 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of anion on Ag(I) meso-helical chains formed with 4,4′-dipyridyl ketone: solvent versus anion bridging and anion effects on the strength of ligand binding†
Komal M.
Patil
,
Scott A.
Cameron
,
Stephen C.
Moratti
and
Lyall R.
Hanton
*
Department of Chemistry (Te Tari Hua-Ruanuku), University of Otago, P. O. Box 56, Dunedin 9054, New Zealand. E-mail: lhanton@chemistry.otago.ac.nz; Fax: +64 3479 7906; Tel: +64 3479 7918
First published on 12th March 2014
Abstract
The synthesis and characterisation by IR spectroscopy and elemental analysis of ten new Ag(I)–L complexes are described. Of these complexes, nine are characterised by single crystal X-ray diffraction: {[Ag(L)](CF3SO3)·1/2H2O}∞ (1), {[Ag(L)](ClO4)·1/2H2O}∞ (2), {[Ag2(L)2(CH3CN)](ClO4)2·2CH3CN·H2O}∞ (3), {[Ag2(L)2(CH3CN)2](ClO4)2·CH3CN}∞ (4), {[Ag2(L)2(CH3CN)2](PF6)2·2CH3CN}∞ (5), {[Ag(L)2](CF3SO3)·1/2H2O}∞ (6), {[Ag(L)2](BF4)}∞ (7), {[Ag(L)2](PF6)}∞ (8) and {[Ag(L)2](PF6)·2CH3CN}∞ (9). The primary structures of 1–6 were meso-helical one-dimensional (1D) polymers, while 7 was a helical 1D polymer and 8 and 9 were (4,4) networks. Complexes 1–5 possessed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal-to-ligand (M:L) ratios, while complexes 6–9 possessed 1:2 M:L ratios. The meso-helical chains of complexes 1 and 2 were di-μ-bridged at the Ag(I) nodes by the counteranions CF3SO3− and ClO4−, respectively, while the meso-helical chains of complexes 3–5 were di-μ-bridged at the Ag(I) nodes by the CH3CN molecules. The effect of counteranions and solvent molecules on delicate anion–Ag, π–π-stacking and argentophilic interactions was studied through complexes 1–5. The 1D chains of complexes 6 and 7 possessed monodentate L ligand side arms. The uncoordinated N-donors of these side arms were inclined towards the Ag(I) centre of the adjacent chains and demonstrated narrower Ag–Npy–Cg(pyridyl) angles. In the case of complexes 8 and 9, wider Ag–Npy–Cg(pyridyl) angles and stronger N⋯Ag interactions resulted in (4,4) nets. The effects of the size and the nature of the counteranions on the topology were studied through complexes 6–9.
:1 metal-to-ligand (M:L) ratios, while complexes 6–9 possessed 1:2 M:L ratios. The meso-helical chains of complexes 1 and 2 were di-μ-bridged at the Ag(I) nodes by the counteranions CF3SO3− and ClO4−, respectively, while the meso-helical chains of complexes 3–5 were di-μ-bridged at the Ag(I) nodes by the CH3CN molecules. The effect of counteranions and solvent molecules on delicate anion–Ag, π–π-stacking and argentophilic interactions was studied through complexes 1–5. The 1D chains of complexes 6 and 7 possessed monodentate L ligand side arms. The uncoordinated N-donors of these side arms were inclined towards the Ag(I) centre of the adjacent chains and demonstrated narrower Ag–Npy–Cg(pyridyl) angles. In the case of complexes 8 and 9, wider Ag–Npy–Cg(pyridyl) angles and stronger N⋯Ag interactions resulted in (4,4) nets. The effects of the size and the nature of the counteranions on the topology were studied through complexes 6–9.
Introduction
1D architectures form an extensively explored area of coordination polymer chemistry. About 40% of the total reported coordination polymers in the last decade are 1D in nature. Yet there have been very few reviews dedicated to 1D coordination polymers between the years 1993 and 2010, because many researchers perhaps consider 1D coordination polymers to be structurally less attractive than their higher dimensional counterparts.1 However, through weaker interactions these simple and seemingly less attractive structures possess the ability to demonstrate unusual and interesting architectures. In this regard, Ag(I) is particularly useful, and a significant number of 1D coordination polymers contain Ag(I), as its low dimensional and accommodating stereochemistry often allows it to interact with additional donor atoms from solvent or a counteranion.2Rigid linear bridging ligands enable the formation of predictable arrays because of their configuration, coordination activity, and relative orientation of the donor groups. Rigid linear linking ligands such as 4,4′-bipyridine and pyrazine have already been extensively studied for designing linear 1D coordination polymers and higher dimensional networks.3 Introduction of a bend in these ligands presents a new variable to the study of the coordinating aspects of these linear linkers. The bend provides an opportunity to study lower dimensional structures such as helical,4meso-helical,5,6 zigzag7–9 chains and other structures of current interest.10 One simple bent ligand is 4,4′-dipyridyl ketone (L) (Fig. 1). In the solid state, L contains a chiral axis passing through the carbonyl group of the ligand. NDDO calculations reported for L determined that the two rotational energy maxima for rotation of both pyridine rings through or orthogonal to the molecular plane are approximately 45 and 20 kJ mol−1, respectively.11 In solution, these two enantiomers readily interconvert from one form to another because of the low energy of conversion, effectively making them appear achiral.12,13 However in the solid state, the conjugating effect between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Py-rings and the hydrogen repulsion in the planar form provide resonance stability to this ligand.11 The process of stabilisation “freezes” the racemates making L behave as a two-bladed chiral molecular propeller in the solid state.14 An analogous ligand, 4,4′-dipyridyl amine, acts in a similar fashion as a two-bladed molecular propeller.15 Surprisingly, there have been a few examples of the use of L in coordination polymer chemistry. LaDuca and co-workers have reported the use of L as a linking ligand with Cu(NO3)2 forming three-fold double helices of [Cu(L)]n12 and with AgNO3 forming zigzag 1D chains of [Ag(L)(NO3)]n.13 The L ligand formed a pair of 1D meso-helical chains di(μ:κ2O,O′)-bridged at the Ag(I) nodes when a CF3COO− counteranion was employed. These chains extended their framework by virtue of weak π–π interactions.17
O and Py-rings and the hydrogen repulsion in the planar form provide resonance stability to this ligand.11 The process of stabilisation “freezes” the racemates making L behave as a two-bladed chiral molecular propeller in the solid state.14 An analogous ligand, 4,4′-dipyridyl amine, acts in a similar fashion as a two-bladed molecular propeller.15 Surprisingly, there have been a few examples of the use of L in coordination polymer chemistry. LaDuca and co-workers have reported the use of L as a linking ligand with Cu(NO3)2 forming three-fold double helices of [Cu(L)]n12 and with AgNO3 forming zigzag 1D chains of [Ag(L)(NO3)]n.13 The L ligand formed a pair of 1D meso-helical chains di(μ:κ2O,O′)-bridged at the Ag(I) nodes when a CF3COO− counteranion was employed. These chains extended their framework by virtue of weak π–π interactions.17
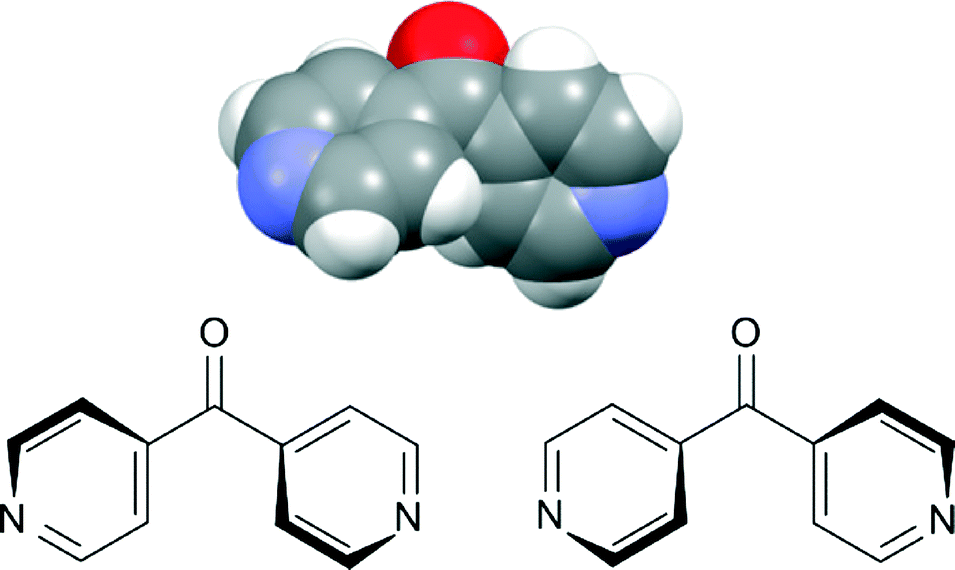 | ||
| Fig. 1 Top: view from the X-ray structure of L showing this ligand as a two-bladed molecular propeller;16 bottom: two enantiomers of L. | ||
Linear and zigzag polymers are widely encountered in the literature. Helical polymers have gained added interest in the past decade because of their inherent chirality,18 while meso-helical polymers remain relatively uncommon. A meso-helix represents an alternative way, compared to a helix, of combining chiral components into an extended structure.19 Thus, a lemniscate (∞) or figure of eight can be converted into a meso-helix by transforming it into the third dimension (Fig. 2).20 This achiral 1D strand consists of alternate linkages of the M- and P-forms of the ligands to the metal centre (M). The chain is thus represented as –M–(M)–P–(M)–M–(M)–P– and can sometimes be misinterpreted as a zigzag chain.21,22
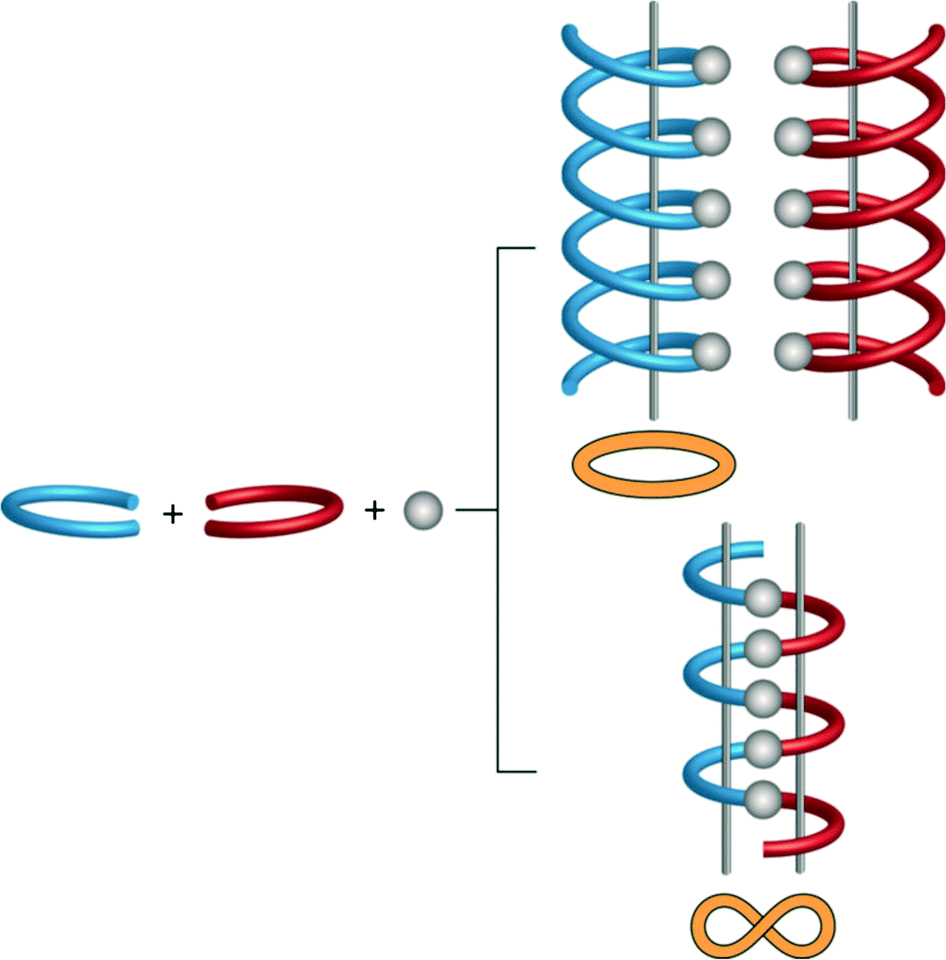 | ||
| Fig. 2 Schematic representation of the formation of the helical and meso-helical chains. Enantiomeric ligands are represented as red and blue forms. | ||
Herein, we describe the use of the bent bridging ligand L to form two series of related coordination polymers of Ag(I) salts with varying M:L ratios (1:1 and 1:2). These coordination polymers were structurally characterised by single crystal X-ray diffraction, IR spectroscopy and elemental analysis. The first Ag(I) series comprised six 1:1 complexes (1–3, 3a, 4, and 5) generated by employing the counteranions CF3SO3−, ClO4−, BF4− and PF6−. Since all of the 1:1 Ag–L complexes were meso-helical 1D chains, the diversity in counteranions did not play a profound role in determining the primary structure. However, these coordination polymers provided an opportunity to study the delicate anion–Ag versus CH3CN–Ag bridging interactions and their consequences for π–π-stacking and argentophilic interactions in this series of related 1D meso-helical chains. The second Ag(I) series comprised four related 1:2 complexes (6–9) generated by employing the counteranions CF3SO3−, PF6− and BF4−. In contrast to the first series, the primary structure of these coordination polymers was influenced by the nature of the counteranion, which moderated the extent of interaction between the N-pyridyl (Npy) donor on the peripheral arms and the Ag(I) ion. The counteranion did not directly interact with the Ag(I) ion.
Results and discussion
Coordination polymers 1–9 were all prepared using the same 1:1 v/v CH3CN:CH3OH solvent system. Reactions were carried out in 1:1 and 1:2 M:L molar ratios. The products formed showed a considerable degree of sensitivity towards the nature of the counteranion and also the M:L ratio. For AgCF3SO3 and AgBF4, two products were isolated and the final products had M:L ratios in agreement with the starting ratios as determined by microanalyses. For AgPF6, regardless of the M:L ratio used, a mixture of products was formed and a 1:1 and two 1:2 pseudo-polymorphic coordination polymers were isolated. For AgClO4, regardless of the M:L ratio, only polymorphic 1:1 products could be isolated. Even when a 2:1 M:L ratio was used, only a 1:1 product was formed which was found to be a pseudo-polymorph of the other two 1:1 AgClO4 products.
Synthesis and structure of {[Ag(L)](CF3SO3)·1/2H2O}∞, 1
The 1:1 molar reaction between AgCF3SO3 and L resulted in a tan solid. The microanalysis was consistent with the 1:1 formulation. Infrared studies of these samples confirmed the presence of L as the peaks at 1682 (ketonic CO group), 3124–3053 (aromatic C–H stretching), 1611 and 1555 (CC bending) and 759–660 cm−1 (aromatic C–H bending) were observed. The peak corresponding to the CO moiety of this complex was lower (1682 cm−1) than that observed in the free ligand (1731 cm−1). The peaks corresponding to the stretching of the SO, C–F, S–O and C–S bonds of the CF3SO3− counteranion were observed at 1330–1271, 1236–1018, 940–844 and 759–572 cm−1, respectively. In infrared studies of AgCF3SO3, Johnston and Shriver have demonstrated that the peak at 1271 cm−1 arises from asymmetric SO3 stretching, at 1236 cm−1 from symmetric CF3 stretching and at 760 cm−1 due to the CF3 angle deformation and the symmetric C–S stretching.23
Complex 1 crystallised in the monoclinic space group C2/c with one Ag(I) cation, one complete L ligand, one CF3SO3− counteranion and half a H2O of crystallisation in the asymmetric unit. Complex 1 formed a 1D meso-helical strand running along the [1 0 1] diagonal axis (Fig. 3). The Ag(I) ion was essentially linear with an N–Ag–N angle of 175.72(6)°. The slight bend was a consequence of weak interactions between the Ag(I) cation and the O-atoms of adjacent CF3SO3− anions (Fig. 3).24 The pyridyl rings of L formed a two-bladed chiral propeller at an angle between the rings of 50.37(9)° and generated 1D strands. From the viewpoint of chirality, these 1D strands consisted of alternate linkages of the M- and P-forms of the ligands with the Ag(I) ions. The chain was thus represented as –M–(Ag)–P–(Ag)–M–(Ag)–P–, resulting in a meso-helical structure.19
 | ||
| Fig. 3 Left: view of the 1D cationic meso-helical polymeric strand of 1 running along the [1 0 1] direction (crystallographic numbering; ellipsoids 50% probability level). Right: view showing weak bridging in 1 by CF3SO3− counteranions. Only the major component of the disordered CF3SO3− counteranion is shown and H2O molecules have been omitted for clarity. | ||
Adjacent meso-helical chains were formed into anti-parallel pairs through a weak π–π interaction [centroid-to-centroid distance 3.803(2) Å; inter-planar dihedral angle 9.80(9)°, minimum interatomic distance 3.615(2) Å; minimum ring slippage between planes 1.664 Å]. The pairs of chains were di(μ:κ2O,O′)-bridged by weak Ag⋯OSO2CF3 interactions which appeared to pull the Ag(I) ions closer together. The Ag⋯Ag distance was found to be 3.4704(16) Å. The bridging CF3SO3− anions on each side of the pairs of chains were linked together by hydrogen-bonding interactions with H2O of solvate [H⋯O distance of 1.94(4) Å corresponding to an O⋯O distance of 2.71(2) Å]. The pairs of chains interact with an adjacent pair of chains through π–π-interactions [centroid-to-centroid distance 3.766(2) Å, inter-planar dihedral angle 0°, minimum interatomic distance 3.746(2) Å; ring slippage between planes 1.680 Å] such that the rings involved in the interaction are registered every fifth pyridyl ring along the chains.
Synthesis and structure of {[Ag(L)](ClO4)·1/2H2O}∞, 2
Evaporation of the solvents from a 2:1 molar reaction between AgClO4 and L resulted in X-ray quality colourless crystals of 2. However, the microanalysis was consistent with a 1:1 formulation. Infrared studies of these samples confirmed the presence of L as the peaks at 1680 (ketonic CO group), 3095 (aromatic C–H stretching), 1612–1555 (CC bending) and 759–657 cm−1 (aromatic C–H bending) were observed. The presence of peaks at 1285, 1055, 952 and 691–619 cm−1 indicated the presence of ClO4−.
Complex 2 crystallised in the monoclinic space group C2/c with one Ag(I) cation, one complete L ligand, one ClO4− counteranion and half a H2O of crystallisation in the asymmetric unit. It formed infinite 1D meso-helical strands running along the [1 0 1] diagonal axis (Fig. 4). The Ag(I) ion was essentially linear with an N–Ag–N angle of 170.12(6)°.24 The slight bend indicated a relatively weak interaction between an adjacent ClO4− anion at 2.727(2) Å and the Ag(I) cation. The Ag⋯OClO3 interactions fell in the middle of the range of Ag⋯O contact lengths [2.291–3.238 Å] for similar two-coordinated Ag⋯OClO3 complexes as indicated in the CSD database (version 5.33).25,26 The pyridyl rings of L formed a two-bladed chiral propeller and registered an angle of 53.37(8)° between the planes of the rings. This complex possessed achiral meso-helical 1D chains similar to 1.
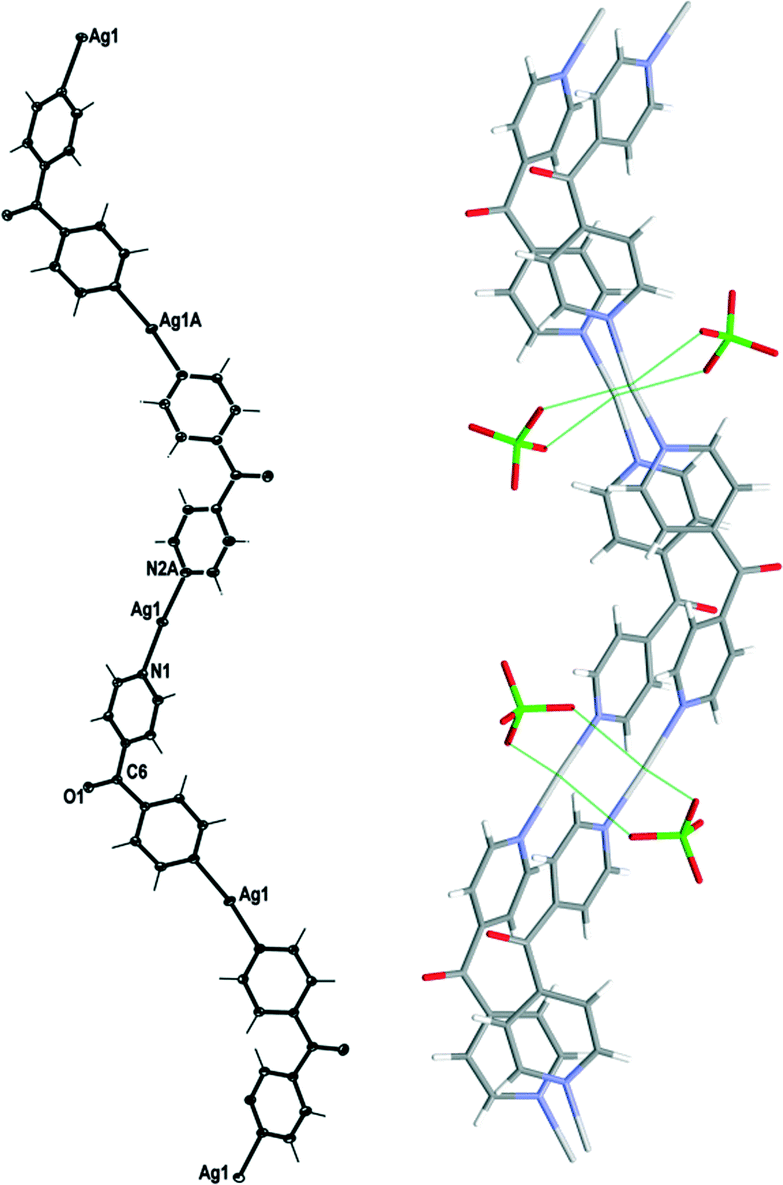 | ||
| Fig. 4 Left: view of the 1D cationic meso-helical polymeric strand of 2 running along the [1 0 1] direction (crystallographic numbering; ellipsoids 50% probability level). Right: view showing weak bridging in 2 by ClO4− counteranions. H2O molecules were omitted for clarity. | ||
The adjacent meso-helical chains were formed into anti-parallel pairs through two complementary interactions. One of which was weak π–π-interactions involving all of the pyridine rings of the adjacent chain [centroid-to-centroid distance 3.763(2) Å, inter-planar dihedral angle 3.62(8)°, minimum interatomic distance 3.593(2) Å; minimum ring slippage between planes 1.589 Å]. The other involved the Ag ions of the chains being di(μ:κ2O,O′)-bridged together by ClO4− anions on each side of the meso-helical pair (Fig. 4). The Ag⋯Ag distance was 3.369(17) Å and the Ag⋯OClO3 distances were 2.727(2) Å and 2.905(2) Å. The bridging ClO4− anions on each side of the pairs of chains were linked together by H-bonding interactions with H2O of solvate [H⋯O distance of 2.37(4) Å corresponding to an O⋯O distance of 3.039(3) Å]. There were no other noteworthy H-bonding interactions observed in the structure.
Synthesis and structure of {[Ag2(L)2(CH3CN)](ClO4)2·2CH3CN·H2O}∞, 3
Slow evaporation of the solvents from a 1:2 molar reaction between AgClO4 and L resulted in X-ray quality colourless crystals of 3. Again, the microanalysis was consistent with a 1:1 formulation. Infrared analysis revealed that the peak corresponding to the CO moiety of this complex was lower (1665 cm−1) than that observed in 2 (1680 cm−1). The peaks corresponding to the stretching and bending of aromatic rings of L and those for ClO4− were similar to 2. Slow evaporation of the solvents from a 1:1 molar reaction between AgBF4 and L resulted in colourless crystals of {[Ag2(L)2(CH3CN)2](BF4)2·CH3CN·H2O}∞ (3a) which were found to be isomorphous with 3 (experimental section).
Complex 3 formed infinite 1D meso-helical strands along the c axis which through bridging interactions were formed into two-dimensional (2D) sheets in the ac plane. It crystallised in the monoclinic space group P21/c with two crystallographically distinct Ag(I) cations, two complete L ligands, two ClO4− counteranions, three CH3CN molecules and a H2O of crystallisation in the asymmetric unit. The crystallographically distinct Ag(I) cations were present in different 1D meso-helical polymeric strands running parallel to each other along the c axis (Fig. 5). One Ag(I) cation possessed a linear geometry by coordinating with two N-donors of the L ligand, while the other Ag(I) cation possessed a T-shaped geometry by exhibiting additional coordination to a CH3CN molecule. The linear Ag(I) cation displayed a slight bend and the Npy–Ag2–Npy angle measured 174.57(14) Å. This bend may have been the consequence of the weak Ag2⋯OClO3 interactions [O23⋯Ag2 contact was 2.742(4) Å]24 and the weak N⋯Ag interactions with the free CH3CN molecules [the N7⋯Ag2 contact was 2.853(5) Å and the N6⋯Ag2 contact was 3.023(4) Å]. The CH3CN⋯Ag contacts for the two-coordinated Ag(I) cations were in the range of 2.555–3.265 Å.25,26 A CH3CN molecule coordinated to Ag1 at nearly perpendicular angles [the N(1)–Ag(1)–N(5) and N(5)–Ag(1)–N(2A) angles were 95.87(14)° and 94.84(14)°, respectively] and generated T-shaped Ag(I) nodes. The NCH3CN–Ag distance was 2.505(4) Å. The Npy–Ag1–Npy angle of the T-shaped Ag(I) cation was 163.77(14)°. The T-shaped Ag1 displayed a distorted geometry and deviated from the plane (N5 N1 N2) by 0.2144 Å. The Ag–Npy bond lengths were found to be 2.171(4) Å and 2.177(4) Å, while the Ag–Npy bond lengths of T-shaped Ag(I) were slightly elongated [the Ag1⋯N1 bond length was 2.203(4) Å and the Ag1⋯N2 bond length was 2.198(4) Å]. The bound CH3CN displayed a bend in the C–N–Ag angle [147.0(4)°] by virtue of the weak interaction of CH3CN with Ag2 of a neighbouring chain [the N⋯Ag2 contact was 3.121(4) Å]. The pyridyl rings of L formed a two-bladed chiral propeller and bridged the Ag(I) cores and formed infinite 1D meso-helical strands running along the c axis. The pyridyl rings of L coordinated to linear Ag(I) registered an angle of 40.3(2)° between the planes of the rings, while the pyridine rings of the other L registered an angle of 46.0(2)° between the planes of the rings.
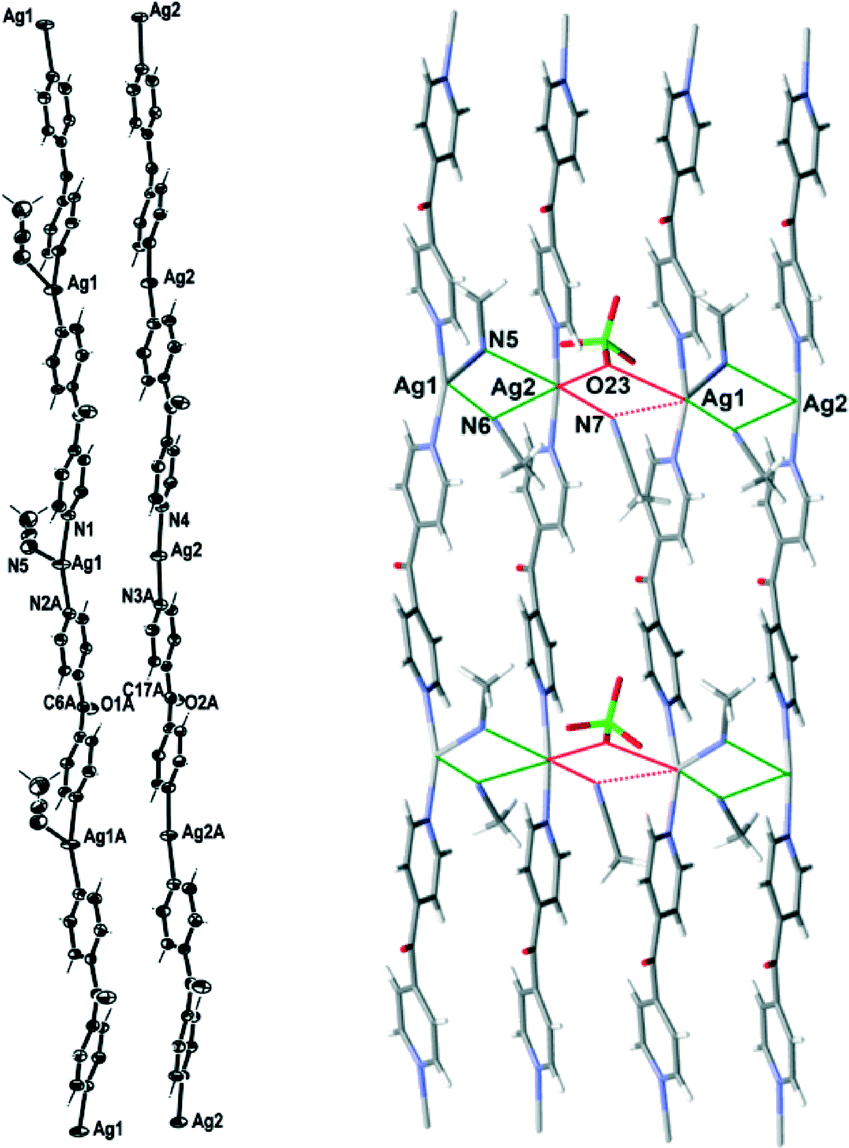 | ||
| Fig. 5 Left: view of the two distinct 1D cationic meso-helical polymeric strands of 3 running along the c axis (crystallographic numbering; ellipsoids 50% probability level). Right: view showing bridging of two distinct meso-helical strands of 3 with CH3CN molecules and weak bridging of the pair of strands with ClO4− and CH3CN molecule. A non-interacting ClO4− counteranion and the H2O molecules were omitted for clarity. | ||
The linear and T-shaped Ag(I) ions were present in separate strands and formed a pair of dissimilar strands. This pair of strands was di(μ:κ2N)-bridged by the CH3CN molecules (Fig. 5) and displayed weak π–π-stacking interactions [the centroid-to-centroid distances were 3.717(3) Å (inter-planar dihedral angle 5.5(2)°, minimum interatomic distance 3.601(3) Å; minimum ring slippage between planes 1.293 Å) and 3.749(2) Å (inter-planar dihedral angle 1.8(2)°, minimum interatomic distance 3.701(2) Å; minimum ring slippage between planes 1.600 Å)]. These distances were registered every alternate pyridine ring throughout the pair of strands. One ClO4− counteranion bridged this pair of strands by virtue of C–H⋯anion interactions [the O11⋯H13 contact was 2.59 Å and the corresponding O11⋯H–C13 contact was 3.222(6) Å; the O14⋯H1 contact was 2.46 Å and the corresponding O14⋯H–C1 contact was 3.143(6) Å]. The Ag⋯Ag distance was 3.4858(6) Å. A pair of strands was linked with an adjacent pair of strands by the weak bifurcated bridging (μ:κ2O) of the ClO4− counteranion on one side [the O3Cl–O23⋯Ag1 and the O3Cl–O23⋯Ag2 contacts were 3.205(5) Å and 2.742(4) Å, respectively] and the weak bifurcated bridging (μ:κ2N) of a CH3CN molecule on the other side [the N7⋯Ag2 and N7⋯Ag1 contacts were 2.854(5) and 3.329(5) Å, respectively] (Fig. 5). This asymmetric bridging was augmented by virtue of weak π–π-stacking interactions [centroid-to-centroid distance was 3.790(2) Å, inter-planar dihedral angle 1.8(2)°, minimum interatomic distance 3.746(2) Å; minimum ring slippage between planes 1.681 Å] and these interactions were registered every second pyridyl ring. The H2O of crystallisation interacted with three ClO4− counteranions by virtue of H-bonding interactions. One interaction was relatively strong [H⋯O distance of 2.17(4) Å corresponding to an O⋯O distance of 2.966(6) Å], while the other two were weaker and interacted in a bifurcated fashion [H⋯O distances of 2.67(7) and 2.81(5) Å corresponding to O⋯O distances of 3.168(7) and 3.468(6) Å, respectively].
Synthesis and structure of {[Ag2(L)2(CH3CN)2](ClO4)2·CH3CN·H2O}∞, 4
Slow evaporation of solvent from a solution of AgClO4 and L in a 1:1 M:L ratio resulted in X-ray quality colourless crystals of 4. The infrared spectrum revealed that the peaks corresponding to the ketonic CO group, stretching and bending of aromatic rings of L and ClO4− were similar to those of 2.
Complex 4 formed infinite 1D meso-helical strands along the c axis which through bridging interactions were formed into 2D sheets in the ac plane. It crystallised in the monoclinic space group P21/c with two crystallographically distinct Ag(I) cations, two complete L ligands, two ClO4− counteranions and three CH3CN molecules in the asymmetric unit. The crystallographically distinct Ag(I) cations were present in different 1D meso-helical polymeric strands running parallel to each other along the c axis (Fig. 6). One Ag(I) cation possessed a linear geometry by coordinating with two N-donors of the L ligand, while the other Ag(I) cation possessed a four-coordinated geometry by exhibiting additional coordination to two CH3CN molecules. The linear Ag(I) cation displayed a slight bend and the Npy–Ag1–Npy angle measured 165.85(18) Å. This bend may have been a consequence of the weak N⋯Ag interactions with the three CH3CN molecules [the N⋯Ag contacts ranged between 2.887(7) and 3.043(6) Å].25,26 The four-coordinated Ag(I) cation adopted a geometry between a seesaw and a trigonal pyramid as evidenced by a τ4 value of 0.70.27 The N5–Ag2 and N7–Ag2 distances were 2.414(6) Å and 2.657(6) Å, respectively. The Npy–Ag2–Npy angle of the four-coordinated Ag(I) cation was 155.14(17)°. The Ag1–Npy bond lengths were found to be equal [2.186(5) Å], while the Ag2–Npy bond lengths were slightly elongated [the Ag2⋯N3 bond length was 2.223(5) Å and the Ag2⋯N4 bond length was 2.222(5) Å]. The bound CH3CN displayed a bend in the C–N–Ag angle [the C23–N5–Ag2 angle was 141.5(6)° and the C27–N7–Ag2 angle was 166.1(5)°] by virtue of the weak interaction of CH3CN with Ag1 of a neighbouring chain [the N5⋯Ag1 and N7⋯Ag1 contacts were 2.887(7) and 3.043(6) Å, respectively]. The pyridyl rings of L formed a two-bladed chiral propeller and bridged the Ag(I) cores and formed infinite 1D meso-helical strands running along the c axis. The pyridyl rings of L coordinated to the linear Ag(I) registered an angle of 45.6(3)° between the planes of the rings, while the pyridine rings of the other L registered an angle of 50.7(3)° between the planes of the rings.
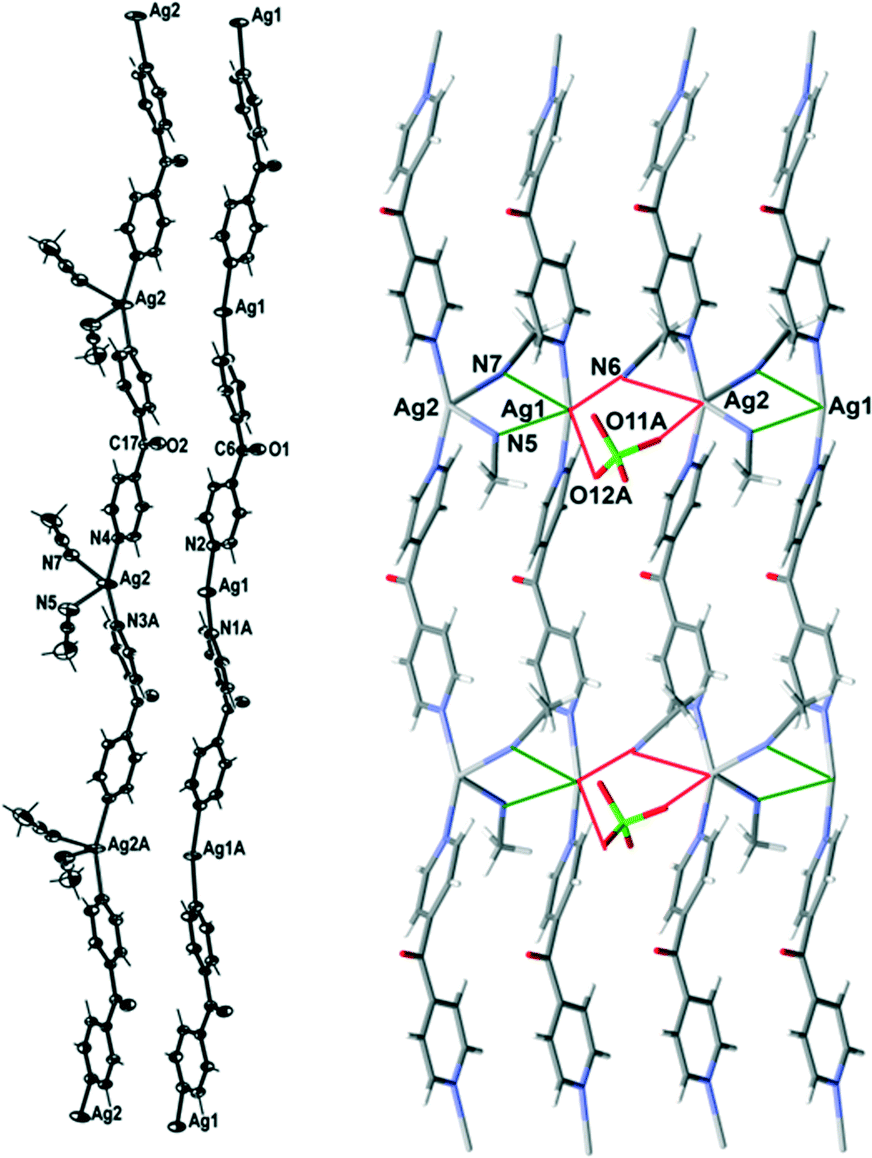 | ||
| Fig. 6 Left: view of the two distinct 1D cationic meso-helical polymeric strands of 4 running along the c axis (crystallographic numbering; ellipsoids 50% probability level). Right: view showing bridging of two distinct meso-helical strands of 4 with CH3CN molecules and weak bridging of the pair of strands with ClO4− and CH3CN molecule. Only the major component of the disordered ClO4− counteranion is shown and the other ClO4− counteranion was omitted for clarity. | ||
The linear and four-coordinated Ag(I) cations were present in separate strands and formed a pair of dissimilar strands. This pair of strands was di(μ:κ2N)-bridged by CH3CN molecules (Fig. 6) and displayed weak π–π-stacking interactions [the centroid-to-centroid distances were 3.884(4) Å (inter-planar dihedral angle 5.8(3)°, minimum interatomic distance 3.770(4) Å; minimum ring slippage between planes 1.254 Å) and 3.945(4) Å (inter-planar dihedral angle 1.5(3)°, minimum interatomic distance 3.935(4) Å; minimum ring slippage between planes 1.939 Å)]. These distances were registered every alternate pyridyl ring throughout the pair of strands. The Ag⋯Ag distance was 3.694(3) Å. A pair of strands was linked to an adjacent pair of strands by the weak μ:κ2O,O′-bridging of the ClO4− counteranion on one side [the O3Cl–O11A⋯Ag2 and the O3Cl–O12A⋯Ag1 contacts were 3.201(16) Å and 2.953(13) Å, respectively] and the weak μ:κ2N-bridging of a CH3CN molecule on the other side [the N6⋯Ag1 and N6⋯Ag2 contacts were 2.886(6) and 3.217(6) Å, respectively] (Fig. 6). This asymmetric bridging was augmented by virtue of weak π–π-stacking interactions [centroid-to-centroid distance was 3.884(4) Å (inter-planar dihedral angle 5.8(3)°, minimum interatomic distance 3.770(3) Å; minimum ring slippage between planes 1.254 Å) and 3.944(4) Å (inter-planar dihedral angle 1.5(3)°, minimum interatomic distance 3.935(4) Å; minimum ring slippage between planes 1.939 Å)] and these interactions were registered every alternate pyridyl ring. The Ag⋯Ag distance between the adjacent pair of strands was 4.139(3) Å.
Synthesis and structure of {[Ag2(L)2(CH3CN)2](PF6)2·2CH3CN}∞, 5
A bulk reaction with a 1:2 M:L ratio of AgPF6 and L resulted in the formation of a brown crystalline precipitate in moderate yield. However, the microanalysis of the bulk sample was consistent with a 1:1 formulation. Infrared studies of these samples confirmed the presence of L as the peaks at 1675 (ketonic CO group), 1612 and 1555 (CC bending) and 757–651 cm−1 (aromatic C–H bending) were observed. The very strong sharp peak at 821 cm−1 and strong sharp peak at 555 cm−1 indicated the presence of PF6− counteranions.28
Complex 5 formed infinite 1D meso-helical strands along the b axis which through bridging interactions were formed into 2D sheets in the ab plane. It crystallised in the monoclinic space group P21/c and the asymmetric unit consisted of two crystallographically distinct Ag(I) cations, two complete L ligands, two PF6− counteranions and four CH3CN molecules. The two crystallographically distinct Ag(I) cations and ligands were alternately present in the same 1D meso-helical strand despite it running along the crystallographic b axis (Fig. 7). The Ag(I) cations displayed a pseudo T-shaped three-coordinated geometry by coordinating with two Npy-donors of two distinct L ligands and a CH3CN molecule. Ag1 deviated from the plane of the donor atoms (N3 N1 N5) by 0.120 Å, while Ag2 deviated from the plane (N2 N4 N7) by 0.160 Å. The Ag(I) nodes were bridged by two L ligands and 1D meso-helical strands were generated. The pyridyl rings of L formed a two-bladed chiral propeller. The pyridyl rings of the two L ligands registered the angles of 48.43(15)° and 48.05(15)° between the planes of the rings.
 | ||
| Fig. 7 Left: view of the 1D cationic meso-helical polymeric strand of 5 running along the b axis (crystallographic numbering; ellipsoids 50% probability level). Right: view showing bridging of two adjacent meso-helical strands of 5 with CH3CN molecules. The PF6− counteranions and two uncoordinated CH3CN molecules were omitted for clarity. | ||
Two CH3CN molecules coordinated to Ag1 and Ag2 at distances of 2.632(3) and 2.664(3) Å, respectively. The bound CH3CN molecules displayed a bend in C–N–Ag angles [the C23–N5–Ag1 angle was 141.7(2) and the C27–N7–Ag2 angle was 143.6(2)°] by virtue of weak interactions with Ag(I) cations of neighbouring chains. These interactions resulted in the di(μ:κ2N)-bridging of the two antiparallel chains at alternate Ag(I) nodes by the bound CH3CN molecules generating a 2D sheet in the ab plane (Fig. 7). The N5⋯Ag1 contact was 2.772(3) Å, while the N7⋯Ag2 contact was 2.751(3) Å. The bridging was augmented by very weak π–π-stacking interactions [the centroid-to-centroid contact was 3.9103(18) Å (inter-planar dihedral angle 8.01(15)°, minimum interatomic distance 3.7430(18) Å; minimum ring slippage between planes 1.175 Å)]. The Ag⋯Ag distances at the bridged nodes [Ag1⋯Ag1 3.5632(4) Å and Ag2⋯Ag2 3.4709(4) Å] were considerably shorter than that at the non-bridged nodes [Ag1⋯Ag1 4.4372(5) Å and Ag2⋯Ag2 4.5448(5) Å]. The uncoordinated CH3CN molecules interacted with the pyridine rings of the anti-parallel chains through weak N⋯H–C interactions that ranged between 2.30 and 2.83 Å. There were no noteworthy H-bonding interactions observed in the structure.
Synthesis and structure of {[Ag(L)2](CF3SO3)·1/2H2O}∞, 6
A 1:2 molar reaction between AgCF3SO3 and L resulted in colourless crystals of 6. The microanalysis was consistent with a 1:2 formulation. Infrared studies of these samples confirmed the presence of L as the peaks at 1675 (ketonic CO group), 3200–3000 (aromatic C–H stretching), 1640–1554 (CC bending) and 756–660 cm−1 (aromatic C–H bending) were observed. The peaks corresponding to the stretching of the SO, C–F, S–O and C–S bonds of the SO3CF3− counteranion were observed at 1328–1284, 1222–1145, 949–830 and 660–634 cm−1, respectively.
Complex 6 crystallised in the monoclinic space group C2/c to form a 1D meso-helical chain decorated with L arms (Fig. 8). The chains ran along the c axis. The asymmetric unit contained one Ag(I) cation, two L ligands, one CF3SO3− counteranion and half a H2O of crystallisation. The Ag(I) ions formed a distorted trigonal planar arrangement with three nitrogens of the pyridyl rings at angles of 112.8(2)°, 116.1(2)° and 127.5(2)° for N3–Ag1–N4, N1–Ag1–N3 and N1–Ag1–N4, respectively, while N2 remained uncoordinated forming the decorating arm of the meso-helix. The Ag(I) ion deviated by 0.251 Å from the plane of three bound N-donors. The weak interaction between OH2O⋯Ag1 [2.825(6) Å] distorted the planarity of the AgN3 moiety. In addition, the H2O molecule was H-bonded to the ketone O atom bridging two adjacent chains. The counteranions did not interact significantly with the polymeric stands.
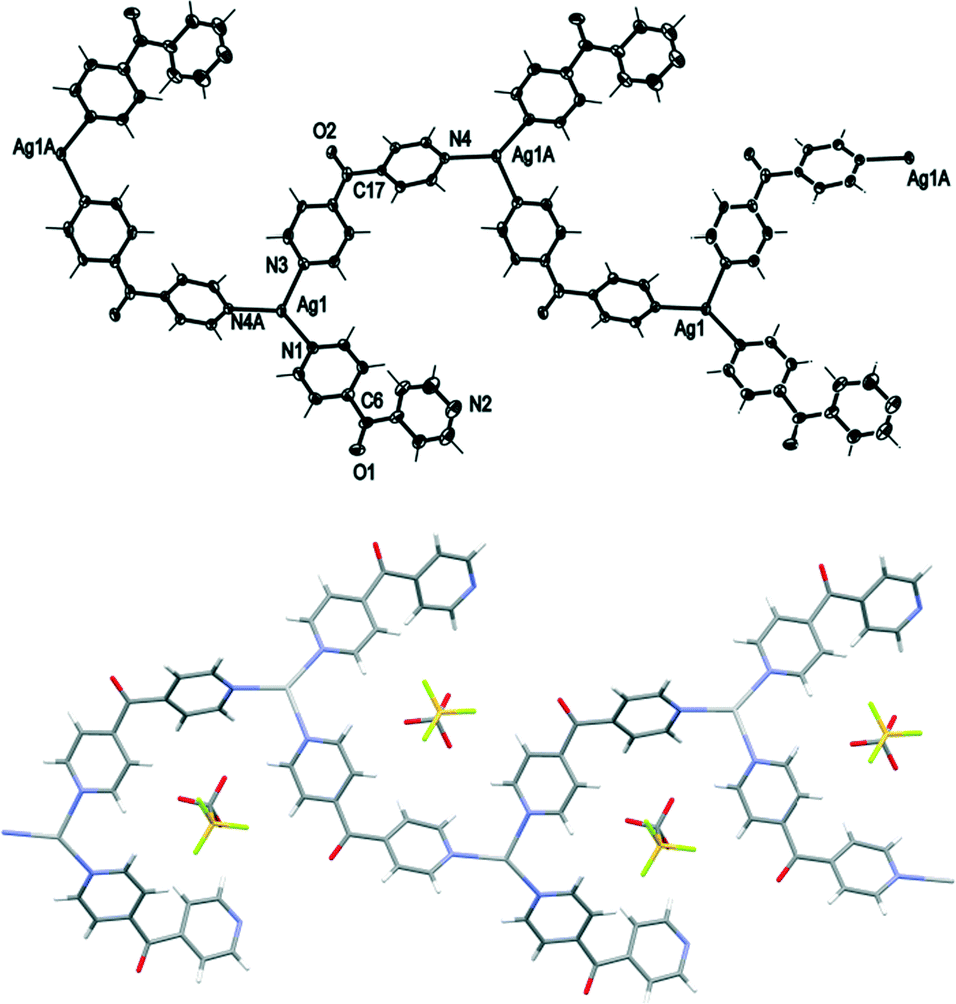 | ||
| Fig. 8 Top: view of the 1D cationic meso-helical polymeric strand of 6 running along the c axis (crystallographic numbering; ellipsoids 50% probability level). Bottom: view of a 1D meso-helical polymeric strand of 6 showing inclusion of CF3SO3− counteranions. Only the major component of the disordered CF3SO3− counteranion is shown and H2O molecules have been omitted for clarity. | ||
The pyridyl rings of L formed a two-bladed chiral propeller. The L ligands which formed the polymeric backbone registered an angle of 48.8(4)°, while the decorated L arms registered an angle of 58.6(4)° between the planes of the pyridine rings. The uncoordinated N2 of the dipyridyl ketonic arms showed a slight inclination towards Ag1 of an adjacent chain with N2⋯Ag1 contact of 3.153(8) Å29,30 and registered an angle of 111.95° between the Ag(I)–N2py–Cgpy. This long contact distance and very narrow angle indicated that there is essentially no interaction between pyridyl N2 and Ag1 (Fig. 9). The arrangement of the decorated arms created a cavity encompassing two CF3SO3− counteranions.
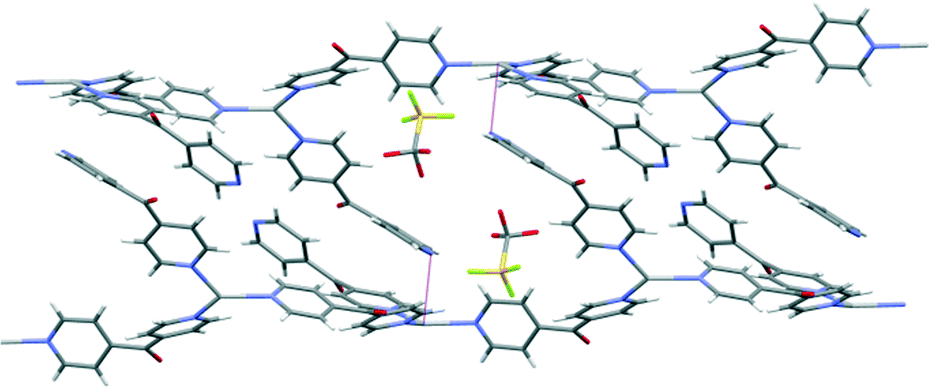 | ||
| Fig. 9 The anti-parallel meso-helical chains of complex 6 showing the disposition of the long N2⋯Ag contact (shown in purple). | ||
Synthesis and structure of {[Ag(L)2](BF4)}∞, 7
Slow evaporation of solvents from a solution of AgBF4 and L in a 1:2 M:L ratio resulted in X-ray quality colourless crystals of 7. The microanalysis of the bulk reaction with similar molar and solvent ratios was consistent with a 1:2 formulation. Infrared studies of these samples confirmed the presence of L as the peaks at 1677 (ketonic CO group), 3106–3054 (aromatic C–H stretching), 1608–1555 (CC bending) and 756–660 cm−1 (aromatic C–H bending) were observed. The peaks at 1032, 756 and 520 cm−1 confirmed the presence of the BF4− counteranion.
Complex 7 crystallised in the monoclinic space group P21/c to form infinite polymeric chains along the b axis. Each asymmetric unit contained one Ag(I) cation, two L ligands and one BF4− counteranion (Fig. 10). One L ligand bridged the three coordinated Ag(I) nodes and extended the polymer to a 1D chain, while the second L ligand interacted with Ag(I) through monodentate interactions and formed decorating side arms of the chain. Surprisingly, the 1D chain of 7 existed as a helix rather than a meso-helix. This was the only example of a helical chain for this entire series of compounds. Of necessity, both M and P forms of the helices were present in the centrosymmetric structure. The uncoordinated N2 of these side arms resided in the vicinity of Ag1 of the adjacent chain. The Ag1 bond distances to the other Npy-donors of 2.2121–2.3861(17) Å were within the normal range. The Ag(I) cation adopted a slightly distorted trigonal-planar geometry with three Npy at the angle of 109.33(6)°, 149.54(6)° and 99.89(6)° for N3–Ag1–N4, N1–Ag1–N3 and N1–Ag1–N4, respectively. The Ag(I) ion deviated by 0.133 Å from the plane of three bound N-donors.
The pyridyl rings of L formed a two-bladed chiral propeller. The L ligands which formed the polymeric backbone registered an angle of 57.50(10)°, while the decorated L arms registered an angle of 65.49(10)° between the planes of the rings. The two crystallographically distinct L ligands were pseudo enantiomers of each other. The uncoordinated N2 of the L side arm showed a significant inclination towards Ag1 with a N2⋯Ag1 contact of 2.6582(18) Å29,30 and registered an angle of 138.80° between the Ag(I)–Npy–Cgpy.31 A search of the CSD database identified 12 complexes with Ag(I)⋯N(pyridine) contacts in the range of 2.60–2.70 Å. For 11 of these complexes, the Ag(I)–Npy–Cgpy angle ranged from 132 to 141°, while one had a value of 125°. The consistency of the angles within this range suggested the presence of a weak interaction between pyridyl N and Ag(I). By virtue of this close contact and the positioning of the decorated arms on the adjacent chains, cavities existed in what appeared to be a pseudo (4,4) rhombic network (Fig. 10). The four Ag1 ions occupied the corners of the rhombus, and the adjacent sides of the network measured 11.102 Å and 11.471 Å. The four ligands of the rhombus were arranged such that two CO groups of the opposite ligands, which were pseudo enantiomers of each other, pointed above the plane of the rhombus and towards each other, while the remaining two CO groups of the ligands, which were also pseudo enantiomers, pointed below the plane and splayed away from each other (Fig. 10). This arrangement caused the pseudo (4,4) network to be flat and achiral. The weak CH–π-interactions between C–H25⋯Cg2 (N2 containing pyridine) [the distance between H–centroid was 2.72 Å]32 and the weak π–π-interactions [the Cg2⋯Cg2 contact was 3.7572(16) Å, inter-planar dihedral angle 0°, minimum interatomic distance 3.736(16) Å; ring slippage between planes 1.525 Å]33 may have helped to facilitate the positioning of N2 near the Ag ion.
 | ||
| Fig. 10 Top: view of the 1D cationic helical polymeric strand of 7 running along the b axis (crystallographic numbering; ellipsoids 50% probability level). Bottom: view of a (4,4) network of 7 formed by virtue of N2⋯Ag1 interactions between the adjacent meso-helical chains. | ||
The BF4− counteranion resided within each cavity of the pseudo network by virtue of two weak attractive forces. A weak anion–π-interaction existed between BF4− and the pyridine ring containing N3 [F3B–F⋯Cg 3.314(9) Å]34 and also weak CH–anion interactions existed between BF4− and pyridyl H atoms [the F⋯H contact distances ranged from 2.35 to 2.61 Å, and the corresponding F⋯C distances ranged from 3.027(2) to 3.203(3) Å]. The cation–anion interactions stacked the adjacent layers of grids in an offset –A–B–A–B– fashion.
Synthesis and structure of {[Ag(L)2](PF6)}∞, 8, and {[Ag(L)2](PF6)·2CH3CN}∞, 9
Slow evaporation of solvents from a solution of AgPF6 and L in a 1:2 M:L ratio resulted in X-ray quality colourless crystals of both 8 and 9. The microanalyses of these samples were consistent with a 1:2 formulation. Infrared studies of these samples confirmed the presence of L as the peaks at 1686 and 1674 (ketonic CO group), 1604 and 1556 (CC bending) and 690–647 cm−1 (aromatic C–H bending) were observed. The environment around the ketonic CO may have caused the CO peak to split in two separate peaks. The very strong sharp peak at 818 cm−1 and strong sharp peak at 556 cm−1 indicated the presence of PF6− counteranions.28
Complex 8 crystallised in the monoclinic space group P21/n to form an infinite 2D network in the ac plane. Each asymmetric unit of this complex contained one Ag(I) cation, two L ligands and one PF6− counteranion. The Ag(I) cation adopted a four-coordinated geometry between a seesaw and a trigonal pyramid as evidenced by a τ4 value of 0.78.27 The Ag1–Npy bonds were in the normal range from 2.244(3) to 2.383(3) Å. The four Npy donors coordinated to the Ag(I) ion at angles between 94.52(11) and 138.04(12)°. These pyridyl rings demonstrated more regular Ag(I)–Npy–Cgpy angles of between 150.51 and 175.69°. The L ligand formed a two-bladed chiral molecular propeller, and the pyridyl rings of L registered angles of 54.44(16)° and 42.16(17)° between the planes of the rings. The two crystallographically distinct L ligands had the same pseudo enantiomeric form. Both the L ligands bridged the four coordinated Ag nodes, and a corrugated (4,4) rhombic network was generated (Fig. 11). The adjacent sides of the network measured 11.070(3) Å and 11.651(3) Å. The four ligands of the rhombus were arranged in an irregular way such that the CO groups of three of the ligands pointed above and one pointed below the plane of the rhombus (Fig. 11). The three ligands with the CO groups which pointed above were of the same pseudo enantiomeric form, while the one with the CO group pointing below was of the other pseudo enantiomeric form. This arrangement caused the (4,4) network to be achiral and extremely corrugated and the planes of the adjacent facing rhomboids oriented themselves at angles of 66.3°. The adjacent sheets of the (4,4) network were interdigitated and stacked on top of each other in a –A–B–A– fashion along the b axis. These sheets interacted with each other by weak OCO⋯π interactions [O⋯Cg5 contact was 3.282(3) Å]35 and weak π–π-interactions [Cg4⋯Cg2 contact was 3.899(3) Å, inter-planar dihedral angle 12.50(17)°, minimum interatomic distance 3.739(3) Å; minimum ring slippage between planes 0.9035 Å].33
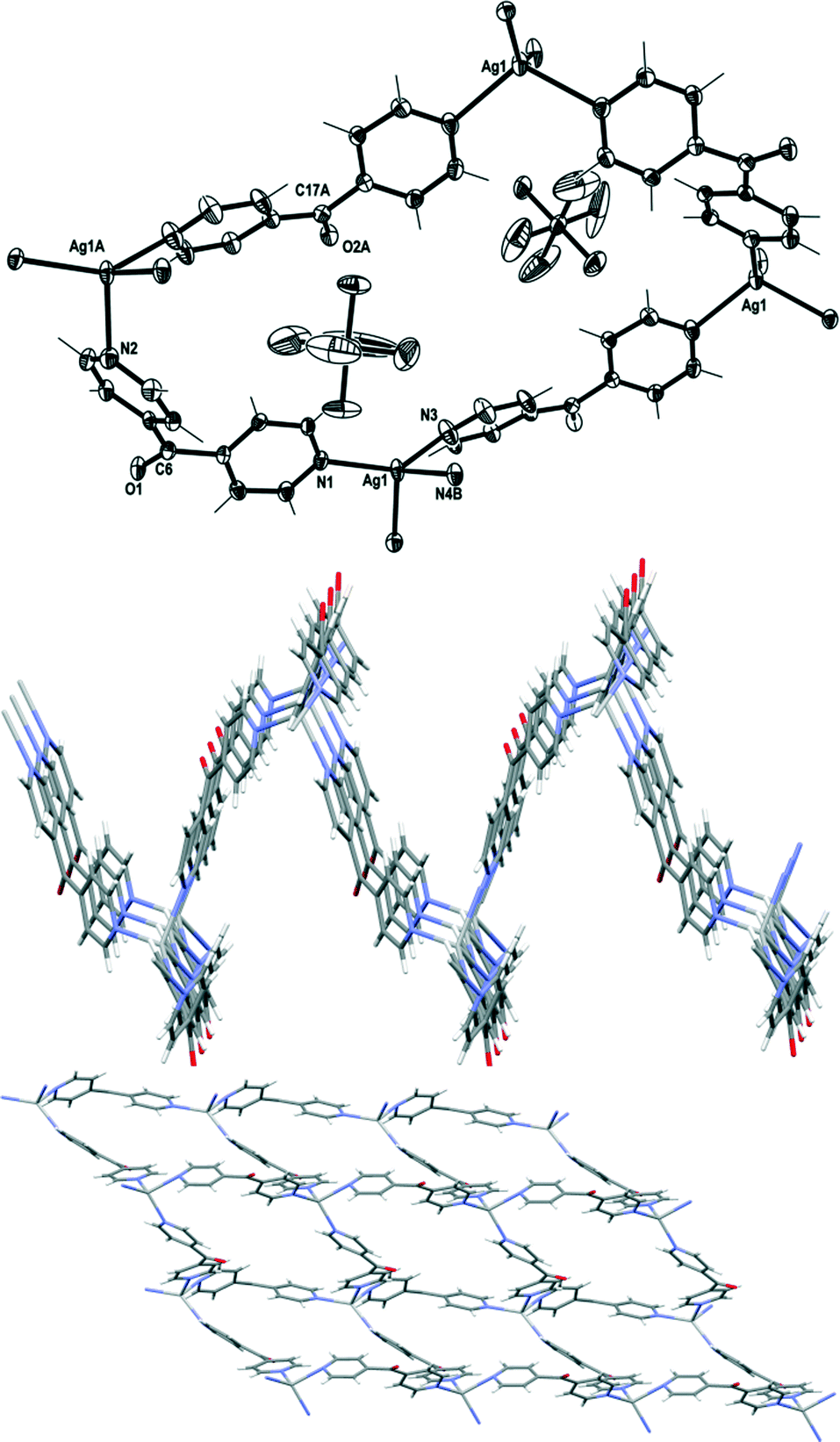 | ||
| Fig. 11 Top: view of a section of the (4,4) net of 8 displaying PF6− counteranions residing above and below the plane of the rhombus (crystallographic numbering; ellipsoids 50% probability level). Middle: view in the ab plane showing a corrugated network of . PF6− counteranions were omitted for clarity. Bottom: view in the ac plane showing a (4,4) network of complex 8. PF6− counteranions were omitted for clarity. | ||
The two PF6− counteranions resided within the cavity being slightly above and below the plane of the rhombus by virtue of several weak anion–CH interactions [H⋯F contact distances between 2.44 and 2.97 Å] and strong anion–carbonyl interactions. The F11⋯C6 contact was 2.874(4) Å and the F14⋯C17 contact was 2.959(4) Å. A search of the CSD database suggested that for complexes containing pyridyl ketone like ligands, there were only two out of 204 observations which displayed a F⋯CCO contact below the van der Waals limit of 3.2 Å.36,37 In total in the CSD there are 422 observations of general OC⋯F–PF5− interactions which range from 2.53 to 3.17 Å with a mean contact of 3.02 Å.26,27
Complex 9 crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) to form an infinite 2D network in the ab plane. Each asymmetric unit comprised one Ag(I) cation, two L ligands, one PF6− counteranion and two CH3CN of crystallisation. The Ag(I) cation adopted a distorted tetrahedral geometry as evidenced by a τ4 value of 0.91.27 The four pyridine N-atoms coordinated to the Ag(I) ion at angles between 93.43(12) and 119.90(12)° and interacted with the Ag(I) ions in the regular range of 2.268(4)–2.352(4) Å. These pyridyl rings demonstrated Ag(I)–Npy–Cgpy angles between 168.46 and 175.58°. The L ligand formed a two-bladed chiral molecular propeller, and the pyridyl rings of L registered angles of 53.78(14)° and 77.2(9)° between the planes of the rings. The two crystallographically distinct L ligands were of the same pseudo enantiomeric form. Both the L ligands bridged the Ag nodes perpendicular to each other and a regular (4,4) rhombic network was generated (Fig. 12). The adjacent sides of the network measured 10.950(7) Å and 11.108(7) Å. These distances corresponded to the length of crystallographic a and b axes. The four ligands of the rhombus, all of the same pseudo enantiomeric form, were arranged such that the CO groups of the two ligands pointed above and two pointed below the plane of the rhombus (Fig. 12). This arrangement gave a more regular network which was also chiral.38,39 The adjacent sheets of the (4,4) network, which were enantiomers of each other, were interdigitated and stacked on top of each other in a –A–B–A– fashion along the c axis.
to form an infinite 2D network in the ab plane. Each asymmetric unit comprised one Ag(I) cation, two L ligands, one PF6− counteranion and two CH3CN of crystallisation. The Ag(I) cation adopted a distorted tetrahedral geometry as evidenced by a τ4 value of 0.91.27 The four pyridine N-atoms coordinated to the Ag(I) ion at angles between 93.43(12) and 119.90(12)° and interacted with the Ag(I) ions in the regular range of 2.268(4)–2.352(4) Å. These pyridyl rings demonstrated Ag(I)–Npy–Cgpy angles between 168.46 and 175.58°. The L ligand formed a two-bladed chiral molecular propeller, and the pyridyl rings of L registered angles of 53.78(14)° and 77.2(9)° between the planes of the rings. The two crystallographically distinct L ligands were of the same pseudo enantiomeric form. Both the L ligands bridged the Ag nodes perpendicular to each other and a regular (4,4) rhombic network was generated (Fig. 12). The adjacent sides of the network measured 10.950(7) Å and 11.108(7) Å. These distances corresponded to the length of crystallographic a and b axes. The four ligands of the rhombus, all of the same pseudo enantiomeric form, were arranged such that the CO groups of the two ligands pointed above and two pointed below the plane of the rhombus (Fig. 12). This arrangement gave a more regular network which was also chiral.38,39 The adjacent sheets of the (4,4) network, which were enantiomers of each other, were interdigitated and stacked on top of each other in a –A–B–A– fashion along the c axis.
 | ||
| Fig. 12 Top: view of a section of the (4,4) net of 9 displaying PF6− counteranion residing in the cavity (crystallographic numbering; ellipsoids 50% probability level). Bottom: view in the ab plane showing a (4,4) network of 9. | ||
The PF6− counteranions were embedded in the cavities of the (4,4) network by virtue of weak anion–π and CH–anion interactions. The distance between F15-to-centroid contact was 3.028(4) Å,34 while the distance between H1⋯F15 was 2.47 Å and the corresponding C9⋯F16 distance was 3.369(5) Å. No other noteworthy π–π-stacking and H-bonding interactions were observed.
Comparison of structures
In structures 1–9, the coordination environment of the Ag(I) ions ranged from linear to trigonal pyramidal. The L ligand bridged the Ag(I) cores and generated the primary structure of 1D meso-helical chains in complexes 1–6, a helical 1D chain in complex 7 and 2D networks in complexes 8 and 9. The 1D meso-helical polymers in 1 and 2 existed as pairs of chains di(μ:κ2O,O′)-bridged with counteranions, while in 3–5 they existed as 2D grids extended by bridging of the Ag(I) nodes of the 1D meso-helical polymers by counteranions and CH3CN molecules. Complex 6 existed as a genuine 1D meso-helical polymer, while complex 7 was a pseudo (4,4) network and complexes 8 and 9 were (4,4) networks. The L ligand also acted as a two-bladed chiral molecular propeller within each solid-state structure such that the planes of the two pyridine rings intersected each other at an angle summarised in Table 1. The average twist in the planes of the pyridine rings was 52.6°. It is interesting to note that the pseudo-polymorphous complexes 3 and 4 and complexes 8 and 9 displayed large variations in these angles for only one of the ligands incorporated in the structures.| Structure | Angle (°) |
|---|---|
| 1 | 50.37(9) |
| 2 | 53.37(8) |
| 3 | 40.3(2) and 46.0(2) |
| 4 | 45.6(3) and 50.7(3) |
| 5 | 48.05(15) and 48.43(15) |
| 6 | 48.8(4) and 58.6(4) |
| 7 | 57.50(10) and 65.49(10) |
| 8 | 54.44(16) and 42.16(17) |
| 9 | 53.78(14) and 77.2(9) |
The influence of the coordinating ability of the anions on the stoichiometry of the resultant Ag(I) complexes was demonstrated by our closely related series of coordination polymers.40–42 The weakly coordinating CF3SO3− counteranion did not have an influence on the variation in stoichiometry as the stoichiometries of both complexes 1 and 6 were consistent with the starting M:L ratios of 1:1 or 1:2, respectively. The ClO4− counteranions demonstrated stronger coordination to Ag(I) in the presence of L as compared to the CF3SO3− counteranion. This prevented the formation of coordination polymers with different M:L ratios, as regardless of the starting M:L ratios, 2:1, 1:2 or 1:1, only the 1:1 complexes 2, 3 and 4, respectively, could be isolated. An excess of ClO4− present in the preparation of 2 resulted in di(μ:κ2O,O′)-bridging of the 1:1 polymer chain with ClO4−, while a deficiency as in the case of 3 and 4 instead resulted in di(μ:κ2N)-bridging of the 1:1 chains with CH3CN molecules. The starting M:L ratio of 1:2 in the reaction of AgPF6 with L resulted in 5 with 1:1 and 8 and 9 with 1:2 M:L ratios. Owing to the non-coordinating nature of the PF6− counteranion, the 1:1 1D chains of 5 were di(μ:κ2N)-bridged by CH3CN molecules.
In the 1:1 M:L complexes, the AgN2 moieties with the linear geometry displayed a slight bend in the Npy–Ag–Npy angles by virtue of weak Ag–anion interactions.24 In the isostructural complexes 1 and 2, the counteranions bridged the Ag(I) nodes of the adjacent meso-helical strands from both sides through di(μ:κ2O,O′)-bridging of the Ag(I) cores of the adjacent strand. The bulky CF3SO3− counteranion of 1 displayed less influence on the bend in the linearity of Ag(I) cation [175.72(7)° vs. 170.12(6)°] and registered higher Ag⋯O distances when compared with the less bulky ClO4− anion (Table 2). The tighter bridging of the Ag(I) nodes by the ClO4− counteranions resulted in a shorter Ag⋯Ag contact distance and stronger π–π-interactions (Table 2). The ClO4− counteranions of the pseudo-polymorphous complexes 2, 3 and 4 revealed various bridging modes. The adjacent strands of 2 were di(μ:κ2O,O′)-bridged by ClO4− counteranions, while the chains of 3 and 4 were weakly di(μ:κ2N)-bridged by CH3CN molecules. However in 3 and 4, the ClO4− counteranions assisted the μ:κ2N bridging of the CH3CN molecules by bridging the adjacent pair of strands through μ:κ2O and μ:κ2O,O′ interactions, respectively. The bridging of ClO4− counteranions along with the bifurcated bridging of a CH3CN molecule from the other side extended the structures of 3 and 4 to 2D grids. In 2 and 3, the meso-helical chains bridged by ClO4− recorded shorter Ag⋯Ag contact distances but weaker π–π-interactions than the meso-helical strands bridged by CH3CN. Surprisingly, the meso-helical strands of 4 demonstrated slightly tighter bridging by CH3CN molecules but higher Ag⋯Ag contact and weaker π–π-interactions than the pseudo-polymorphous 3. The CH3CN molecules of 5 bridged the pair of meso-helical chains more tightly as compared to the bridging observed in 3. This was evidenced by a shorter Ag⋯Ag contact in the case of 5 as compared to 3. However, the π–π-interactions were found to be weaker in 5. In 5, the Ag⋯Ag contact at the bridged nodes was considerably shorter than that at the unbridged nodes, thus highlighting the effect of CH3CN bridging.
| Complexes | π–π | Ag⋯Ag | Bridging |
|---|---|---|---|
| 1 | 3.803(2) Å | 3.4704(16) Å | 2.927(2) and 3.160(4) Å |
| 2 | 3.763(2) Å | 3.369(17) Å | 2.727(2) and 2.905(2) Å |
| 3 | 3.717(3) and 3.749(2) Å | 3.4858(6) Å | 2.742(4) and 3.023(4) Å; 2.505(4) and 3.121(4) Å |
| 4 | 3.884(4) Å and 3.945(4) Å | 3.694(3) Å | 2.414(6) and 2.887(7) Å; 2.657(6) and 3.043(6) Å |
| 5 | 3.9103(18) Å | 3.5632(4) and 3.4709(4) Å | 2.632(3) and 2.772(3) Å; 2.751(3) and 2.664(3) Å |
A search of the CSD database (version 5.33) for Ag–anion interactions shorter than the sum of the van der Waals radii suggested that out of 1182 reported AgClO4 complexes about 22% of the complexes demonstrate μ:κ2O,O′-bridging of the counteranion, while in about 7% of the complexes the ClO4− counteranions displayed di(μ:κ2O,O′)-bridging of the Ag(I) ions. Similarly, out of 1038 reported AgCF3SO3 complexes about 13% of the complexes demonstrate μ:κ2O,O′-bridging of the counteranion, while in about 5% of the complexes, the CF3SO3− counteranions displayed di(μ:κ2O,O′)-bridging of the Ag(I) ions. Out of 959 reported AgCH3CN complexes, only 2.2% of the complexes demonstrate μ:κ2N-bridging of CH3CN molecules, while in 1.6% of the complexes, the CH3CN molecules displayed di(μ:κ2N)-bridging of the Ag(I)ions (Fig. 13).25,26
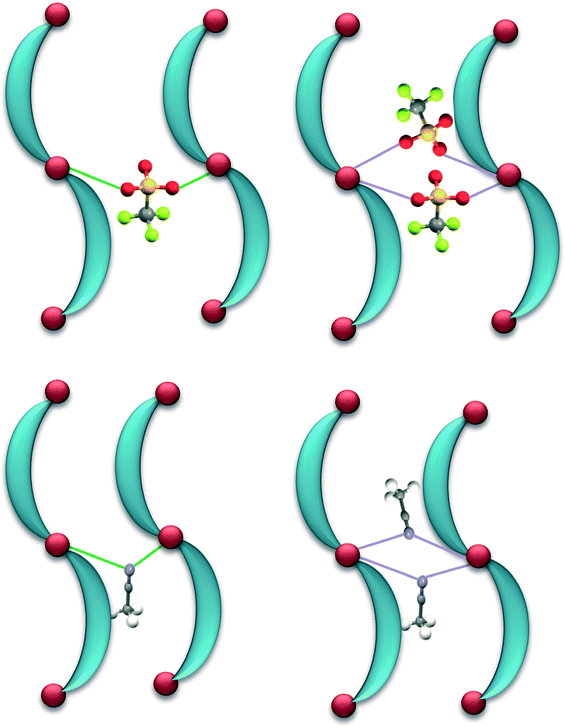 | ||
| Fig. 13 Top: schematic representation of μ:κ2O,O′-bridging (in green) and di(μ:κ2O,O′)-bridging (in purple) of pairs of 1D strands by CF3SO3 counteranions at Ag(I) nodes. Bottom: schematic representation of μ:κ2N-bridging (in green) and di(μ:κ2N)-bridging (in purple) of pairs of 1D strands by CH3CN molecules at Ag(I) nodes. | ||
In the 1:2 M:L complexes 6 and 7, the uncoordinated N2 donor of the decorating L side arms of the strands displayed inclination towards the Ag(I) centre on the adjacent polymeric chain and forced an unusual geometry on that metal centre. The counteranions in both these complexes did not coordinate to the Ag(I) ion. However, the size of the counteranion played a critical role in elaborating the dimensionality of the chains. In the former complex, the bulkier CF3SO3− counteranion reduced the Ag(I)–N2py–Cgpy angle [111.95°] and thus restricted the coordination of N2 with the Ag(I) cation [the N2⋯Ag contact was 3.152(7) Å]. By contrast, the less bulkier BF4− counteranion displayed less interference in the N⋯Ag interactions [the N2⋯Ag contact was 2.6595(18) Å and the Ag(I)–N2py–Cgpy angle was 138.80°] and thus facilitated the generation of a pseudo (4,4) network. The pyridyl rings of the decorating L side arms of 7 displayed more flexibility (as evident from Table 1) and thus assisted the formation of the pseudo (4,4) network. The pyridyl rings of 8 displayed wider Ag(I)–N2py–Cgpy angles [150.51–175.69°] and shorter Ag–N contacts [2.244(3)–2.384(4) Å] and thus facilitated in the formation of a corrugated (4,4) network. The pyridyl rings of 9 displayed further wider Ag(I)–N2py–Cgpy angles [168.46–175.58°] and shorter Ag–N contacts [2.267(4)–2.353(4) Å] and thus helped form a regular (4,4) network. Out of 2523 examples in the CSD database (version 5.33) for a search of Ag(I)–Npy–Cgpy angles, 125 complexes were observed to be in the range of 89.8–140.8°. Scrutinizing the Ag(I)⋯Npy contact distances [ranging between 2.512 and 3.280 Å] in these complexes, it was observed that the distances are within the sum of the van der Waals radii for Ag–N [3.25 Å].25,26 There were 2427 observations with Ag(I)–Npy–Cgpy angles between 144.6 and 180° and their Ag(I)⋯Npy distances range within 2.084–2.399 Å. This shows that the wider the Ag(I)–Npy–Cgpy angle, the stronger the Ag(I)⋯Npy interaction.
The CO groups of the four ligands of the rhombus of 6 splayed outwards. In 7, the CO groups of the four ligands of the rhombus were arranged such that two CO groups of the opposite ligands, which were pseudo enantiomers, pointed above the plane of the rhombus and towards each other, while the two CO groups of the remaining ligands, which were also pseudo enantiomers, pointed below the plane and were splayed away from each other. This arrangement produced an achiral sheet. The rhombus of the 2D network of 9 has a regular orientation with two CO groups pointing up and two pointing down and two CH3CN molecules in each cavity. All four ligands of the rhombus were of the same pseudo enantiomeric form producing a chiral sheet. By contrast, the rhombus of the 2D network of 8 has an irregular orientation with three CO groups pointing up and one pointing down and no CH3CN molecules in the structure. Three ligands of the rhombus had the same pseudo enantiomeric form, while the remaining ligand was of the other pseudo enantiomeric form. This gave rise to an achiral sheet. Thus, the irregular orientation of the CO groups in 8 appeared to make the structure corrugated rather than flat, while the vacillations of the CO groups prevented the formation of true 2D networks in 6 and 7. These differences may have been the cause of the embedding of the counteranions and the solvent molecules in the network cavities.
Conclusions
In conclusion, we have described two series of related coordination polymers of Ag(I) salts and L ligand with varying M:L ratio (1:1 and 1:2). The primary structure of the first Ag(I) series was not sensitive to the counteranion. However, the delicate anion–Ag and CH3CN–Ag bridging interactions showed a subtle effect on π–π-stacking and argentophilic interactions. Owing to these delicate interactions, a transition from 1D meso-helical chains to 2D grids was observed. The second Ag(I) series displayed a remarkable sensitivity to the counteranion showing a transition from ordered 1D meso-helical chains to 2D (4,4) nets.
Experimental section
Commercially available 4,4′-dipyridyl ketone was acquired from Chem Bridge. All chemicals were used as received without further purification. All solvents were of LR grade or above. The samples for microanalysis studies were dried under vacuum to remove volatile sample residues. Elemental microanalyses were carried out at the Campbell Microanalytical Laboratory, University of Otago. All measured microanalysis results were within an uncertainty of 0.4%. Infrared (IR) spectra were recorded on a Perkin-Elmer Spectrum BX FT-IR system.Caution! Although no problems were encountered in this work, transition metal perchlorates are potentially explosive. They should be prepared in small amounts and handled with care.
Reaction of L with AgCF3SO3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 ratio
:1 ratio
Solid AgCF3SO3 (13.94 mg, 0.054 mmol) dissolved in CH3CN (0.8 mL) was added dropwise to a methanolic solution of L (10 mg, 0.054 mmol). The resultant solution was sonicated and allowed to evaporate slowly for a week to yield X-ray quality colourless crystals of {[Ag(L)](CF3SO3)·1/2H2O}∞ (complex 1) which on drying in vacuo converted into a tan powder. Yield: 11 mg, 46%; analysis found: C 32.95, H 1.89 and N 6.29; calculated for the formula C12H8O4N2SF3Ag: C 32.67, H 1.83 and N 6.35; selected IR/cm−1: 3124–3053 (w, br), 1682 (m, sh), 1611 (w, sh), 1555 (w, sh), 1423 (m, sh), 1330–1271 (s, br), 1236 (s, br), 1216 (s, br), 1150 (s, br), 1105 (s, br), 1018 (s, sh), 940 (m, sh), 878–844 (w, br), 759 (m, sh), 660 (m, sh), 630 (s, sh), 572 (m, sh), 515 (s, sh).
Reaction of L with AgClO4·H2O in a 1:2 ratio
Solid AgClO4·H2O (22.5 mg, 0.108 mmol) dissolved in CH3CN (1.5 mL) was added dropwise to a methanolic solution of L (10 mg, 0.054 mmol). The resultant solution was sonicated and allowed to stand for a week to yield X-ray quality colourless crystals of {[Ag(L)](ClO4)·1/2H2O}∞ (complex 2) which on drying in vacuo converted into a tan powder. Yield (based on L): 10 mg, 47%; analysis found: C 33.51, H 2.25 and N 7.61; calculated for the formula C11H8O5N2ClAg: C 33.75, H 2.06 and N 7.16; selected IR/cm−1: 3095 (w, br), 1680 (m, sh), 1612 (w, sh), 1555 (w, sh), 1285 (m, br), 1055 (s, br), 952 (m, sh), 759 (m, sh), 691 (m, sh), 657 (s, sh) and 619 (s, sh).
Reaction of L with AgClO4·H2O in a 2:1 ratio
Solid AgClO4·H2O (11.2 mg, 0.054 mmol) dissolved in CH3CN (1 mL) was added dropwise to a methanolic solution of L (20 mg, 0.108 mmol). The resultant solution was sonicated and allowed to evaporate slowly for a week to yield X-ray quality colourless crystals of {[Ag2(L)2CH3CN](ClO4)2·2CH3CN·H2O}∞ (complex 3) which on drying in vacuo converted into a tan powder. Yield (based on Ag(I)): 11 mg, 50%; analysis found: C 34.26, H 2.51 and N 7.06; calculated for the formula C11H8O5N2ClAg·1/2CH3OH: C 33.89, H 2.47 and N 6.87; selected IR/cm−1: 3101 (w, br), 1665 (m, sh), 1609 (w, sh), 1556 (w, sh), 1286 (m, br), 1056 (s, br), 950 (m, sh), 758 (m, sh), 688 (m, sh), 659 (s, sh) and 620 (s, sh).
Reaction of L with AgBF4 in a 1:1 ratio
Solid AgBF4 (21.2 mg, 0.108 mmol) dissolved in CH3CN (1.5 mL) was added dropwise to a methanolic solution of L (20 mg, 0.108 mmol). The resultant solution was sonicated and allowed to stand for a week to yield colourless crystals of {[Ag2(L)2(CH3CN)2](BF4)2·CH3CN·H2O}∞ (complex 3a) which on drying in vacuo converted into a tan powder. The crystals were twinned and of poor quality. However, these crystals were found to be isomorphous to complex 3 [a = 7.5055(8) Å, b = 19.063(3) Å and c = 23.000(3) Å; α = 90°, β = 91.312(4)° and γ = 90°; V = 3290(1) Å3]. Yield: 21 mg, 51%; analysis found: C 35.04, H 2.03 and N 7.42; calculated for the formula C11H8ON2BF4Ag: C 34.87, H 2.13 and N 7.39; selected IR/cm−1: 3108 (w, br), 1682 (m, sh), 1609 (w, sh), 1554 (w, sh), 1417 (m, sh), 1282 (m, br), 1162 (w, sh), 1034 (s, br), 879 (m, sh), 760 (m, sh), 658 (s, sh) and 520 (s, sh).
Reaction of L with AgClO4·H2O in a 1:1 ratio
Solid AgClO4·H2O (11.3 mg, 0.054 mmol) dissolved in CH3CN (1.5 mL) was added dropwise to a methanolic solution of L (10 mg, 0.054 mmol). The resultant solution was sonicated and allowed to stand for a week to yield X-ray quality colourless crystals of {[Ag2(L)2(CH3CN)2](ClO4)2·CH3CN}∞ (complex 4) which on drying in vacuo converted into a tan powder. Yield: 15 mg, 71%; analysis found: C 33.51, H 2.25 and N 7.61; calculated for the formula C11H8O5N2ClAg: C 33.75, H 2.06 and N 7.16; selected IR/cm−1: 3102 (w, br), 1675 (m, sh), 1609 (w, sh), 1554 (w, sh), 1281 (m, br), 1061 (s, br), 951 (m, sh), 758 (m, sh), 688 (m, sh), 656 (s, sh) and 620 (s, sh).
Reactions of L with AgPF6 in a 2:1 ratio
Reaction 1: solid AgPF6 (13.2 mg, 0.054 mmol) dissolved in CH3CN (4 mL) was added dropwise to a 4 mL methanolic solution of L (20 mg, 0.108 mmol). The resultant clear solution was stirred overnight and concentrated in volume to 2 mL. Addition of 0.5 mL of diethyl ether yielded a brown crystalline precipitate which was filtered, washed with diethyl ether and dried in vacuo. Yield (based on Ag(I)): 13 mg, 55%. X-Ray quality colourless crystals of {[Ag2(L)2(CH3CN)2](PF6)2·2CH3CN}∞ (complex 5) were grown by slow evaporation of solvents (1:1 v/v CH3CN:CH3OH) from the solution of AgPF6 (6.9 mg, 0.027 mmol) and L (10 mg, 0.054 mmol). Analysis found: C 33.43, H 2.34 and N 7.81; calculated for the formula C11H8ON2PF6Ag·CH3OH·CH3CN: C 32.96, H 2.96 and N 8.24; selected IR/cm−1: 3629 (w, br), 1675 (m, sh), 1612 (w, sh), 1555 (w, sh), 1418 (m, sh), 1284 (m, sh), 1160 (w, sh), 1134 (w, sh), 881 (m, sh), 821 (vs, sh), 757 (m, sh), 690 (m, sh), 651 (s, sh) and 555 (s, sh).
Reaction 2: solid AgPF6 (13.2 mg, 0.054 mmol) dissolved in CH3CN (1 mL) was added dropwise to a methanolic solution of L (20 mg, 0.108 mmol). The resultant solution was sonicated and allowed to evaporate slowly for a week to yield X-ray quality colourless crystals of {[Ag(L)2](PF6)}∞ (complex 8) and {[Ag(L)2](PF6)(CH3CN)2}∞ (complex 9) which on drying in vacuo converted into a tan powder. Yield (based on Ag(I)): 20 mg, 60%. Analysis found: C 42.65, H 2.69 and N 9.11; calculated for the formula C22H16O2N4PF6Ag: C 42.54, H 2.60 and N 9.02; selected IR/cm−1: 1686 (m, sh), 1674 (m, sh), 1604 (w, sh), 1556 (w, sh), 1494 (w, sh), 1413 (m, sh), 1281 (m, sh), 1158 (w, sh), 949 (w, sh), 818 (vs, sh), 690 (m, sh), 647 (s, sh) and 556 (s, sh).
Reaction of L with AgCF3SO3 in a 2:1 ratio
Solid AgCF3SO3 (13.94 mg, 0.054 mmol) dissolved in CH3CN (1 mL) was added dropwise to a methanolic solution of L (20 mg, 0.108 mmol). The resultant solution was sonicated and allowed to evaporate slowly for a week to yield X-ray quality colourless crystals of {[Ag(L)2](CF3SO3)·1/2H2O}∞ (complex 6) which on drying in vacuo converted into a tan powder. Yield (based on Ag(I)): 22 mg, 65%; analysis found: C 44.47, H 2.84, N 9.00, and S 4.91; calculated for the formula C23H16O5N4SF3Ag: C 44.18, H 2.58, N 8.96, and S 5.13; selected IR/cm−1: 3200–3000 (w, br), 1675 (m, sh), 1640 (w, sh), 1604 (w, sh), 1554 (w, sh), 1410 (m, sh), 1328 (m, sh), 1284 (m, sh), 1263 (s, sh), 1222 (m, sh), 1145 (m, sh), 1058 (s, sh), 1029 (s, sh), 949 (m, sh), 879 (m, sh), 830 (m, sh), 756 (m, sh), 660–634 (s, br).
Reaction of L with AgBF4 in a 2:1 ratio
Under a blanket of Ar gas, methanolic solution (4 mL) of L (40 mg, 0.216 mmol) was added to 4 mL of a CH3CN solution of AgBF4 (21.2 mg, 0.108 mmol). The resultant solution was allowed to react overnight. Addition of 0.5 mL of diethyl ether to this solution resulted in colourless crystalline solid which was filtered and dried in vacuo. Yield (based on Ag(I)): 28 mg, 43%; analysis found: C 48.17, H 3.07 and N 10.18; calculated for the formula C22H16O2N4F4BAg·CH3CN: C 47.72, H 3.17 and N 11.59; selected IR/cm−1: 3106–3054 (w, br), 1677 (m, sh), 1608 (w, sh), 1555 (w, sh), 1409 (m, sh), 1282 (m, sh), 1160 (w, sh), 1032 (s, br), 756 (m, sh), 660 (s, sh), 520 (m, sh).
Solid AgBF4 (10.6 mg, 0.054 mmol) dissolved in CH3CN (1 mL) was added dropwise to a methanolic solution of L (20 mg, 0.108 mmol). The resultant solution was sonicated and allowed to evaporate slowly for a week to yield X-ray quality colourless crystals of {[Ag(L)2](BF4)}∞ (complex 7).
X-Ray data collection and structure solution
Crystallographic data are summarised in Table 3. Selected bond lengths and angles of complexes 1–9 are available in the ESI along with a description of how the disordered components of the complexes were treated. X-Ray diffraction data were collected at the University of Otago on a Bruker APEX II CCD diffractometer with graphite monochromated Mo-Kα (λ = 0.71073 Å) radiation. Intensities were corrected for Lorentz and polarisation effects and multiscan absorption corrections were applied to all structures. The structures were solved by direct methods such as SHELXS43,44 or SIR-9745 and refined on F2 using all data by full-matrix least-squares procedures such as SHELXL 97.43 Non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms were placed in ideal positions except for the hydrogen atoms of the H2O molecules in 1, 2, 3, 4 and 6 which were located from the Fourier synthesis maps. In 1, the CF3SO3− anion was disordered over two sites with site occupancy of 0.36 and 0.64. This CF3SO3− anion and H2O molecule were modelled using DFIX constraints. In 4, both ClO4− anions were disordered (50%). A very disordered CH3CN of solvent was removed from the structure using the SQUEEZE procedure of PLATON46 as it could not be modelled. Analysis of the X-ray data indicated that crystals might carry a well-defined twin. However, a twin law that superimposed all or half of the reflections could not be found. The twinning was evident in both data collections for 4. As a result, the precision of the data for 4 was not high. The crystals of 5 were of poor quality. Two different data sets for 5 were collected and solved, both of which were of poor quality because of weakly diffracting crystals. The solution reported herein represents the best quality solution. A CH3CN molecule in 5 was disordered and the C and N atoms of this molecule were refined isotropically and the hydrogen atoms of this molecule were not placed. The disordered CH3CN molecule was modelled with site occupancy of 0.6 and 0.4 and additional restraints were used to maintain the linearity. A CF3SO3− anion was disordered with site occupancy of 0.51 and 0.49. The F and O atoms of this molecule were refined isotropically. In 8, the PF6− anion was disordered on a four-fold axis over two sites with site occupancy of 0.35 and 0.65. This PF6− anion was modelled using DFIX constraints. All calculations were performed using the WinGX47 interface. Detailed analyses of the extended structure were carried out using PLATON46 and MERCURY (version 3.0)26,48 crystallographic data are listed in the appendix.| Structure | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Formula | C24H18O9N4S2F6Ag2 | C22H18Ag2Cl2N4O11 | C28H27Ag2Cl2N7O11 | C28H25Ag2Cl2N7O10 | C30H28Ag2F12N8O2P2 | C46H34O11N8S2F6Ag2 | C22H16AgBF4N4O2 | C22H16AgF6N4O2P | C26H22AgF6N6O2P |
| Formula weight | 900.28 | 801.04 | 924.21 | 906.19 | 1038.26 | 1268.67 | 563.07 | 621.23 | 703.34 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | C2/c | C2/c | P21/c | P21/c | P21/c | C2/c | P21/c | P21/n |
P |
| a/Å | 13.7574(9) | 12.4586(5) | 7.5345(3) | 7.8231(5) | 7.9890(5) | 27.5782(15) | 11.020(11) | 11.0709(7) | 10.950(7) |
| b/Å | 11.2371(8) | 9.8846(4) | 19.1191(10) | 18.8411(11) | 22.9536(15) | 10.6328(5) | 18.3216(17) | 18.5174(12) | 11.1081(7) |
| c/Å | 19.0368(12) | 21.2874(9) | 23.0434(11) | 22.8612(13) | 20.1547(15) | 18.6286(9) | 10.7558(10) | 11.6388(8) | 12.1273(9) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 85.248(2) |
| β/° | 95.529(2) | 102.7935(18) | 92.045(1) | 90.874(3) | 91.046(3) | 117.5172(17) | 93.782(4) | 96.473(3) | 86.205(3) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 83.680(3) |
| V/Å3 | 2929.3(5) | 2556.6(18) | 3317.4(3) | 3369.3(5) | 3695.3(6) | 4844.8(6) | 2167.4(5) | 2370.8(3) | 1458.7(2) |
| Z | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 2 |
| T/K | 90(2) | 90(2) | 90(2) | 90(2) | 90(2) | 90(2) | 90(2) | 90(2) | 90(2) |
| μ/mm−1 | 1.577 | 1.811 | 1.412 | 1.387 | 1.249 | 0.987 | 0.994 | 0.993 | 0.819 |
| Total reflections | 22092 |
39118 |
39984 |
38976 |
58540 |
26131 |
26132 |
21026 |
17617 |
| Unique reflections (Rint) | 8857 (0.0312) | 9878 (0.0325) | 9643 (0.0411) | 9961 (0.0394) | 9915 (0) | 9998 (0.0538) | 9900 (0.0347) | 9898 (0.0210) | 9112 (0.0289) |
| R 1 indices [I > 2σ(I)] | 0.0199 | 0.0285 | 0.0495 | 0.0687 | 0.0694 | 0.0596 | 0.0221 | 0.0340 | 0.0446 |
| wR2 (all data) | 0.0522 | 0.1269 | 0.1158 | 0.1792 | 0.2414 | 0.1368 | 0.0559 | 0.0846 | 0.1247 |
| Goodness-of-fit | 1.088 | 1.209 | 1.069 | 1.066 | 1.149 | 1.150 | 1.083 | 1.145 | 1.082 |
Acknowledgements
We acknowledge the Department of Chemistry for the award of Master's Scholarship and thank the University of Otago for publication bursary (to KMP). We thank Professor Geoff Jameson for assistance with the twinning in 4. Lisa Bucke is thanked for her assistance in preparing Fig. 2.Notes and references
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688–764 CrossRef CAS PubMed.
- C.-L. Chen, B.-S. Kang and C.-Y. Su, Aust. J. Chem., 2006, 59, 3–18 CrossRef CAS.
- D. Visinescu, M. Andruh, A. Müller, M. Schmidtmann and Y. Journaux, Inorg. Chem. Commun., 2002, 5, 42–45 CrossRef CAS.
- M.-L. Tong, X.-M. Chen, B.-H. Ye and S. W. Ng, Inorg. Chem., 1998, 37, 5278–5281 CrossRef CAS.
- L. Hou, W.-J. Shi, Y.-Y. Wang, B. Liu, W.-H. Huang and Q.-Z. Shi, CrystEngComm, 2010, 12, 4365–4371 RSC.
- Y. Xia, S. Li, B. Wu, Y. Liu and X.-J. Yang, CrystEngComm, 2011, 13, 5763–5772 RSC.
- F. Ma, D.-B. Wei and Z.-M. Cao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m611–m612 CAS.
- C.-Q. Wan, S. A. Al-Thabaiti, X.-D. Chen and T. C. W. Mak, Eur. J. Inorg. Chem., 2013, 30, 5265–5273 CrossRef.
- C.-Q. Wan, Z.-W. Wang, G. Wang, A.-M. Li, Q.-H. Jin, Y.-H. Deng and T. C. W. Mak, Eur. J. Inorg. Chem., 2013, 14, 2591–2600 CrossRef.
- K. J. Wei, Y. S. Xie, J. Ni, M. Zhang and Q. L. Liu, Inorg. Chem. Commun., 2006, 9, 926–930 CrossRef CAS PubMed.
- R. Benedix, Z. Anorg. Allg. Chem., 1984, 24, 303 CAS.
- M. A. Braverman, J. H. Nettleman, R. M. Supkowski and R. L. LaDuca, Inorg. Chem., 2009, 48, 4918–4926 CrossRef CAS PubMed.
- G. A. Farnum, W. R. Knapp and R. L. LaDuca, Polyhedron, 2009, 28, 291–299 CrossRef CAS PubMed.
- E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochem. Organomet. Inorg. Compd., 1994, 1156 Search PubMed.
- D. B. Cordes, L. R. Hanton and M. D. Spicer, Inorg. Chem., 2006, 45, 7651–7664 CrossRef CAS PubMed.
- A. D. Clark, Z. Kristallogr. - New Cryst. Struct., 1998, 213, 382 CAS.
- X. Z. Sun, H. J. Yan and C. Q. Wan, Z. Kristallogr. - New Cryst. Struct., 2013, 228, 375–376 CAS.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed.
- D. A. McMorran, Inorg. Chem., 2007, 47, 592–601 CrossRef PubMed.
- L. Han and M. Hong, Inorg. Chem. Commun., 2005, 8, 406–419 CrossRef CAS PubMed.
- R. Horikoshi, T. Mochida, N. Maki, S. Yamada and H. Moriyama, J. Chem. Soc., Dalton Trans., 2002, 28–33 RSC.
- R. Carballo, B. Covelo, E. García-Martínez, A. B. Lago and E. M. Vázquez-López, Polyhedron, 2009, 28, 923–932 CrossRef CAS PubMed.
- D. H. Johnston and D. F. Shriver, Inorg. Chem., 1993, 32, 1045–1047 CrossRef CAS.
- M. A. M. Abu-Youssef, V. Langer and L. Ohrstrom, Dalton Trans., 2006, 2542–2550 RSC.
- I. R. Thomas, I. J. Bruno, J. C. Cole, C. F. Macrae, E. Pidcock and P. A. Wood, J. Appl. Crystallogr., 2010, 43, 362–366 CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- R. G. Serres, C. A. Grapperhaus, E. Bothe, E. Bill, T. Weyhermüller, F. Neese and K. Wieghardt, J. Am. Chem. Soc., 2004, 126, 5138–5153 CrossRef CAS PubMed.
- E. C. Constable, C. E. Housecroft, M. Neuburger, S. Reymann and S. Schaffner, Eur. J. Inorg. Chem., 2008, 22, 3540–3548 CrossRef.
- E. C. Constable, C. E. Housecroft, B. M. Kariuki, M. Neuburger and C. B. Smith, Aust. J. Chem., 2003, 56, 653–655 CrossRef CAS.
- P. L. Caradoc-Davies, L. R. Hanton and W. Henderson, J. Chem. Soc., Dalton Trans., 2001, 2749–2755 RSC.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- C. A. Black, L. R. Hanton and M. D. Spicer, Chem. Commun., 2007, 3171–3173 RSC.
- C.-Q. Wan, X.-D. Chen and T. C. W. Mak, CrystEngComm, 2008, 10, 475–478 RSC.
- T. Bark and R. P. Thummel, Inorg. Chem., 2005, 44, 8733–8739 CrossRef CAS PubMed.
- G. Yang, S. L. Zheng, X. M. Chen, H. K. Lee, Z. Y. Zhou and T. C. W. Mak, Inorg. Chim. Acta, 2000, 303, 86–93 CrossRef CAS.
- X. Hou, M. Schober and Q. Chu, Cryst. Growth Des., 2012, 12, 5159–5163 CAS.
- R. Singh, X. Hou, O. Molly, M. Schober and Q. Chu, CrystEngComm, 2012, 14, 6132–6135 RSC.
- A. Bellusci, M. Ghedini, L. Giorgini, F. Gozzo, E. I. Szerb, A. Crispini and D. Pucci, Dalton Trans., 2009, 7381–7389 RSC.
- A. Bellusci, A. Crispini, D. Pucci, E. I. Szerb and M. Ghedini, Cryst. Growth Des., 2008, 8, 3114–3122 CAS.
- R. P. Feazell, C. E. Carson and K. K. Klausmeyer, Inorg. Chem., 2005, 45, 935–944 CrossRef PubMed.
- G. M. Sheldrick, SHELX97. Programs for Crystal Structure Analysis, Release 97-2, Institüt für Anorganische Chemie der Universität, Tammanstrasse 4, D-3400, Göttingen, Germany, 1997 Search PubMed.
- Siemens, SHELXTL. Structure Determination Software, Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA, 1995 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- A. L. Spek, PLATON. A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1999 Search PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond lengths and angles for complexes 1–9. CCDC 982462–982470. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00155a |
| This journal is © The Royal Society of Chemistry 2014 |