 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFine tuning of crystal architecture by intermolecular interactions: synthon engineering
Petra
Bombicz
*a,
Tobias
Gruber
b,
Conrad
Fischer†
b,
Edwin
Weber
b and
Alajos
Kálmán
a
aResearch Group of X-ray Diffraction, Institute of Organic Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, P.O. Box 286, H-1519 Budapest, Hungary. E-mail: bombicz.petra@ttk.mta.hu
bInstitut für Organische Chemie, TU Bergakademie Freiberg, Leipziger Str. 29, Sachs, D-09596 Freiberg, Germany. E-mail: edwin.weber@chemie.tu-freiberg.de; Fax: +49 3731 392386
First published on 13th January 2014
Abstract
There has been a long time effort to influence or favourably fine tune structural properties by introduction of substituents or guest molecules of different sizes, shapes and chemical composition to consequently alter physico-chemical properties of the respective crystals. These attempts require the recognition, understanding and application of intermolecular interactions, crystallographic and, in case of occurrence, non-crystallographic symmetries. It brings us to the field of crystal engineering, which aims to produce new substances with required properties based on the knowledge of the structural properties of already characterised solids. A series of calixarene crystal structures are presented where the crystal packing is determined by spatial or by electrostatic effects. A series of laterally substituted calixarenes where both steric requirements and electrostatic forces play a role in the crystal architecture shows how the supramolecular synthon can be engineered.
 Petra Bombicz | Petra Bombicz received a B.Sc. in 1987, an M.Sc. in organic synthesis in 1989, a “Dr. univ.” in organosilicon chemistry in 1993 and a PhD in crystallography in 1997 from the Budapest University of Technology and Economics. She joined the Laboratory of X-ray diffraction of the Research Centre for Natural Sciences of the Hungarian Academy of Sciences in 1993, where twenty years later she became the head. Her research interests include supramolecular chemistry, crystal engineering, intermolecular interactions, polymorphy and isostructurality. |
 Tobias Gruber | Tobias Gruber received his PhD in 2008 from the University of Freiberg (Saxony) on calixarenes and related compounds. After a year in the group of Manfred Jung (Freiburg/Breisgau), he moved to Oxford for a postdoctoral stay in the group of Christopher Schofield. In 2011, he started his independent research career in Braunschweig and moved later back to Freiberg. His research interests include research on artificial receptors for biologically active compounds, crystal engineering and chemical biology. |
 Conrad Fischer | Conrad Fischer obtained his Dipl.-Chem./MSc (2003–2008) in Organic Chemistry from the University of Freiberg/Germany and was awarded with the Georgius-Agricola Medal for the best thesis. In the research group of Prof. Dr E. Weber, he received his PhD in 2012 for the structural characterization of lateral substituted calixarenes. Currently, he is working on bioanalytical approaches for in vitro MS detection at the University of Calgary/Canada. |
 Edwin Weber | Edwin Weber received his diploma in 1973 and a PhD in 1976 at the University of Würzburg. He carried out postdoctoral studies at University in Bonn and as a NATO-fellow at University of Wisconsin. He became a Lecturer in 1984 and a Professor in 1986 of Organic Chemistry at the University of Bonn. From 1992–1993, he temporarily held a Full Professorship at the University of Kiel. He is a full Professor of Organic Chemistry at the Technical University Bergakademie Freiberg since 1993. His current research fields focus on different aspects of supramolecular chemistry including molecular recognition and inclusion, porous framework design, crystal engineering, chemical sensing and electronic materials properties. |
 Alajos Kálmán | Alajos Kálmán has received MSc in 1958, PhD in 1968 from Eötvös Loránd University Budapest; and DSc in 1975. He has been a Professor of crystal chemistry from 1977. He is a Member of the Hungarian and the European Academy of Sciences since 2001 and since 2003. Until his retirement in 2005, he headed the Department of X-ray Diffraction in the Research Centre for Natural Sciences, Hungarian Academy of Sciences. He was a member of the Executive Committee of the Hungarian Chemical Society 1985–2007, Vice-President 1990–1997, and President 1997–2007. He was a member of the Executive Committee of IUCr 1984–1993, Vice-President 1990–1993, and co-editor of Acta Crystallographica 1990–1999. His current research interests are in X-ray crystallography is supramolecular chemistry, polymorphism, isostructurality, morphotropy (non-crystallographic rotations). |
Introduction
The properties of materials depend on the properties of the building blocks and the interactions among them.1 The non-covalent interactions have an influence on the molecular conformation, the packing arrangement and the molecular recognition processes. They affect the stability of inclusion compounds and clathrates, as well as the biological activity of macromolecules. Crystal forces are often sufficient to influence intramolecular torsion angles of a flexible molecule enhancing the importance of secondary weak interactions. The conformational variability makes calixarenes ideal target compounds both in the field of crystal engineering and supramolecular complex formation.Polymorphs, the different crystal structures with the same chemical composition, can be thought of as supramolecular isomers, species in which the relative positioning of the same molecule is different.2 In turn, a given packing arrangement may tolerate small changes caused either by the gradual change in site and/or size of substitution or by the gradual change of guest molecules incorporated into a host lattice. How much can a crystal structure tolerate small molecular changes? The packing coefficient3 for organic crystals is around 0.6–0.7. It means that there is ca. 30–40% free space in close packed organic structures. This free space offers the opportunity to tolerate small molecular changes in a given structural arrangement.
The investigation of isostructurality is a useful tool for a deeper understanding of close packing principles. It is an advantage, in contrast to polymorphy, that the degree of similarity can be described by numerical descriptors.4–6 The cell similarity index (π)4–6 is for the comparison of the unit cells, calculated using the orthogonalized lattice parameters of the related crystals. In the event of great similarity between the two unit cells, π is close to zero. The isostructurality index (I(s))4–6 indicates the distance differences between the crystal coordinates of identical non-H atoms within the same section of the related structures. I(s) takes into account both the differences in the geometry of the molecules and the positional differences in the same asymmetric unit caused by rotation and translation. The molecular isometricity index (I(m))4–6 is introduced for the comparison of the molecular conformation, it is a direct measure of the degree of approximate isomorphism of two molecules. I(m)4–6 is calculated by least-squares fitting7 of the positions occupied by the identical non-H atoms of the two molecules resulting from the superposition of the molecules. The I(m) value is space group independent. The volumetric isostructurality index (I(v))8 gives the percentage ratio of the overlapping volume of molecules. The advantage of the calculation of I(v) is that weakly isostructural structures, even with different guest molecules, or with disordered atomic positions can be compared, and hydrogen atoms are included in the calculation. For the quantification of similarity relationships “supramolecular constructs”, e.g. sub-components of complete crystal structures are identified by XPac9 by consistent geometrical features in crystal structures implying only geometrical similarity. A reference (kernel) molecule is defined and a coordination shell is generated on the basis of some distance criteria in the calculation.
When no more chemical change can be supported by a crystal structure, either a different packing arrangement and/or a different molecular conformation appears. When the tolerance is terminated and a different packing arrangement is developed, packing motifs may still be kept but the motifs are moved relative to each other. The relationships among the remaining structural motifs in the new crystal packing compared to the commencing crystal arrangement are often non-crystallographic symmetries. They may be non-crystallographic rotation and translation. This phenomenon is called morphotropy, a link between the isostructurality and polymorphy.10,11
During the effort of fine-tuning the structural properties with the application of substituents or guest molecules, we influence intermolecular interactions and crystallographic symmetries. With regard to this, calixarenes represent one of the most investigated compound types due to their easy availability, conformational flexibility, tunable amphiphilic properties, and versatile inclusion features.12–19 A series of calixarene crystal structures are presented where spatial requirements have a determining role in the crystal packing over the electrostatic interactions and vice versa where the electrostatic effects are the driving forces of the conformation and packing behaviour. A series of laterally substituted calixarenes demonstrate how the well balanced spatial requirements and electrostatic forces play a role in the arrangement of packing motifs in the crystals. The directed manipulation of the supramolecular packing architecture20 can be perceived as a case of crystal engineering, for which “synthon engineering” might be an appropriate designation.
Discussion
Structures having similar chemical composition can be isostructural. The challenge is more complex if we have not monomolecular solids but multi-component host–guest systems, where the guest is a carefully selected solvent or a small molecule. However, there is a limit at which the changes in the size and properties of the host or guest molecules are not tolerated by the crystal anymore, and thus a different crystal structure appears. Packing motifs may still remain in the different crystal structures. There is a morphotropic relationship10,11 between the structures if these packing motifs are related with non-crystallographic symmetries. The determining forces of the packing arrangement can be the spatial requirements or the electrostatic effects or their combination in a well-balanced manner.Spatial driving force
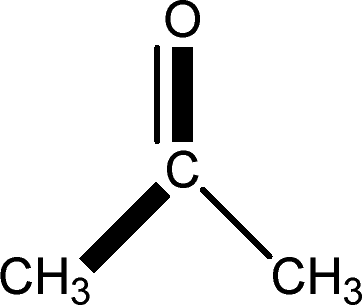
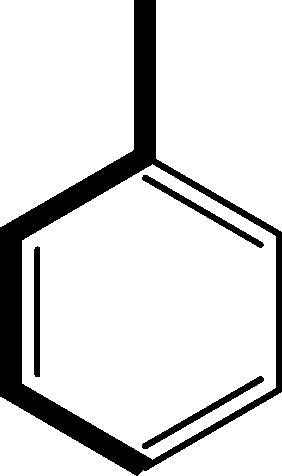
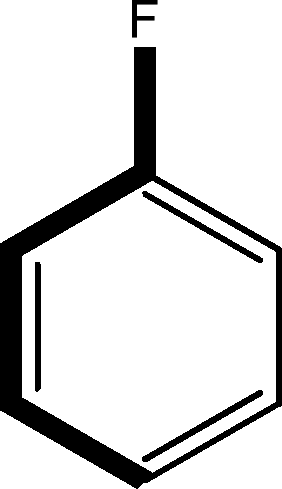
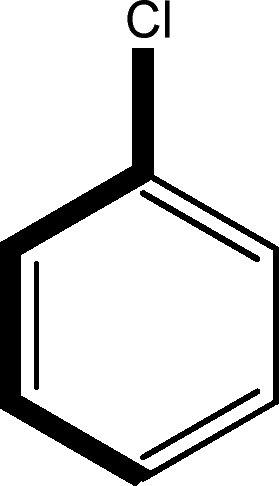
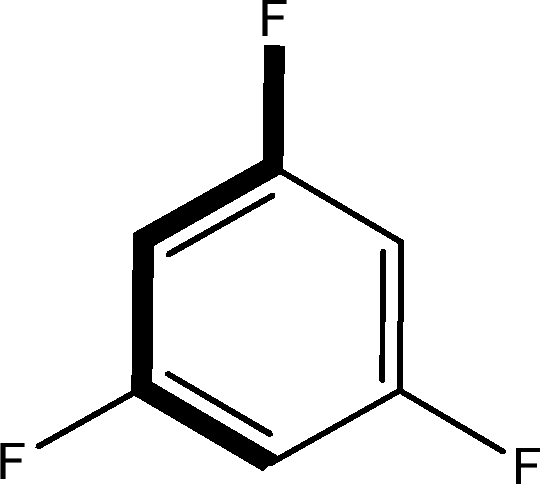
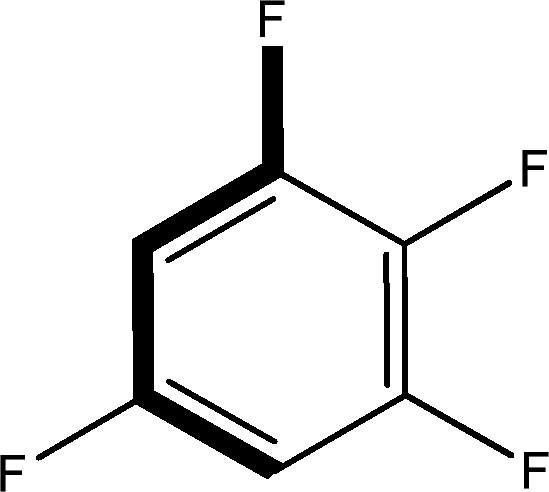
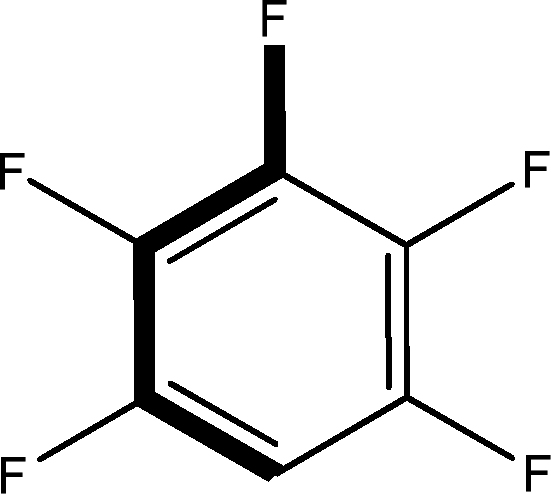
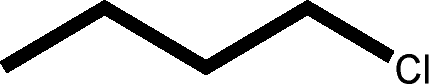
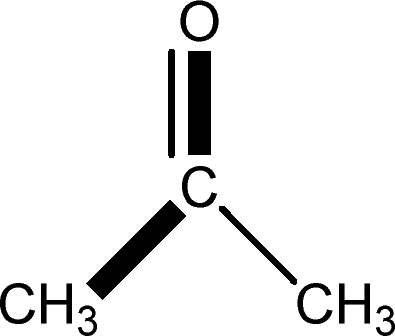
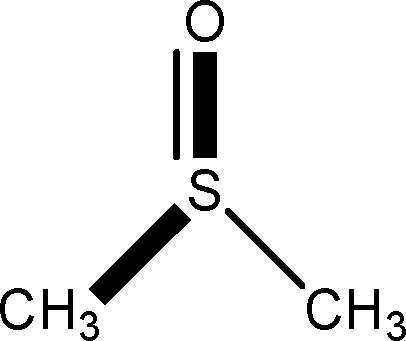
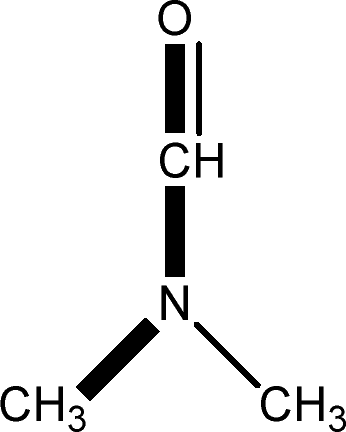
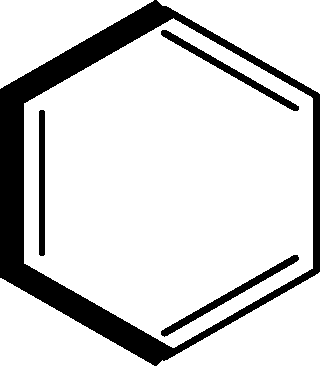
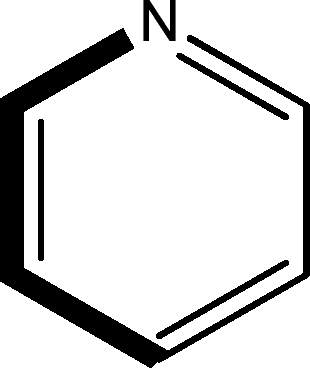
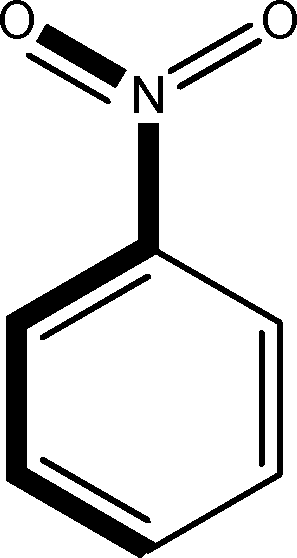
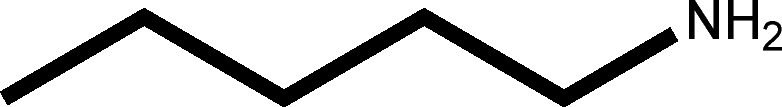
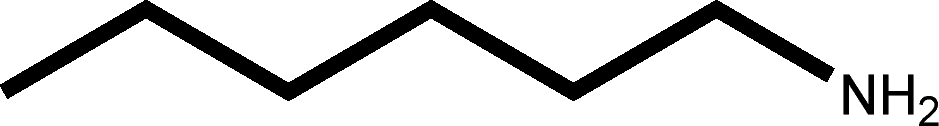
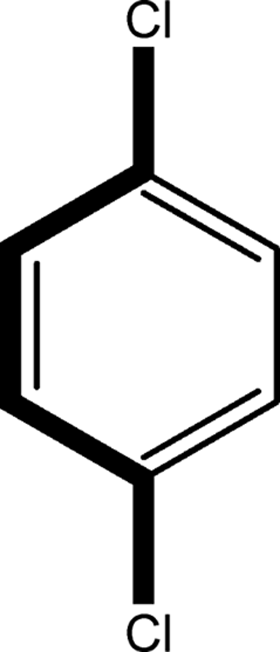
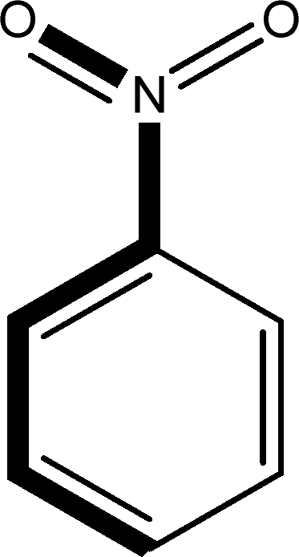
There are 53 individual structures of the inclusion compounds of 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetrahydroxycalix[4]arene in the Cambridge Structural Database,21 version 5.34 where 3D coordinates are free of redundancy and errors. Most of the structures crystallize in three space groups: 7 structures in the space group P2/n (13), 18 structures in the space group P4/n (85) and 11 structures in the space group P4/nnc (126). The arrangements of the molecules in the space groups P2/n and P4/n are identical, the host and guest stoichiometry is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the guest is nested in the cavity of the calixarene molecule (Fig. 1a). The divergence of the P2/n monoclinic unit cells in the 7 structures from the higher symmetry P4/n tetragonal one is small. The maximum difference in the P2/n cell dimensions is 0.74% in length a and c (BEGYIW01) and 0.26° in β angle from 90° (EZUMUH). The host and guest stoichiometry is 2:1 in the P4/nnc structures: two host molecules encapsulate one guest (Fig. 1b). The guest molecules are often found to be disordered in the cavity of the calixarenes. Irrespective of the chemical functionality of the guest molecule, it is the size of the guest molecule which determines the packing arrangement in these inclusion compounds (Table 1). For example, pentane, n-butanol and benzene inclusion compounds crystallize in the space group P4/n, but n-dodecane, n-heptanol and nitrobenzene inclusion compounds crystallize in the space group P4/nnc. If the longitudinal length of the guest molecule is 3, 4, 5 or incidentally 6 atoms, then the 1:1 stoichiometry and the P2/n or P4/n space groups are realised. If the length of the guest molecule is 5 atoms or more, 6, 7, 8 atoms and so on, then the 2:1 stoichiometry and the space group P4/nnc appear. The inclusion compounds with n-butylamine and nitrobenzene, where the 1D length of the guest molecule is 5 and 6 atoms, represent the subset of the two groups, and they are able to crystallise in both space groups. This calixarene inclusion system succeeds to be a fine tuned molecular switch based purely on the spatial requirements.
:1, and the guest is nested in the cavity of the calixarene molecule (Fig. 1a). The divergence of the P2/n monoclinic unit cells in the 7 structures from the higher symmetry P4/n tetragonal one is small. The maximum difference in the P2/n cell dimensions is 0.74% in length a and c (BEGYIW01) and 0.26° in β angle from 90° (EZUMUH). The host and guest stoichiometry is 2:1 in the P4/nnc structures: two host molecules encapsulate one guest (Fig. 1b). The guest molecules are often found to be disordered in the cavity of the calixarenes. Irrespective of the chemical functionality of the guest molecule, it is the size of the guest molecule which determines the packing arrangement in these inclusion compounds (Table 1). For example, pentane, n-butanol and benzene inclusion compounds crystallize in the space group P4/n, but n-dodecane, n-heptanol and nitrobenzene inclusion compounds crystallize in the space group P4/nnc. If the longitudinal length of the guest molecule is 3, 4, 5 or incidentally 6 atoms, then the 1:1 stoichiometry and the P2/n or P4/n space groups are realised. If the length of the guest molecule is 5 atoms or more, 6, 7, 8 atoms and so on, then the 2:1 stoichiometry and the space group P4/nnc appear. The inclusion compounds with n-butylamine and nitrobenzene, where the 1D length of the guest molecule is 5 and 6 atoms, represent the subset of the two groups, and they are able to crystallise in both space groups. This calixarene inclusion system succeeds to be a fine tuned molecular switch based purely on the spatial requirements.
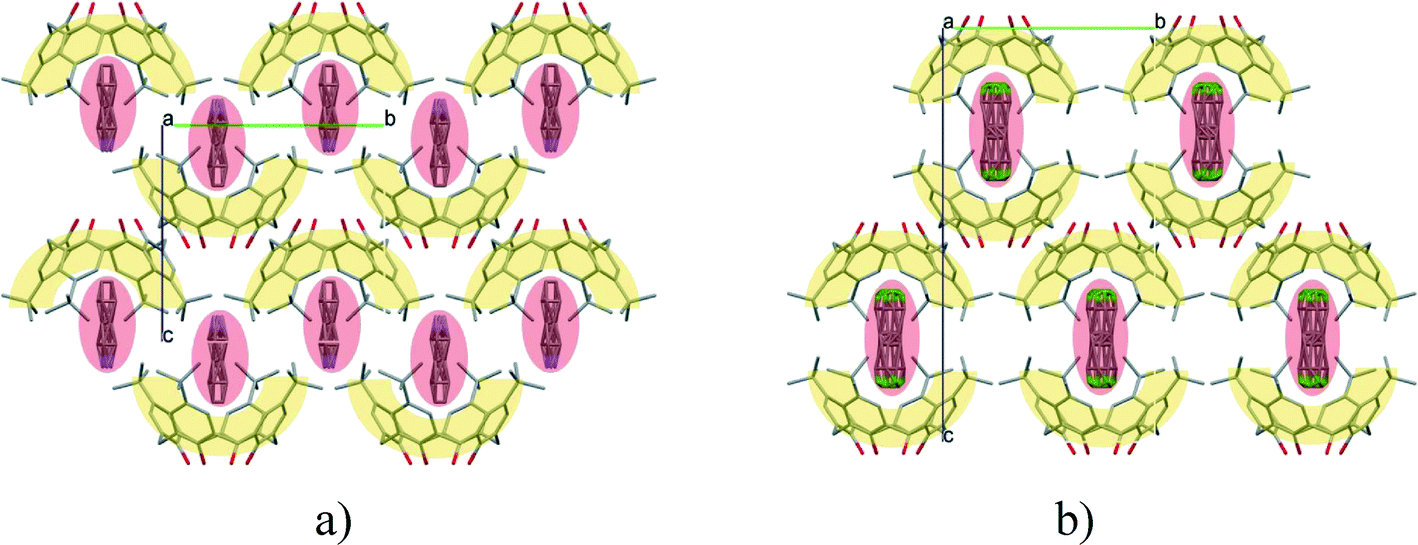 | ||
| Fig. 1 The two most often occurring packing arrangements of the inclusion compounds of 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetrahydroxycalix[4]arene in the space groups of a) P2/n and P4/n with 1:1 stoichiometry (MUBHUM, P4/n) as well as b) P4/nnc with 2:1 stoichiometry (QUSGOA). | ||
:1 stoichiometry, as well as of P4/nnc with 2:1 stoichiometry. The guest molecule is indicated only, the CSD refcode referring to the inclusion compound is given. The 1D length of the guest molecule is highlighted
| P2/n | ||||||
|
|
|
|
|
|
|
| BEGYIW01 | BHPMZC04 | BOCZUO02 | UWIVEC | EZUMER | EZUMIV | EZUMUH |
| P4/n | ||||||

|

|

|
|

|
— | — |
| NILCAM | MUBHUM | NILCOA | NILCEQ | NILCIU | — | — |

|

|
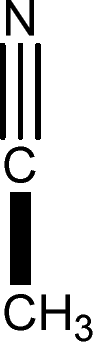
|
|
|
|
— |
| MOVMAM | LODNOH | ZAHMOK | BEGYIW | VEGPIG | BUWLOV | — |
|
|
|
|
|
|
|
| GOKPEB | GOKQUS | BHPMYC | BOCZUO01 | UWIWIH | CUPWAL | NAPCIQ |
| P4/nnc | ||||||

|

|
|

|

|
||
| MUBJAU | QUSFOZ | TESZAT | QUSFIT | QUSGOA | ||
|

|
|
|
|||
| TESYUM | QUSGAM | |||||

|

|
|||||
| QUSGIU | QUSGEQ | QUSFUF | JEFTOD | |||
Electrostatic driving force
In most of the crystal structures, the role of the guest molecule is not only to be present and to fill voids but also to participate actively in the construction of the structure by intermolecular interactions. The manner of the guest recognition seriously affects the crystal structures.The upper rim dinitro and the lower rim ethyl ester substituted calix[4]arene molecules crystallized from selected polar aprotic and protic solvents: acetone, DMF, DMSO and n-BuOH as guests have identical space group (P21/n) and the calculated cell similarity indices (π)4–6 show only slight differences in the unit cell dimensions.12 The upper rim of the host is involved in the complexation of the non-protic guests (cavitate type), whereas the protic butanol molecule forms favourable secondary interactions at the lower-rim site (clathrate type) (Fig. 2). The location of the calix[4]arene molecule in the unit cell of the n-BuOH containing system is related to the host position in all other three complexes by a non-crystallographic twofold axis (Fig. 3). Due to the different locations and orientations of the calix[4]arene host within the unit cell of the n-BuOH clathrate, no reasonable isostructurality I(s) indices4–6 are calculated for the involved pair of host molecules. The rotation of the host in the unit cell of the n-BuOH inclusion compound is a consequence of the change in the supramolecular (host–guest) recognition mode. It is a morphotropic change induced by supramolecular interactions indicated by the end of isostructurality but keeping molecular isometricity at the same time.
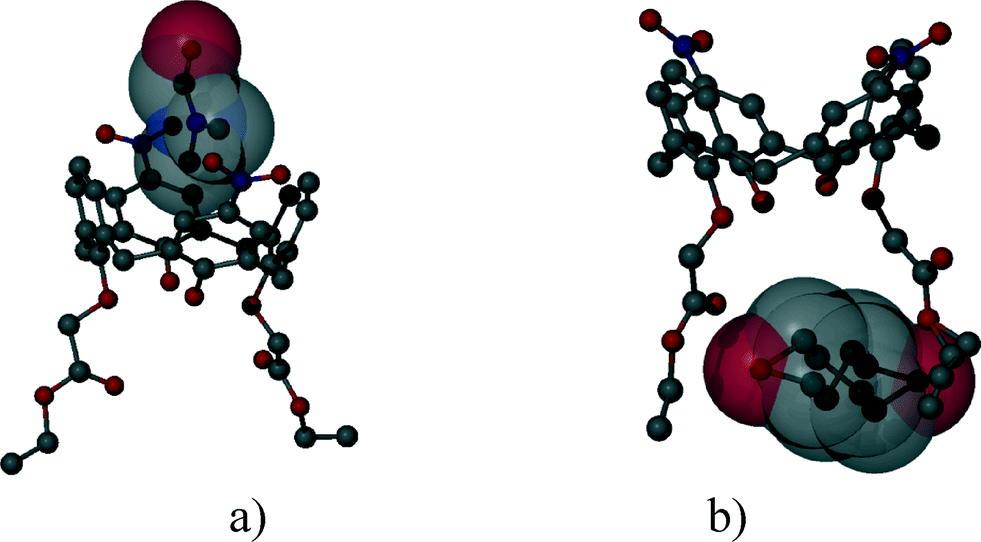 | ||
| Fig. 2 The location of the a) DMF and b) n-BuOH guest molecules related to the calix[4]arene host molecules.12 | ||
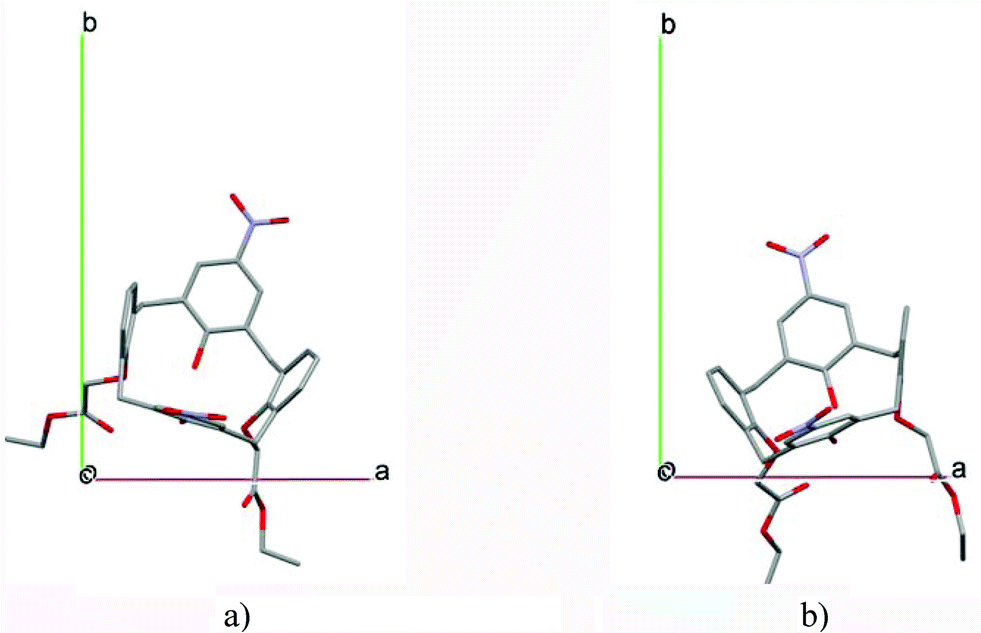 | ||
| Fig. 3 The position of the calix[4]arene host molecules in the unit cells of the inclusion compounds containing a) DMF and b) n-BuOH guest molecules viewed along the c axis. The locations of the hosts are related by a virtual, non-crystallographic twofold symmetry thus indicating a morphotropic relationship. | ||
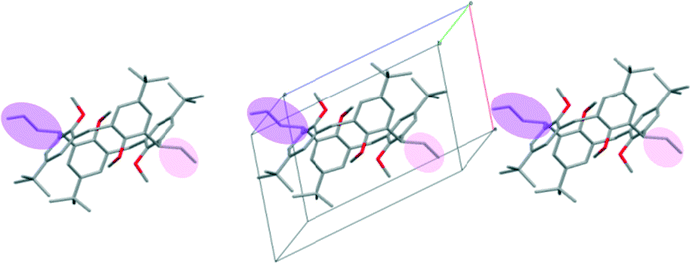
The introduction of n-hexanoyl substituents to the upper rim of a calix[4]arene enlarges the cavity volume and enables complex formation with keto and hydroxylic terpenes, guest molecules such as (−)-menthone, (−)-menthol.16 The host:guest stoichiometry 2:2 and the space group P21 are identical, and the low cell similarity index (π)4–6 indicates a high structural similarity. There is a substantial change in the placement of the molecules in the unit cell by the complementary size and polarity relationships between the cone shaped calixarene cavity and the guest molecules (Fig. 4). The two structures cannot be transformed to each other by crystallographic symmetries determined by the space group without an enantiomeric change. The host positions in the (−)-menthone and (−)-menthol complexes are related by non-crystallographic twofold axes parallel to the b crystallographic axis in a and c at 1/4 and 3/4 (Fig. 5). However, there is no twofold axis in the space group P21. The host frameworks have a morphotropic relation described by non-crystallographic motion as the result of close packing requirements. The rotation or translation of the common motif may transform one specific pattern into another with denser packing. Morphotropy is the phenomenon where the structural motifs can be correlated with non-crystallographic symmetries.10,11 Morphotropy was described first for monomolecular crystals. These structures are the first examples of morphotropy initiated by supramolecular effects in multicomponent crystals.
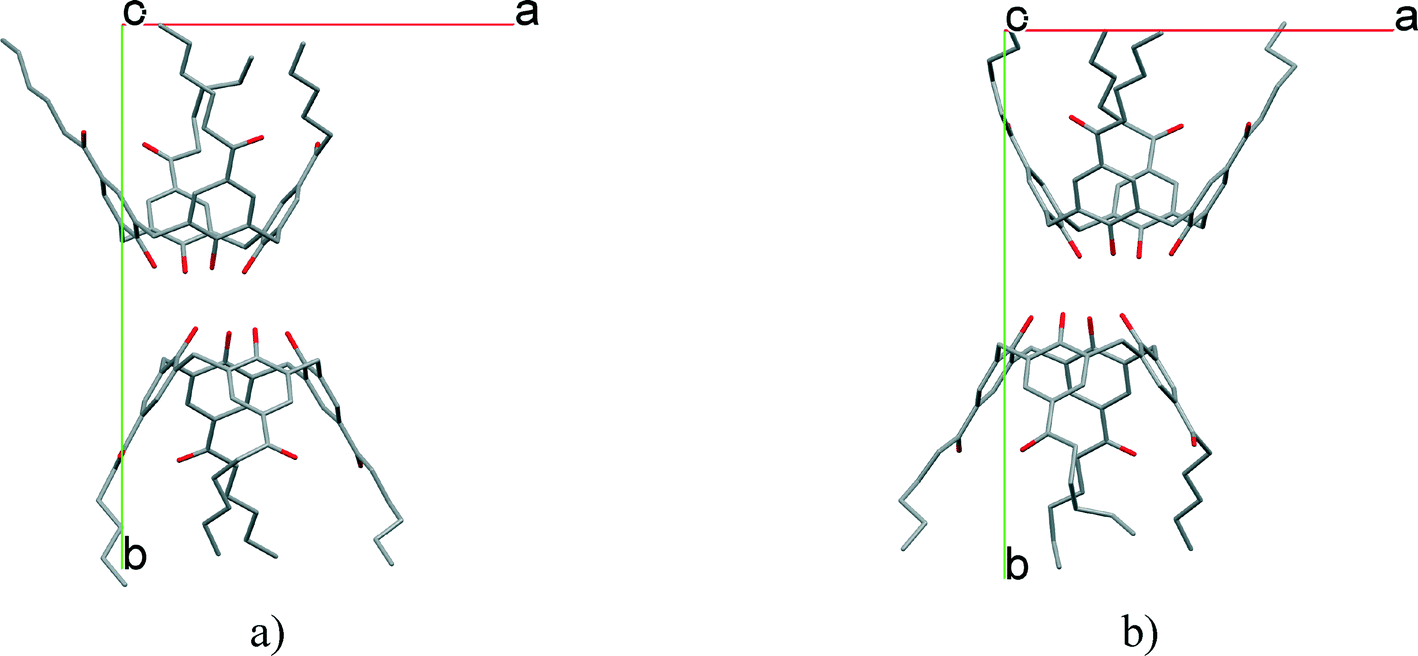 | ||
| Fig. 4 The position of the calix[4]arene host molecules in the unit cells of the inclusion compounds containing a) menthone and b) menthol guest molecules viewed from the c axis. There are two inclusion complex molecules in the asymmetric unit. The locations of the hosts are related by a virtual, non-crystallographic twofold symmetry thus indicating a morphotropic relationship.16 | ||
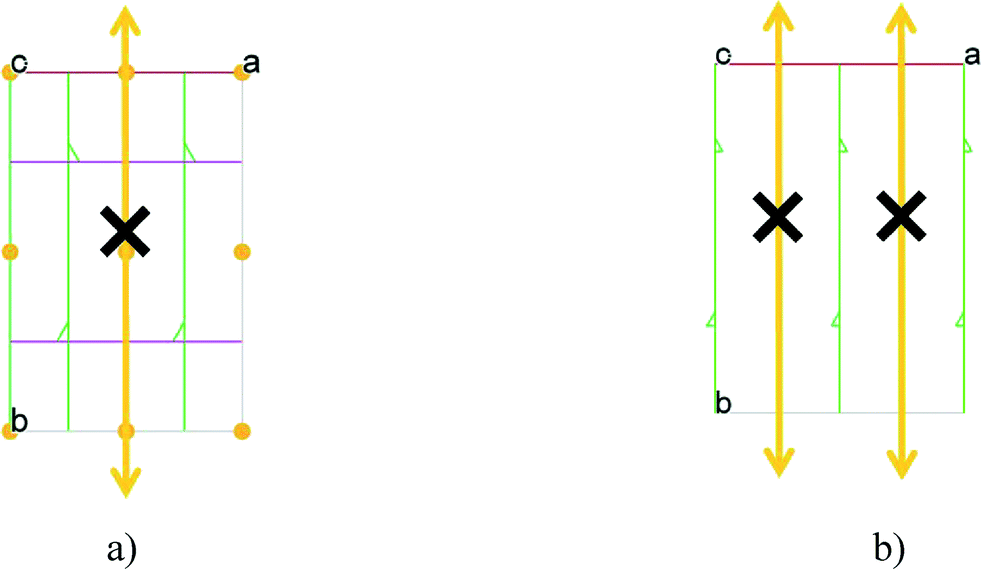 | ||
| Fig. 5 The space groups a) P21/n and b) P21 indicating the existing symmetry elements and the virtual twofold axes (see also Fig. 3 and 4) which organize the morphotropic relation between the morphotropic pairs.12,16 (Adapted from ref. 16.) | ||
Lateral substitution; balanced spatial and electrostatic driving forces
Packing motifs may remain in chemically fine tuned systems, where the crystal systems and the space groups are different. In order to study the effect of bridge modification on the supramolecular properties, series of laterally non-, mono- or disubstituted tetra-tert-butyltetramethoxycalix[4]arene compounds were synthesised (Scheme 1).17–19,22,23 Either one or two bridge substituents on opposite methylene units of the parent tetramethoxycalix[4]arene exercise a distinct influence on the molecular conformation of the host as well as on the supramolecular architecture of the crystal packing.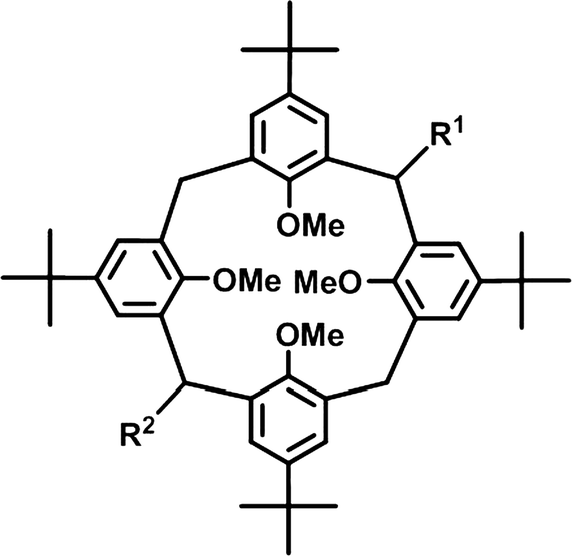 | ||
| Scheme 1 Bridge non- (R1 = H, R2 = H), mono- (R2 = H) and disubstituted calix[4]arenes. The lateral substituents act as spacers. | ||
No substitution
The laterally non-substituted host inclusion compound,22 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetramethoxycalix[4]arene, crystallised with THF has the partial cone conformation (monoclinic, P21/c, host:guest stoichiometry is 2:2) (Fig. 6a). The guest molecules are positioned interstitially in channels bonded by weak C–H⋯π interactions. There are serpentine like channels disposable to the solvent guest molecules in the crystal structure.
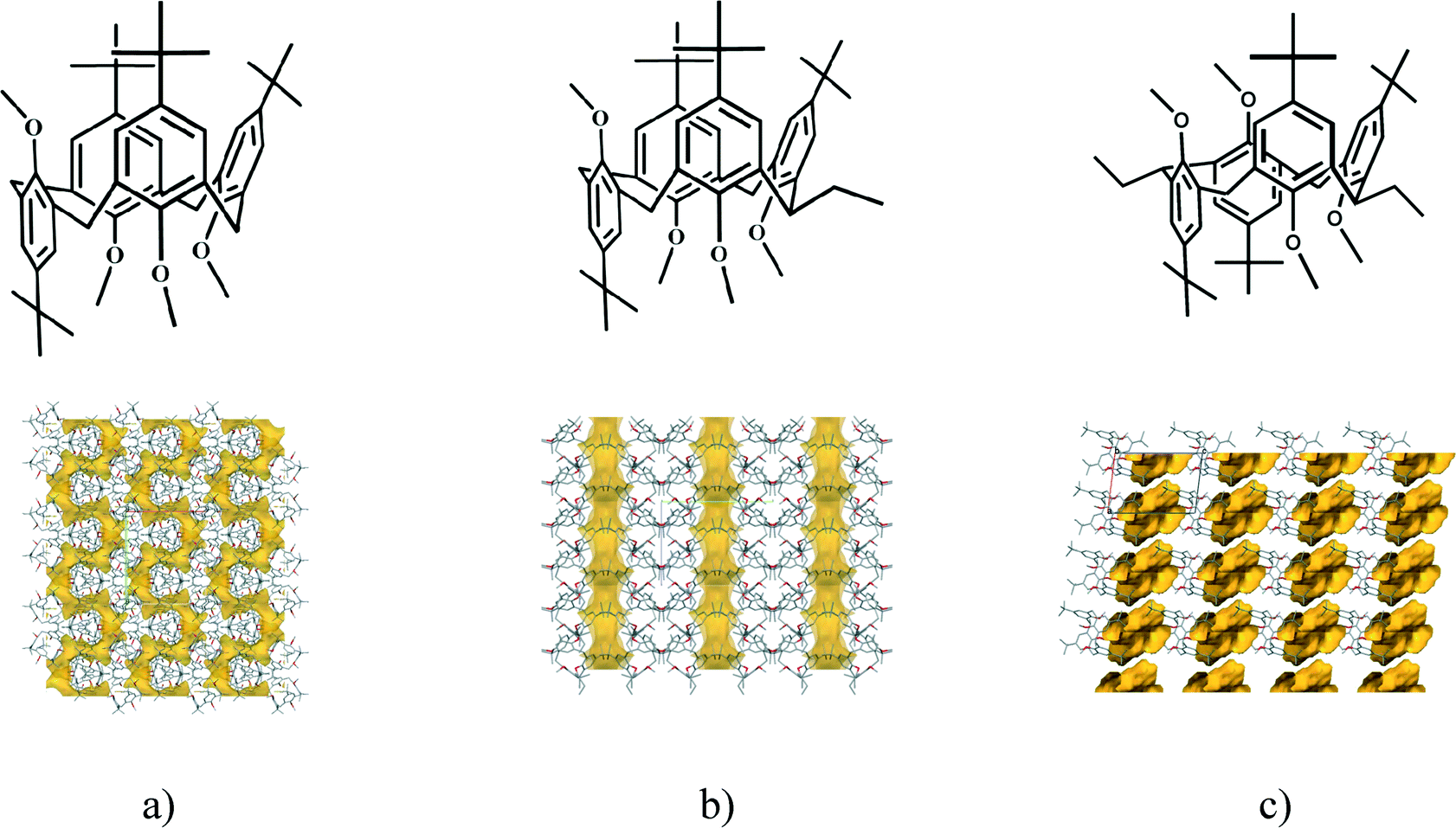 | ||
| Fig. 6 Laterally a) non-22 b) mono-17 or c) disubstituted18,19 tetra-tert-butyltetramethoxycalix[4]arene compounds: formula diagrams and crystal packing arrangements showing the serpentine like channels, the straightened channels and the individual voids, respectively. (With kind permission from Springer Science and Business Media17 (packing diagrams of Fig. 6a and b).) | ||
Monosubstitution
The calixarene molecules in the laterally monosubstituted structures17,23 all adopt the partial cone conformation. It has already been calculated to be the lowest energy conformer.23 All methoxy groups point out of the cavity. The bridge mono-ethyl substituted 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetramethoxycalix[4]arene crystallizes in the monoclinic crystal system, space group P21/n, without inclusion of any molecule.17 The presence of different solvent molecules17 induces the formation of the higher symmetry orthorhombic structures of the laterally substituted calix[4]arene (Pca21, host:guest stoichiometry is 1:1), being isostructural irrespective of the included guest molecule (THF, CHCl3 and CH2Cl2). The guest molecules are always positioned interstitially in channels bonded by weak C–H⋯π interactions in the vicinity of the aromatic ring neighbouring the lateral ethyl substituent. The introduction of a lateral substituent to the host decreases the packing ability of the inclusion crystals, it may expand the total space available for guest molecule inclusion by nearly 30%. Due to the consequence of bridge monosubstitution, the serpentine like channels observed in the laterally unsubstituted calixarene structure are straightened to linear channels (Fig. 6b). It looks like the crystal is breathing as the effect of chemical modification of the calixarene host at the lateral position. Being increasingly in possession of supramolecular chemical information, proper crystal engineering has nearly been achieved: chemical tools were applied to change the crystal structure – the serpentine like channels of a calixarene crystal were straightened.
Disubstitution
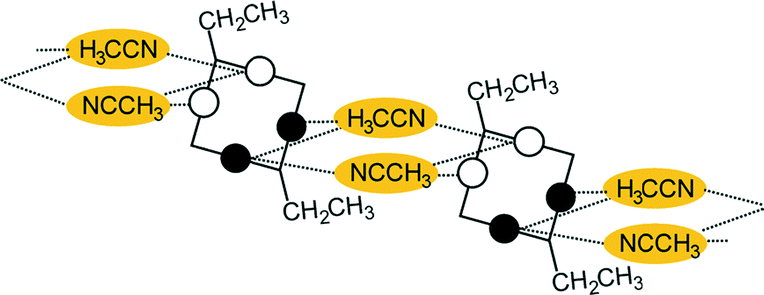
A conformational change upon attachment of a second lateral substituent to the opposite methylene bridge site in the diametrically bridge-disubstituted calix[4]arenes transforms the molecular geometry from partial cone to 1,2-alternate,18,19 resulting in dense packing with highly reduced solvent accessible voids (Fig. 6c). Different types of packing arrangement occur in the acetonitrile solvates of the diversely bridge-disubstituted 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetramethoxycalix[4]arenes (Table 2). In all the examined structures, the lateral substituents act as spacers. The main packing motif consists of molecular chains in each structure along the axis of the bridge substitution irrespective of the varying crystal systems and space groups. Perfect centrosymmetry can only be realized if the two bridge substituents are the same. The bridge substitution clearly influences the packing arrangement and the inclusion behaviour of the host molecules, which can be fine-tuned very sensitively by size and functionality of the substituents (Table 2). The chain formation is the determining packing motif irrespective of the chemical functionality of the lateral substituents. The chain is assembled even if the lateral substituent is a nonpolar alkyl chain or a polar carboxyl group. If the size limit is exceeded, the bridge substituents are forced to participate in the supramolecular bonding system. The increase in the sterical demand of the bridge substituents results in a more efficient utilisation of the crystal volume and a gradual reduction of residual empty voids of the lattice. The size of the residual voids as well as the total potential solvent accessible volume are well correlated with the packing coefficients. The C–H⋯π interactions change systematically with the increasing size of the lateral substituents contributing to the stability of the crystal structures. In this bridge disubstituted calixarene system, both spatial and electrostatic forces are finely balanced. The mastering of the molecular packing arrangement via the secondary interactions could be understood as a kind of “synthon engineering”.| No | R1, R2 | Space group | H:G stoichiometry |
Motif |
|---|---|---|---|---|
| 1 | R1 = R2 = CH3 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
0.5:2 |
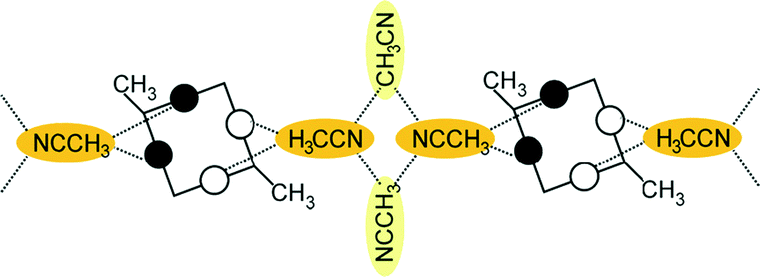
|
| 2, 6 | R1 = CH2CH3 | P21 | 1:2 |
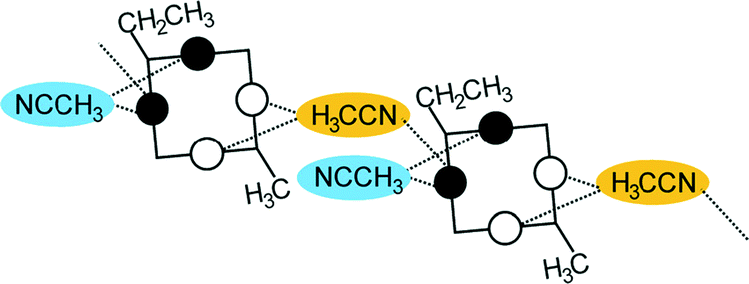
|
| R2 = CH3 | ||||
| R1 = CH2CH3 | ||||
R2 = CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
||||
| 3, 4 | R1 = R2 = CH2CH3; | P21/n | 0.5:1 |
|
| R1 = R2 = CH2CHCH2 |
||||
| 5 | R1 = CH2CH3 | P1 | No guest |
|
| R2 = CH2CHCH2 |
||||
| 7 | R1 = R2 = CH2Ph | P21/c | 0.5:1 |
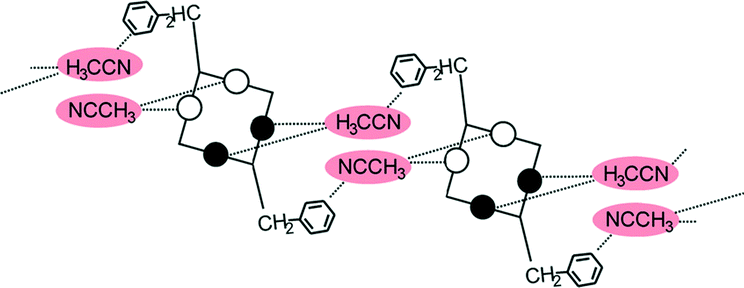
|
| 8 | R1 = CH2CH3 | Pc | 1:3 |
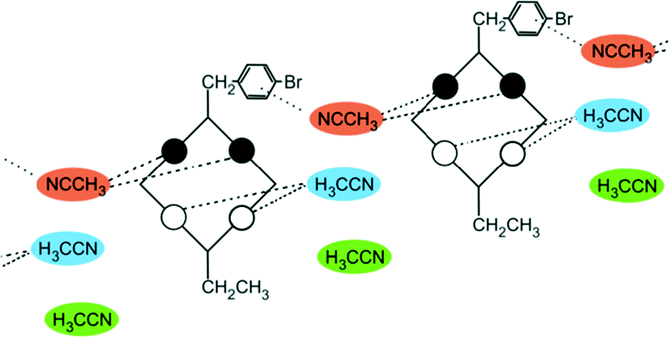
|
| R2 = CH2PhBr | ||||
| 9 | R1 = CH3 and R2 = COOH | Pc | 1:2:2 |
|
Conclusion
The fine tuning of the crystal architecture, e.g. structural arrangement and intermolecular interactions, is described using calix[4]arenes consisting of substituents and guest molecules of different sizes, shapes and chemical composition. The fine tuning requires the recognition, understanding and application of intermolecular interactions, crystallographic and in case of occurrence, non-crystallographic symmetries. The morphotropy is the link between isostructurality and polymorphy, where structural motifs remain but they are related by non-crystallographic symmetries. Morphotropy can be induced by supramolecular effects. There are calixarene crystal structures where either the spatial or the electrostatic effects have the determining role in the self-assembly of the molecules. The lateral substitution of calix[4]arenes affects the packing arrangement. In the case of bridge monosubstitution, the serpentine like channels observed in the laterally unsubstituted calixarene structure are straightened to linear channels expanding the total space available for the guest molecules. With this transformation of the crystal packing arrangements, a genuine crystal engineering effort comes true. The lateral disubstitution of the calix[4]arenes with gradual change of the size and functionality of the substituents resulted in a very sensitively spatially and electrostatically fine-tuned system. It shows that the supramolecular synthon can be engineered. Therefore, we suggest the term “synthon engineering” for the directed manipulation of the molecular packing architecture including the system of secondary interactions caused by the respectively tuned synthons.Acknowledgements
P. B. and A. K. acknowledge the support from the National Scientific Research Foundation (OTKA K-100801).References
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- G. R. Desiraju, Nature, 2001, 412, 397–400 CrossRef CAS PubMed.
- A. I. Kitaigorodskii, Organic Chemical Crystallography, Consultants Bureau, New York, 1961, pp. 106–110 Search PubMed.
- A. Kálmán, L. Párkányi and G. Argay, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 1039–1049 CrossRef.
- A. Kálmán and L. Párkányi, Isostructurality of Organic Crystals in Advances in Molecular Structure Research, ed. M. Hargittai and I. Hargittai, JAI Press Inc., Greenwich, Conn., 1997, vol. 3, pp. 189–226 Search PubMed.
- A. Kálmán, in Fundamental Principles of Molecular Modeling, ed. Gans, W., Plenum Press, New York, 1996, p. 209 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- L. Fábián and A. Kálmán, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 1099–1108 CrossRef PubMed.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2005, 7, 324–336 RSC.
- A. Kálmán, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 536–547 CrossRef PubMed.
- A. Kálmán and L. Fábián, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 411–417 CrossRef PubMed.
- T. Gruber, E. Weber, W. Seichter, P. Bombicz and I. Csöregh, Supramol. Chem., 2006, 18, 537–547 CrossRef CAS.
- T. Gruber, M. Peukert, D. Schindler, W. Seichter, E. Weber and P. Bombicz, J. Inclusion Phenom. Macrocyclic Chem., 2008, 62, 311–324 CrossRef CAS.
- T. Gruber, P. Bombicz, W. Seichter and E. Weber, J. Struct. Chem., 2009, 50, 522–531 CrossRef CAS PubMed.
- T. Gruber, M. Gruner, C. Fischer, W. Seichter, P. Bombicz and E. Weber, New J. Chem., 2010, 34, 250–259 RSC.
- T. Gruber, C. Fischer, W. Seichter, P. Bombicz and E. Weber, CrystEngComm, 2011, 13, 1422–1431 RSC.
- C. Fischer, G. Lin, P. Bombicz, W. Seichter and E. Weber, Struct. Chem., 2011, 22, 433–439 CrossRef CAS.
- C. Fischer, P. Bombicz, W. Seichter, F. Katzsch and E. Weber, Cryst. Growth Des., 2012, 12, 2445–2454 CAS.
- C. Fischer, P. Bombicz, W. Seichter and E. Weber, Struct. Chem., 2013, 24, 535–541 CrossRef CAS PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- C. Fischer, T. Gruber, W. Seichter and E. Weber, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o4572–4573 CAS.
- M. Gruner, C. Fischer, T. Gruber and E. Weber, Supramol. Chem., 2010, 22, 256–266 CrossRef CAS.
Footnote |
| † Present address: Department of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, AB T2N 1N4, Canada. |
| This journal is © The Royal Society of Chemistry 2014 |