior of binary mixtures of dioxane and water was investigated using the Kamlet–Taft parameter α and was found to approach that of pure water above 25 M concentration of water.57–59
Our calculations, using the DFT-B3LYP method with a 6-311++G(2d,p) basis set, show that 5 water molecules are sufficient to link the polar sites in 6HQ. The structures shown in Fig. 4 were produced by adding water molecules in close proximity to the polar sites and then fully optimizing the geometries. The produced water network could provide a means to facilitate hydrogen transfer from oxygen to nitrogen. However, in line with the PCM calculations, the relative energies of these structures suggest that even if such a network were formed, hydrogen transfer is still energetically prohibited: the cis-E:5H2O complex was calculated to be more stable than the Z:5H2O complex by 11.25 kcal mol−1.
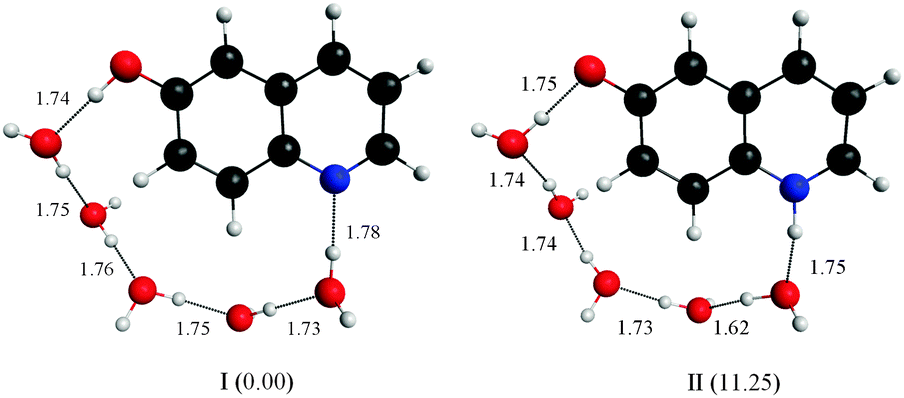 | ||
| Fig. 4 Structures of the most stable ground-state minimum configurations of the cis-E and Z tautomers of 6HQ with networks of five water molecules. The structures were obtained from DFT calculations described in the text. The values in parentheses are relative energies in kcal mol−1. The lengths of the hydrogen bonds are shown (in Å). | ||
Further insight into the solvation of 6HQ by water can be obtained from MD simulations. Simulations were performed in explicit solvent for both the E and Z tautomers embedded in a 12 Å deep layer of TIP4PEW water molecules and equilibrated to room temperature and pressure using standard procedures. Parameterization of the 6HQ was done using the gaff forcefield and AM1BCC charges were assigned. After equilibration, 10 ns of unrestrained MD simulations were conducted and the ptraj module of Amber was used to produce the water density maps shown in Fig. 5. The maps show well-defined hydrogen bonds about the N-heteroatom and much less defined hydrogen bonds about the hydroxyl group in the E tautomer. For the Z tautomer, the results show well-defined hydrogen bonds around both polar sites. The results show clearly that the OH group adapts an average position to the H atom in which no clear cis/trans isomer is favored. This result agrees with the above DFT calculations in which the OH group possesses a free rotation.
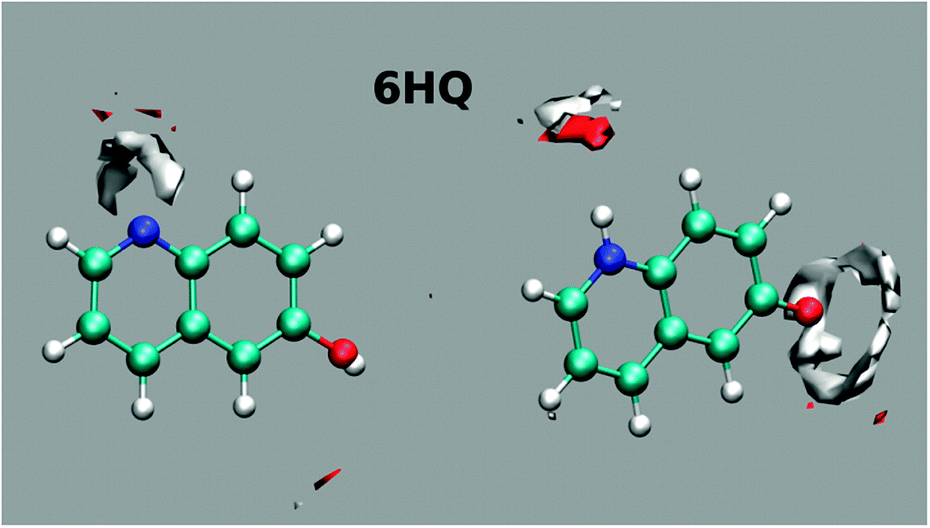 | ||
| Fig. 5 Mapping the water density around the polar sites of the E tautomer (left) and the Z tautomer (right) of 6HQ. The calculations were performed using MD simulations as described in the text. Dark (red) areas indicate increased oxygen density (H-bond acceptor position), and light (white) areas indicate hydrogen density (H-bond donor regions). The average structures of E and Z are superimposed onto the maps. | ||
Additionally, H-bond networks can be analyzed directly from MD trajectory structures. We count the number of structural snapshots in which one to four water molecules connect the polar sites of 6HQ by a continuous H-bond network, based on standard structural parameters of the Amber ptraj module. Very few such water wires were found, with a water chain of length 4 occurring in 3% of snapshots and 0% of shorter chains observed. This is in good agreement with the above results, where five water molecules are needed to connect the polar sites.
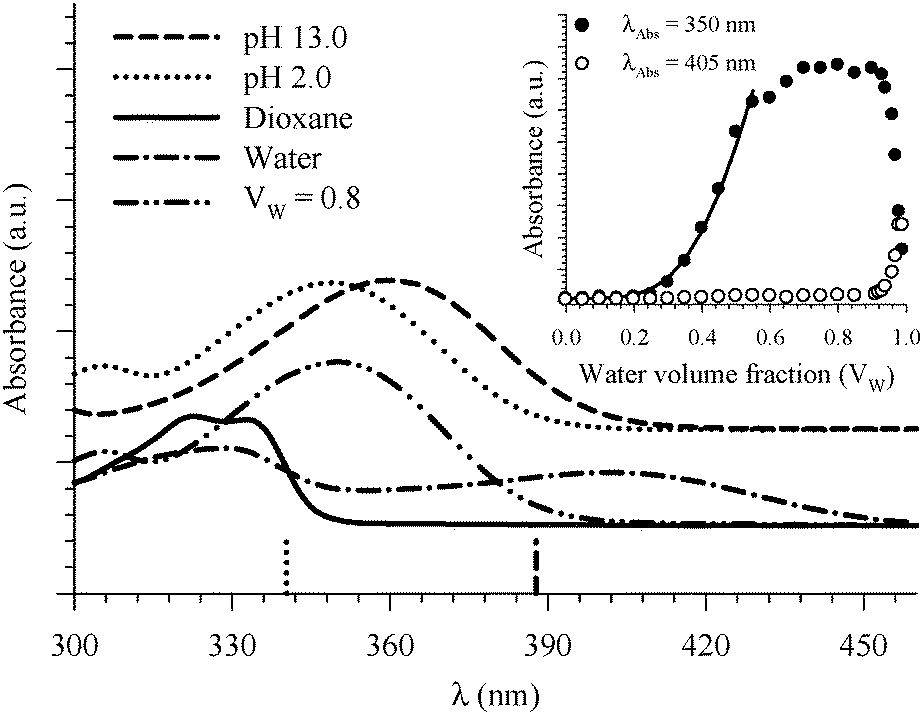 | ||
| Fig. 6 Absorption spectra of 7HQ in different media as indicated in the graph. Vertical lines represent the DFT calculations (PCM) for the Franck–Condon S1 ← S0 transitions in the protonated (dotted line) and deprotonated (dashed line) species. The inset shows the absorbance change of the peaks at 350 and 405 nm as a function of water volume fraction in the binary mixtures of dioxane and water. The solid line represents the best nonlinear regression fit to eqn (3). | ||
Fitting the absorbance change of the protonated species to eqn (3) gives n = 4 (Keq = (1.7 ± 0.2) × 10−7 M−4), which is an estimate for the number of water molecules needed to solvate the polar sites (the fit is shown in the inset of Fig. 6). This may indicate that the local solvation of the neutral and protonated forms is subtly different as we have estimated recently the value of n to be three for 7HQ from the fluorescence change of the Z tautomer as a function of water concentration in the binary mixtures.9 Our DFT calculations indicate that three water molecules are needed to maintain the planar structures of the cis-E and the Z tautomers with a stretched water network that reaches the two polar regions in each tautomer. The optimized structures are shown in Fig. 7 with the corresponding hydrogen bonding lengths (I for cis-E:3H2O and II for Z:3H2O). As shown in the figure, the water network (or water wire) forms strong hydrogen bonds with each polar center in each tautomer. The dihedral angles between the molecular plane and the hydrogen bonds at each polar center in 7HQ are less than 2° in all cases and all XOX angles are close to linearity (≥176°).The cis-E:3H2O complex was calculated to be more stable than the Z:3H2O complex by 6.44 kcal mol−1.
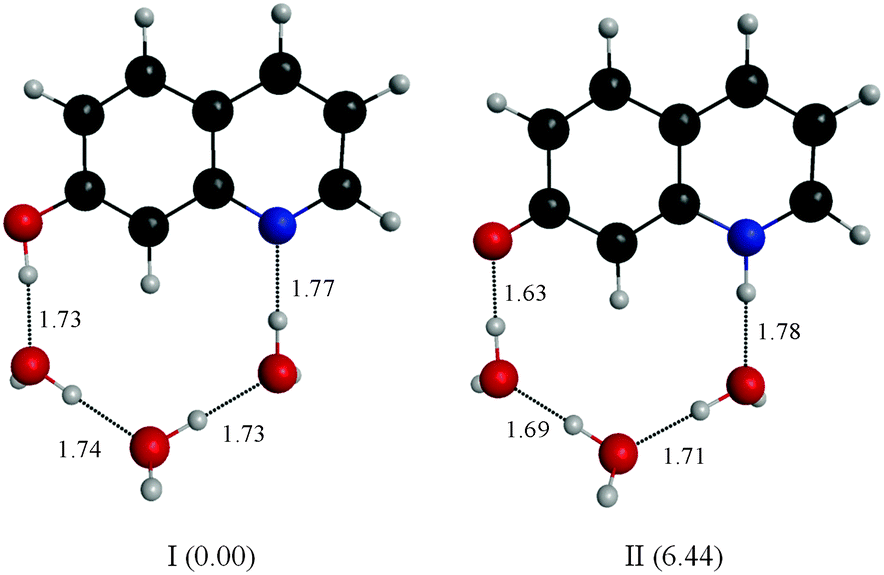 | ||
| Fig. 7 Structures of the most stable ground-state minimum configurations of the cis-E and Z tautomers of 7HQ with three water molecules. The structures were obtained from DFT calculations described in the text. The values in parentheses are relative energies in kcal mol−1. The lengths of the hydrogen bonds are shown (in Å). | ||
The results of mapping the water density around the polar sites in 7HQ are shown in Fig. 8. The maps show well-defined hydrogen bonds about the N-heteroatom, but no directional hydrogen bonds around the hydroxyl group. Free rotation of the OH group is evident from the average structure location of the H-atom being nearly collinear with the C–O bond, with no preference for either the cis- or the trans-isomer. Although the OH group is free to rotate during simulations, interestingly, conducting the 7HQ parameterization with the cis-isomer gives atomic partial charges that are slightly different from those obtained when parameterizing the trans-isomer. In our previous results, we started parameterization from the trans-isomer and while the results showed essentially the same average molecular structure a well-defined hydrogen bond in one direction was observed.9 Since the cis-isomer of 7HQ was found to be the more stable here, we reparameterized 7HQ using this starting structure for the present work. No other HQ isomers exhibited this strong starting structure bias effect.
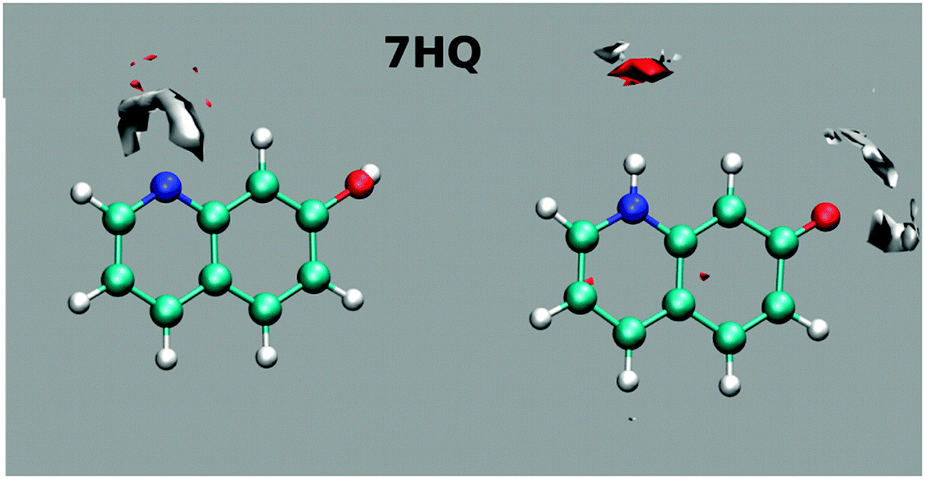 | ||
| Fig. 8 Mapping the water density around the polar sites of the E tautomer (left) and the Z tautomer (right) of 7HQ. The calculations were performed using MD simulations as described in the text. Dark (red) areas indicate increased oxygen density (H-bond acceptor position), and light (white) areas indicate hydrogen density (H-bond donor regions). The average structures of E and Z are superimposed onto the maps. | ||
For the Z tautomer, the maps show well-defined hydrogen bonds around the N-heteroatom and slightly less well-defined hydrogen bonds around the oxygen atom.
An H-bond network analysis as described above found that water chains of three and four molecules connected the polar groups of 7HQ in 4% and 11% of structural snapshots (no shorter chains were found), again in good agreement with the values of n = 3 or 4 found here and in previous work.
Finally, the absorption signature of the Z tautomer starts to emerge at 405 nm (Fig. 6) in the binary mixtures only when Vw > 0.70, and increases sharply for Vw ≥ 0.90. Both E and Z are not formed when the protonated form is present in high concentration in the binary mixtures. This is expected because any E or Z tautomers will be protonated in an acidic environment. It is only when the local environment is neutral that both should be stabilized.
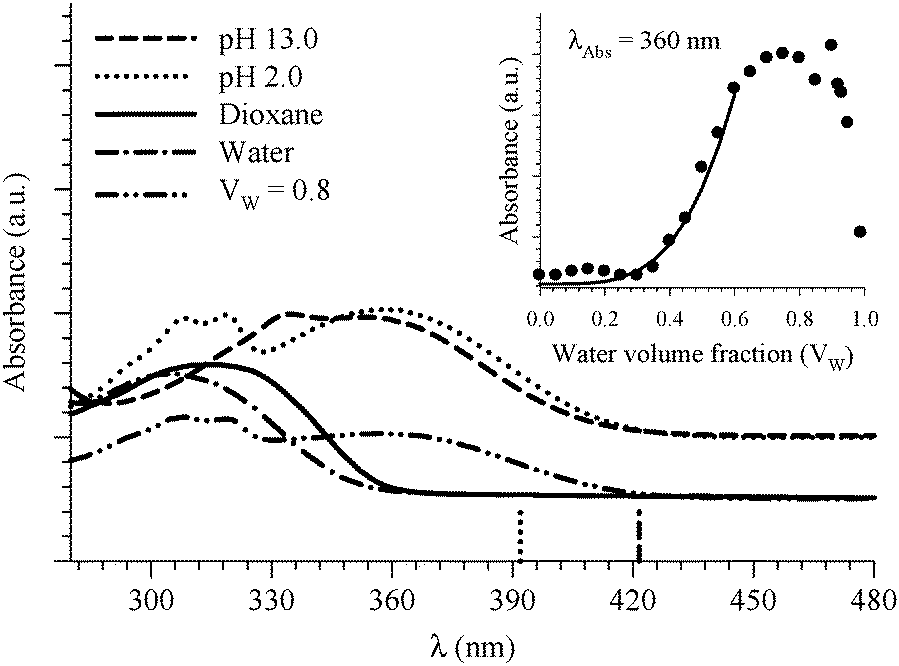 | ||
| Fig. 9 Absorption spectra of 8HQ in different media as indicated in the graph. Vertical lines represent the DFT calculations (PCM) for the Franck–Condon S1 ← S0 transitions in the protonated (dotted line) and deprotonated (dashed line) species. The inset shows the absorbance change of the peak at 360 nm as a function of water volume fraction in the binary mixtures of dioxane and water. The solid line represents the best nonlinear regression fit to eqn (3). | ||
Four water molecules seem to be a large number if the E tautomer is in the cis form. We calculated structures of both the cis-E and the Z tautomers with one water molecule linking the polar sites using the DFT method (Fig. 10). The results show that one water molecule is enough to maintain the planarity of the molecule and to connect the polar sites in each tautomer. The calculations show that there are two intermolecular H-bonds where H2O acts as a bridge between the ring nitrogen atom and the OH group. The cis-E:(H2O) complex was calculated to be more stable than the Z:(H2O) complex by 8.74 kcal mol−1. Similar results were obtained by B3LYP calculations in which the intermolecular hydrogen bond distances are found to be shorter (stronger) in the 8HQ:H2O complex than the intramolecular hydrogen bonds, indicating that the proton transfer process takes place more easily in the complex.60
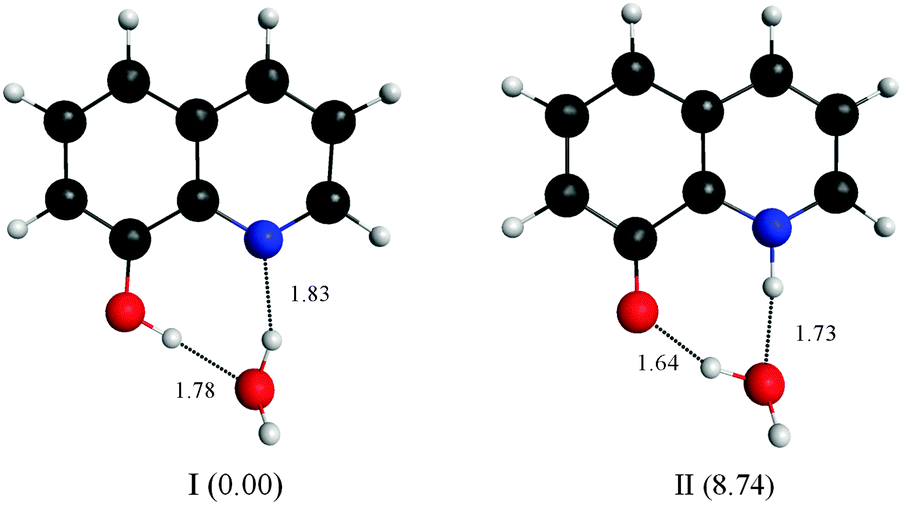 | ||
| Fig. 10 Structures of the most stable ground-state minimum configurations of the cis-E and Z tautomers of 8HQ with one water molecule linking the polar sites. The structures were obtained from DFT calculations described in the text. The values in parentheses are relative energies in kcal mol−1. The lengths of the hydrogen bonds are shown (in Å). | ||
MD simulation results clearly indicate that the E tautomer is stable in the cis isomer (Fig. 11). Mapping the water density around the polar sites in 8HQ shows well-defined hydrogen bonds near the polar sites in each of the tautomers (cis-E and Z).
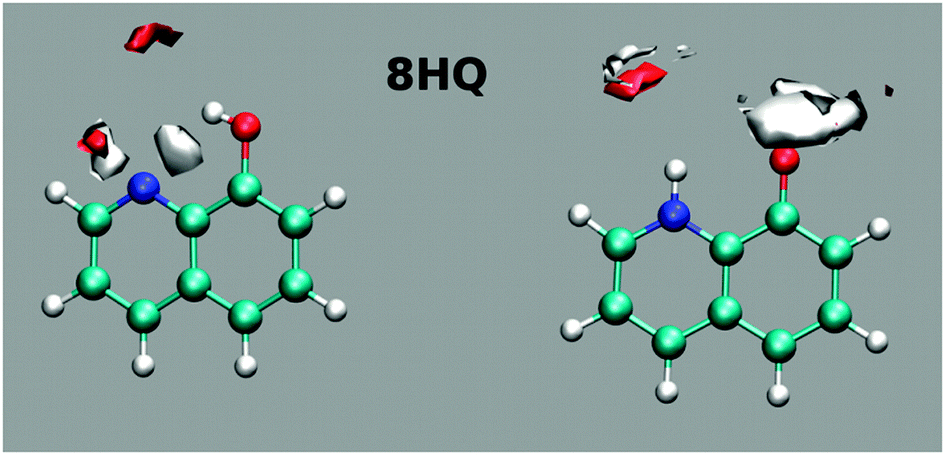 | ||
| Fig. 11 Mapping the water density around the polar sites of the cis-E tautomer (left) and the Z tautomer (right) of 8HQ. The calculations were performed using MD simulations as described in the text. Dark (red) areas indicate increased oxygen density (H-bond acceptor position), and light (white) areas indicate hydrogen density (H-bond donor regions). The average structures of E and Z are superimposed onto the maps. | ||
For the neighboring polar groups of 8HQ, the analysis of the H-bond network from MD simulations indicated that a single water molecule often connects the two sites (8% of snapshots) with longer water chains observed frequently as well, in 3%, 6% and 11% of structural snapshots respectively. The differences in these water H-bond chains found for the three HQ molecules correlate nicely with the increasing distance between O and N. Few water molecules are needed to form a chain for 8HQ, 3–4 molecules for 7HQ and more than four for 6HQ.
Participation of two water molecules in the intermolecular proton transfer process is shown to increase the fraction of the trans-E tautomer.35 The existence of higher aggregates of 8HQ:(H2O)n has been postulated for low concentrations of 8HQ in chlorinated solvents.12 Amati et al. reported the probability distribution of the torsion angle between the OH group and the aromatic rings of 8HQ and observed a doublet around the cis-isomer.35 The two peaks of the doublet are shifted by about +3° and −3° from 180°. The authors correlate this observation with their calculated cis-conformer that shows a similar torsion angle only when the 8HQ is solvated by four water molecules. Our experimental estimate of four water molecules (vide supra) may then be correlated with the existence of this conformer.
4 Conclusions
In this work, we studied the ground state tautomeric equilibria of 6, 7, and 8HQs in neat water and dioxane, and in their binary mixtures. The results indicate that the enol form is the only tautomer for the three HQs in dioxane and water, with the exception of 7HQ in which both the enol and the zwitterion tautomers exist in equilibrium in water. The different tautomers were assigned using DFT calculations performed in the gas phase and using PCM. The existence of a protonated species in each molecule is detected in the absorption spectra when the water concentration is high in the mixture (maximum protonation was observed for Vw between 60–80%). The existence of protonated species was confirmed by comparing the absorption spectra in binary solvents with those in acidic and basic aqueous solutions, and by DFT calculations of the Franck–Condon S1 ← S0 transitions. We determined the number of water molecules solvating the polar sites in each HQ molecule by fitting the absorbance change to a binding isotherm, and the structures were calculated by DFT. MD simulations indicate that the water density is well-defined around the N-heteroatom in all HQs, whereas the water density is only well-defined around the hydroxyl group in 8HQ. The results from the MD simulations point also to the presence of free rotation of the OH group in 6HQ and 7HQ, and the stability of the cis-isomer in 8HQ.The current study clarifies the effect of various polarities of the environment on the formation and stability of different tautomers of HQs in the ground state and should help in understanding the nature of binding sites in macromolecules when HQs are used as local reporters. One possible application of HQs is utilizing them to detect acidic media in binding sites. The unique spectral signatures of 7HQ in water also make this molecule a potential probe to detect the presence of water in nanocavities of macromolecules.
Acknowledgements
The authors would like to thank Sultan Qaboos University for financial support (Grant No. IG/SCI/CHEM/12/01), and Dr Muataz Al-Barwani for assistance with calculations carried out at the University's High Performance Computing Facility.References
- R. A. Copeland, Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, Wiley, New York, 2000 Search PubMed.
- Methods in Molecular Biology: Protein–Ligand Interactions, Methods and Applications, ed. G. U. Nienhaus, Humana Press, New Jersey, 2005 Search PubMed.
- (a) C. Albrecht, K. Blank, M. Lalic-Mülthaler, S. Hirler, T. Mai, I. Gilbert, S. Schiffmann, T. Bayer, H. Clausen-Schaumann and H. E. Gaub, Science, 2003, 301, 367 CrossRef CAS PubMed; (b) J. Kim, C.-Z. Zhang, X. Zhang and T. A. Springer, Nature, 2010, 466, 992 CrossRef CAS PubMed; (c) C. Grashoff, B. D. Hoffman, M. D. Brenner, R. Zhou, M. Parsons, M. T. Yang, M. A. McLean, S. G. Sligar, C. S. Chen, T. Ha and M. A. Schwartz, Nature, 2010, 466, 263 CrossRef CAS PubMed.
- G. Weber and F. J. Farris, Biochem., 1979, 18, 3075 CrossRef CAS.
- R. Narazaki, T. Maruyama and M. Otagiri, Biochim. Biophys. Acta, 1997, 1338, 275 CrossRef CAS.
- O. K. Abou-Zied, N. Al-Lawatia, M. Elstner and T. B. Steinbrecher, J. Phys. Chem. B, 2013, 117, 1062 CrossRef CAS PubMed.
- O. K. Abou-Zied and N. Al-Lawatia, ChemPhysChem, 2011, 12, 270 CrossRef CAS PubMed.
- N. Al-Lawatia, T. Steinbrecher and O. K. Abou-Zied, Proc. SPIE-Int. Soc. Opt. Eng., 2012, 82331, 82331D CrossRef PubMed.
- N. Al-Lawatia, J. Husband, T. Steinbrecher and O. K. Abou-Zied, J. Phys. Chem. A, 2011, 115, 4195 CrossRef CAS PubMed.
- A. Khedr, A. El-Bindary and A. M. A. El-Gawad, Chem. Pap., 2005, 59, 336 CAS.
- M. S. Mehata, H. C. Joshi and H. B. Tripathi, Spectrochim. Acta, Part A, 2002, 58, 1589 CrossRef CAS.
- E. Bardez, I. Devol, B. Larrey and B. Valeur, J. Phys. Chem. B, 1997, 101, 7786 CrossRef CAS.
- S. F. Mason, J. Philp and B. E. Smith, J. Chem. Soc. A, 1968, 3051 RSC.
- P. J. Thistlethwaite, Chem. Phys. Lett., 1983, 96, 509 CrossRef CAS.
- H. J. Park, O.-H. Kwon, C. S. Ah and D.-J. Jang, J. Phys. Chem. B, 2005, 109, 3938 CrossRef CAS PubMed.
- M. Itoh, T. Adachi and K. Tokumura, J. Am. Chem. Soc., 1984, 106, 850 CrossRef CAS.
- S.-Y. Park and D.-J. Jang, J. Am. Chem. Soc., 2010, 132, 297 CrossRef CAS PubMed.
- O.-H. Kwon, H. Doo, Y.-S. Lee and D.-J. Jang, ChemPhysChem, 2003, 4, 1079 CrossRef CAS PubMed and references therein.
- O.-H. Kwon, T. G. Kim, Y.-S. Lee and D.-J. Jang, J. Phys. Chem. B, 2006, 110, 11997 CrossRef CAS PubMed.
- O. Poizat, E. Bardez, G. Buntinx and V. Alain, J. Phys. Chem. A, 2004, 108, 1873 CrossRef CAS.
- S.-I. Lee and D.-J. Jang, J. Phys. Chem., 1995, 99, 7537 CrossRef CAS.
- W. K. Kang, J. S. Cho, M. Lee, D. H. Kim, R. Ryoo, K. H. Jung and D.-J. Jang, Bull. Korean Chem. Soc., 1992, 13, 140 CAS.
- W.-H. Fang, J. Am. Chem. Soc., 1998, 120, 7568 CrossRef CAS.
- A. Lavin and S. Collins, Chem. Phys. Lett., 1993, 204, 96 CrossRef CAS.
- M. R. Nimlos, D. F. Kelley and E. R. Bernstein, J. Phys. Chem., 1987, 91, 6610 CrossRef CAS.
- R. Rautela, N. K. Joshi, H. C. Joshi, N. Tewari and S. Pant, J. Mol. Liq., 2010, 154, 47 CrossRef CAS PubMed.
- H. Yu, O.-H. Kwon and D.-J. Jang, J. Phys. Chem. A, 2004, 108, 5932 CrossRef CAS.
- A. Bach, J. Hewel and S. Leutwyler, J. Phys. Chem. A, 1998, 102, 10476 CrossRef CAS.
- I. García-Ochoa, P. Bisht, F. Sánchez, E. Martinez-Ataz, L. Santos, H. Tripathi and A. Douhal, J. Phys. Chem. A, 1998, 102, 8871 CrossRef.
- M. P. Bratzel, J. J. Aaron, J. D. Winefordner, S. G. Schulman and H. Gershon, Anal. Chem., 1972, 44, 1240 CrossRef CAS.
- R. E. Ballard and J. W. Edwards, J. Chem. Soc., 1964, 4868 RSC.
- S. G. Schulman and Q. Fernando, Tetrahedron, 1968, 24, 1777 CrossRef.
- S. G. Schulman and Q. Fernando, J. Phys. Chem., 1967, 71, 2668 CrossRef CAS.
- G. M. Badger and A. G. Moritz, J. Chem. Soc., 1958, 3437 RSC.
- M. Amati, S. Belviso, P. L. Cristinziano, C. Minichino, F. Lelj, I. Aiello, M. La Deda and M. Ghedini, J. Phys. Chem. A, 2007, 111, 13403 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- M. J. Frisch, et al., GAUSSIAN-03, Rev. B.01, Gaussian Inc., Pittsburgh, PA, 2003 Search PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossvary, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M. J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P. A. Kollman, AMBER 11, Univeristy of California, San Francisco, 2010 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- H. W. Horn, W. C. Swope, J. W. Pitera, J. D. Madura, T. J. Dick, G. L. Hura and T. Head-Gordon, J. Chem. Phys., 2004, 120, 9665 CrossRef CAS PubMed.
- T. Steinbrecher, D. L. Mobley and D. A. Case, J. Chem. Phys., 2007, 127, 214108 CrossRef PubMed.
- S. Coussan, A. Bach and S. Leutwyler, J. Phys. Chem. A, 2000, 104, 9864 CrossRef CAS.
- N. Markova, V. Enchev and I. Timtcheva, J. Phys. Chem. A, 2005, 109, 1981 CrossRef CAS PubMed.
- A. Kyrychenko and J. Waluk, J. Phys. Chem. A, 2006, 110, 11958 CrossRef CAS PubMed.
- L. Carballeira and I. Perez-Juste, THEOCHEM, 1996, 368, 17 CrossRef CAS.
- Y.-C. Tian and W.-H. Fang, J. Phys. Chem. A, 2006, 110, 1170 Search PubMed.
- R.-Z. Liao, W.-J. Ding, J.-G. Yu, W.-H. Fang and R.-Z. Liu, J. Phys. Chem. A, 2007, 111, 3184 CrossRef CAS PubMed.
- O. K. Abou-Zied, Chem. Phys., 2007, 337, 1 CrossRef CAS PubMed.
- O. K. Abou-Zied and O. I. K. Al-Shihi, Phys. Chem. Chem. Phys., 2009, 11, 5377 RSC.
- O. K. Abou-Zied, J. Photochem. Photobiol., A, 2006, 182, 192 CrossRef CAS PubMed.
- E. C. C. Melo, S. M. Costa, A. L. Maçanita and H. Santos, J. Colloid Interface Sci., 1991, 141, 439 CrossRef CAS.
- M. Belletête, M. Lachapelle and G. Durocher, J. Phys. Chem., 1990, 94, 5337 CrossRef.
- M. Belletête, G. Lessard and G. Durocher, J. Lumin., 1989, 42, 337 CrossRef.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, 3rd edn, VCH, Weinheim, Germany, 2003 Search PubMed.
- A. H. Fainberg and S. J. Winstein, J. Am. Chem. Soc., 1956, 78, 2770 CrossRef CAS.
- K. A. Connors, Binding Constants. The Measurements of Molecular Complex Stability, Wiley, New York, 1987 Search PubMed.
- S. M. Andrade and M. B. Costa, Phys. Chem. Chem. Phys., 1999, 1, 4213 RSC.
- E. Casassas, G. Fonrodona and A. de Juan, J. Solution Chem., 1992, 21, 147 CrossRef CAS.
- W. J. Cheong and P. W. Carr, Anal. Chem., 1988, 60, 820 CrossRef CAS.
- Q. S. Li and W. H. Fang, Chem. Phys. Lett., 2003, 367, 637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption spectra of HQs in dioxane–water binary mixtures; absorption spectra of 6HQ in dioxane–methanol and methanol–water binary mixtures. See DOI: 10.1039/c3cp52811a |
| This journal is © the Owner Societies 2014 |